

Abstract
The restriction endonuclease fold (a 3-layer α-β sandwich containing variations of the PD-(D/E)XK nuclease motif) has been greatly diversified during evolution, facilitating its use for many biological functions. Here we characterize DNA binding and cleavage by the PD-(D/E)XK homing endonuclease I-Ssp6803I. Unlike most restriction endonucleases harboring the same core fold, the specificity profile of this enzyme extends over a long (17 basepair) target site. The DNA binding and cleavage specificity profiles of this enzyme were independently determined, and found to be highly correlated. However, the DNA target sequence contains several positions where binding and cleavage activities are not tightly coupled: individual DNA basepair substitutions at those positions that significantly decrease cleavage activity have minor effects on binding affinity. These changes in the DNA target sequence appear to correspond to substitutions that uniquely increase the free energy change from the ground-state and transition state, rather than simply decreasing the overall DNA binding affinity. The specificity of the enzyme reflects constraints on its host gene and limitations imposed by the enzyme’s quaternary structure, and illustrate the highly diverse repertoire of DNA recognition specificities that can be adopted by the related folds surrounding the PD-(D/E)XK nuclease motif.
Introduction
Homing endonucleases are highly specific endonucleases that are encoded as open reading frames within microbial intervening sequences (either introns, which are spliced post-transcriptionally from the flanking RNA products encoded by their host genes, or inteins, which are spliced post-translationally from their host gene’s protein product). Homing endonucleases promote the genetic mobility of the intron or intein (and their own coding sequences) by generating double strand breaks (DSBs) in homologous alleles that lack the intervening sequence. These lesions are repaired by homologous recombination, using the intron- or intein-containing allele as a template, which leads to transfer of the intervening sequence to the new host gene 1–4. Homing endonucleases are wide spread throughout all branches of life, including phage, bacteria, archaea and eukaryotes 4.
Homing endonucleases associated with group I introns tend to be small proteins of less than 30 kDa per peptide chain, due to constraints imposed by their surrounding intron or intein sequences, which must fold into structures capable of splicing. Despite their small size, these proteins recognize long DNA target (14 to 40 base pairs), which reduces their toxicity towards the host organism. However, within these long targets homing endonucleases tolerate sequence variation at individual base pairs; this attenuated fidelity enables them to adapt to sequence polymorphisms in their target and increases their potential for horizontal transfer and further spread.
The majority of characterized homing endonucleases have evolved from protein scaffolds containing one of three catalytic sites and surrounding protein folds, termed the ‘LAGLIDADG’, ‘HNH’ and ‘GIY-YIG’ motifs 4; 5. The LAGLIDADG endonucleases are the most prevalent family in eukaryotes and archaea. This endonuclease family features a highly compact structure with an extended DNA binding surface formed by concave anti-parallel β strands. Two closely juxtaposed active sites located near the domain or subunit interface of the enzyme, leading to cleavage at the center of the target site 6. In contrast, proteins containing the HNH or GIY-YIG motifs consitute the majority of homing endonucleases in phage. These proteins typically exhibit bipartite domain organizations in which their small, relatively nonspecific catalytic domains are tethered to separate DNA binding domains 7; 8. As well, the His-Cys box homing endonucleases (usually found in protists, and typified by the endonuclease I-PpoI) also contain the HNH active site, but in a more compact homodimeric scaffold 9. Regardless of their origin and genomic location, all of these protein families are extremely well-adapted to recognition of long DNA target sites.
More recently, analyses of the intron-encoded protein ‘I-Ssp6803I’ (also called ‘I-SspI’ for simplicity) have demonstrated that certain group I introns found in cyanobacteria harbor endonucleases that display an additional structural fold and catalytic motif 10; 11. Encoded by an intron found within the fMet-tRNA gene in Synechocystis (Figure 1a), I-Ssp6803I displays the same functional properties observed in other homing endonucleases. It is a small protein (150 residues) that recognizes a long DNA target site (23 bp) 11 (Figure 1b and 1c). However, I-Ssp6803I contains a core ‘PD-(D/E)-XK’ catalytic motif and surrounding protein fold that is most typically found in enzymes that act at short DNA targets with strict fidelity, including most known restriction endonucleases, many Holliday junction resolvases, and a wide variety of DNA repair enzymes 12; 13.
Figure 1. I-Ssp6803I and its target site.

(a) The secondary structure of tRNAfMet of cyanobacteria Synechocystis strain 6803 (adapted from11) is shown with the intron insertion site indicated by an arrow. Bases are numbered by conventional tDNA/tRNA numbering system. The corresponding regions for I-Ssp6803I target site are colored and labeled consistently with the depiction of the DNA target site.
(b) The DNA construct used in this study, labeled according to the symmetry of the DNA to either side of the central A:T basepair. The region of this target and displays significant binding and cleavage specificity in our study is outlined by dashed-box. Small arrows indicate the cleavage sites of I-Ssp6803I on each strand. (c) Quaternary and tertiary structure of DNA-bound I-Ssp6803I, and schematic of contacts made by each region of the protein to its DNA target. Only half of the DNA target is represented. Residues contacting DNA bases or backbone are labeled as follows: across the DNA, contacts in the minor groove are indicated on the left of each base while contacts in the major groove are indicated on their right. For the protein, residues observed making identical contacts in both monomers (A and B) are labeled in black, while residues observed making contacts in individual monomers A and B are labeled in green and blue respectively. Contacts made by protein to DNA are colored as follows: blue lines indicate direct contacts between bases and protein sidechains, blue dashed lines indicate direct contacts between bases and protein mainchains, and red lines indicate non-specific contacts to DNA backbone.
This protein fold (SCOP superfamily 3.72.1) corresponds to a α/β/α sandwich structure containing a five-stranded β-sheet with mixed topology flanked on both faces by an α helix 14; 15. A substructure of this domain, consisting of one α helix surrounded by four β strands, is thought to be the conserved core nuclease fold for this superfamily, corresponding to the most highly conserved structural elements in the structures solved to date of various type II restriction endonucleases containing this motif 14; 15. All known variants of this fold position at least two acidic residues, and often (but not always) at least one additional basic residue in the nuclease active site, forming the ‘PD-(D/E)XK’ motif that catalyzes phosphoryl transfer reactions.
In contrast to homing endonucleases, the specificity profile for restriction endonucleases are usually short, generally consisting of 4 to 8 basepairs positioned either in a single contiguous sequence or interruped by several DNA basepairs that do not contribute to target recognition. Their short target site (which ensures its presence in exogenous DNA, such as a typical phage genome) is combined with strict fidelity of cleavage under physiological conditions, allowing an endonuclease to avoid undesired activity against the host genome when coupled with a methylase of comparable specificity. The specificity profile of a typical restriction endonuclease is made possible by its ability to completely saturate the target site nucleotides with a network of sequence-specific contacts to protein atoms (particularly hydrogen bonds formed between protein side chains and DNA bases within the major groove of the target site). As well, many restriction endonucleases induce significant bends in the DNA target as part of their cognate recognition mechanism, which can further elevate specificity through the indirect influence of DNA sequence on its conformational preferences. Finally, many restriction endonucleases utilize dimeric or tetrameric structures to facilitate cooperativity and/or allosteric communication between neighboring target sites, again as a mechanism to increase specificity of DNA cleavage.
The discovery of I-Ssp6803I has provided the opportunity to study the use of the restriction fold and its PD-(D/E)-XK nuclease motif for recognition of a long DNA target, as well as the ability of such an enzyme to adapt its specificity to the constraints imposed by a tRNA host gene. In this study, we describe the production of catalytically active I-Ssp6803I and the determination of its profiles for binding specificity and cleavage specificity. The enzyme generates transient nicked intermediates during DNA cleavage. Its binding and the cleavage specificity profiles (corresponding to the selectivity of the enzyme at the initial substrate-bound state of the reaction, versus its transition state) are similar, but DNA basepair mutations at several positions in the target site cause significant reductions in cleavage with little effect on binding affinity. The specificity of the enzyme reflects sequence constraints on its host tRNAfMet gene and limitations imposed by the enzyme’s symmetric quaternary structure.
Results
The cleavage activity of wild-type I-Ssp6803I
A series of mutations made in the active site of I-Ssp6803I, one of which (K51M) was made specifically for this study, imply that the enzyme utilizes a metal-dependent mechanism of phosphoryl hydrolysis that is similar to other members of the PD-(D/E)XK nuclease superfamily. Several conserved residues (D8, E11, D36 and Q49 as shown in Supplementary Figure S1) are all properly positioned and/or observed crystallographically to bind divalent metal ions 13; all are essential for catalysis. In addition, a neighboring lysine residue (K51) is located appropriately to interact with a metal-associated water that can carry out an in line attack on the scissile phosphate group; mutation of this residue also blocks enzyme activity.
While mutation of the metal-binding residue Glu 11 into Gln (E11Q) generates a protein construct that displays a 350 nM dissociation constant against the cognate DNA target in the presence of calcium 13, mutation of the lysine 51 residue to methionine (K51M) results in a higher DNA binding affinity, with a lower dissociation constant of approximately 50 nM under the same conditions (Supplementary Figure S2). One simple interpretation of these is that glutamate 11 might facilitate high affinity DNA binding (via the involvement an associated metal ion), while lysine 51 might be involved in proton transfer chemistry during DNA hydrolysis.
I-Ssp6803I displays pattern of DNA digestion in which differential rates of individual strand cleavage can be observed (Figure 2). At low protein concentrations (8 and 80 nM enzyme tetramer, in the presence of 10 nM plasmid DNA substrate) significant amounts of early nicked DNA intermediates are observed, that are eventually resolved into fully cleaved product at later time-points in the reaction. At the most elevated concentration of enzyme used in this study (800 nM enzyme tetramer, again in the presence of 10 nM plasmid DNA) the appearance of nicked intermediates during the timecourse of the reaction is reduced at the timepoints collected and monitored during the digest.
Figure 2. Cleavage activity of wild-type I-Ssp6803I.
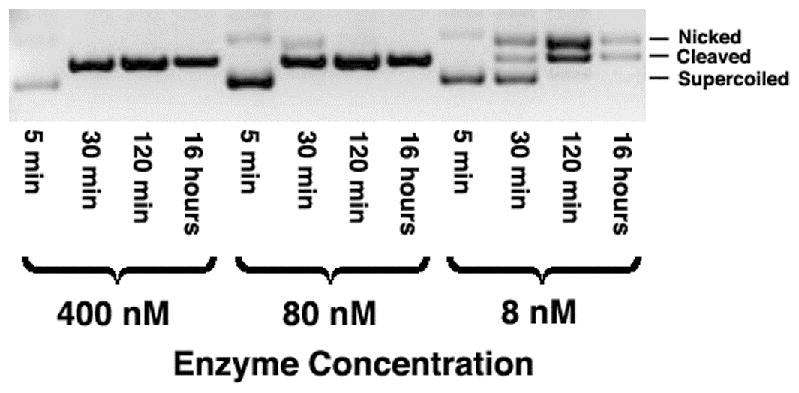
(a) Time courses of wild-type target digestions, using 10 nM DNA plasmid substrates inclubated with variable enzyme concentrations. At low enzyme concentrations, a nicked intermediate accumulates, indicating preferential cleavage of one DNA target strand. (b) Cleavage time-courses (10 nM DNA and 8 nM enzyme) of the wild-type DNA target (middle) and two related substrates, each consisting of a pseudo-palindrome containing an inverted repeat of the left half-site (‘PalL’) or the right half-site (‘PalR’). The PalR target is cleaved more readily than PalL (in agreement with quantitative binding assays that demonstrate tightest binding to the PalR target).
The specificity profiles of I-Ssp6803I
Overview
The sequence specificity of DNA binding and DNA cleavage by I-Ssp6803I were independently measured using an identical series of target site variants, systematically mutated at each position (Supplementary Table S1). For the cleavage specificity assay, these target sites were all cloned into the Invitrogen pTopo2.1 plasmid vector (Invitrogen) and subjected to parallel enzyme digests. Reaction conditions were chosen based on the results of wild-type cleavage assays described above, that would result in approximately 50% linearization of substrate when using an enzyme concentration (700 nM) at least 15-fold above the dissociation constant for the wild-type interaction (assuming that the wild-type KD is no greater than the 50 nM KD measured for K51M). In the analogous determination of binding specificity, a parallelized fluorescence competion assay was devised in which the ability of each target site variant to compete against labeled wild-type target for protein binding was measured, using immobilized his-tagged protein (Figure 3). By direct comparison of these two assays, we hoped to determine the specificity profile of a PD-(D/E)-XK homing endonuclease, and the identify of those positions across the target site at which cleavage activity and binding affinity are closely coupled (or conversely, positions in the target site where binding and cleavage are separable).
Figure 3. High-throughput fluorescence competition-binding assay.
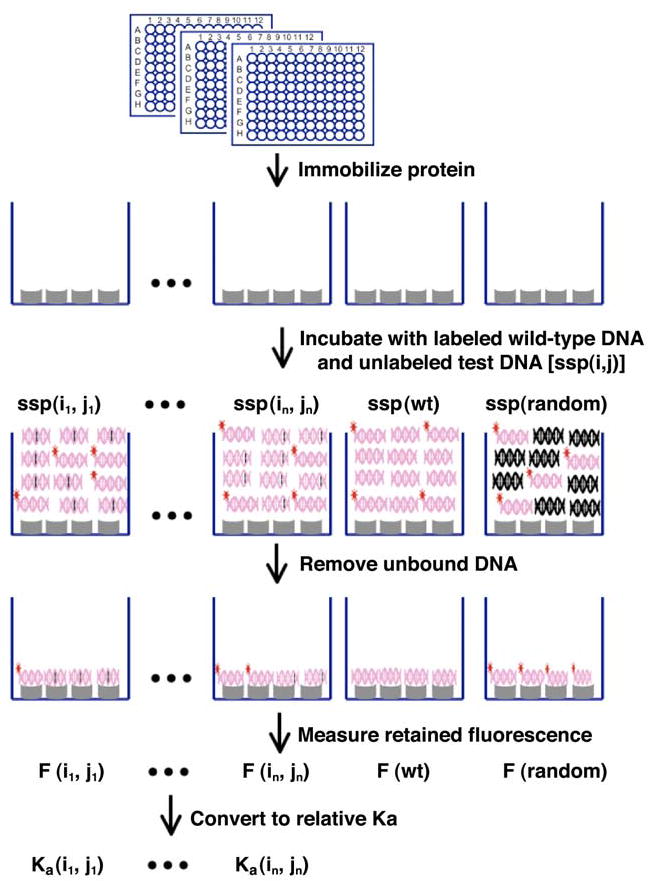
Histidine-tagged protein is immobilized onto nickel coated 96 well micro-plates. A mixture of fluorescently labeled DNA duplex containing the wild-type target site, and one of the single-basepair substitution variants is incubated with the protein, allowing determination of the abilityof each sequence to compete for binding against the wild-type DNA target. In order to calcuate relative affinities (Ka values), the experiment is also conducted using unlabeled wild-type DNA target and a nonspecific, random DNA sequence. Details of this method are provided in the Results section and in Supplementary Figures S1 and S2.
Cleavage Specificity
DNA cleavage specificity extends over a 17 basepair region of the target sequence, corresponding to positions −8 to +8, and is most pronounced at positions +/− 4, 5 and 6 (Figure 4a). The endonuclease is there quite specific with an estimated frequency of cleavage of approximately 1 in 3×10−6 random DNA sequences. This estimate corresponds to the number of potential target site variants that could possibly contain one or more substitutions that individually allow greater than 20% cleavage (4.7 × 104), divided by the total number of possible unique sequences of the same length (417 = 1.7 × 1010). This number is almost certainly an underrepresentation of the enzyme’s actual cleavage specificity, as it assumes that multiple basepair substitutions in the target site that each slightly reduce cleavage activity can be uniformly tolerated by the endonuclease in any number and combination.
Figure 4. Cleavage and binding specificity profiles of I-Ssp6803I.
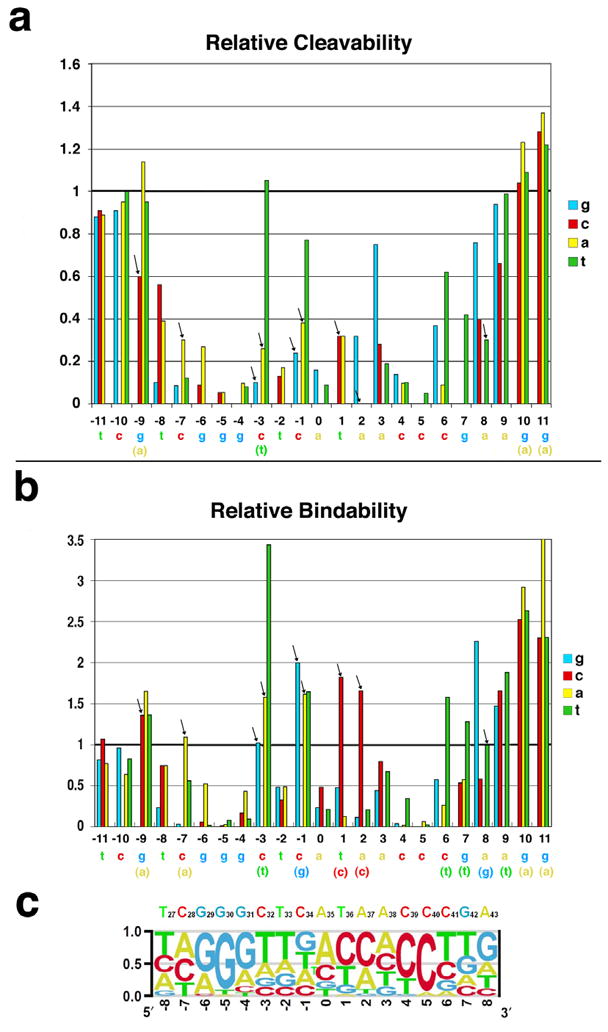
The wild-type target site of I-Ssp6803I is shown below both specificity profiles. Bases shown in parentheses below those targets correspond to those basepair substitutions that are observed to increase either cleavability or binding affinity. Arrows denote basepair substitutions that significantly decrease cleavage, while causing no apparent decrease (or an increase) in substrate binding affinity. Note that (1) the cleavage specificity profile, while similar to the binding specificity profile, is more tightly constrained to the physiological target site of the endonuclease, and (2) the majority (6 of 11) of the basepair substitutions indicated by arrows, display 50% or greater increase in rKa. (a) The relative cleavage of target sites harboring single basepair substitutions at each position of the substrate. The cleavability of the wild-type target is normalized to ‘1’. The digests were run under conditions where the wild-type substrate is cleaved to approximately 50% products, in order to visualize any substrates that are cleaved more efficiently under similar conditions. (b) Relative association constants (rKa(i,j)) for the same target site variants as those shown in panel a above. (c) Specificity probability plot for I-Ssp6803I. The function used to calculate the relative probability of each base at each position (BP(i,j)) is described in methods and in Supplementary Figure S2.
The enzyme’s cleavage specificity profile is very well optimized to its physiological wild-type target sequence. Only five basepair substitutions (−3C to T, −1C to T, +3A to G, +6C to T and +8A to G) out of 51 that are possible within this region allow greater than 50% cleavability as compared to the wild-type target sequence, and only one (−3C to T) is cleaved as well as the wild-type site. Of these substitutions, three (at positions −3, +3 and +8) increase the overall symmetry of the target site by converting a basepair to the same identity as its related position in the opposite half-site. The greatest cleavage specificity is observed at positions +/− 5, where all three possible basepair substitutions reduce cleavability to less than 10% of the wild-type substrate.
Binding specificity
Wild-type I-Ssp6803I (‘I-Ssp6803I’) is extremely toxic to E. coli, preventing its expression as a his-tagged protein and requiring us to examine the DNA binding specificity profile of catalytically inactive point mutants using a high-throughput, competitive binding assay as described in methods and in Figure 3 and Supplementary Figures S3 and S4. Therefore, two separate mutant enzyme constructs, both catalytically inactive, were used in this study in order to avoid generalizations about the binding specificity profile that might be caused by unique effects of a mutation relative to the wild-type enzyme scaffold. The first construct, E11Q, alters an acidic active site residue thought to be required for divalent metal binding 13. The second mutant (K51M) removes a basic amino acid from the active site. The binding specificity of the tighter-binding construct (K51M) is described below and in the accompanying figures, while binding specificity of E11Q (which was found to correspond closely to K51M) is provided in Supplementary Figure S5.
The raw binding data using the DNA target site matrix, consisting of measured fluorescent intensities ‘F’ corresponding to the retention of bound labeled wild-type DNA probe, was internally consistent and reproducible, both in separate experiments using different protein preparations, and when assayed with the two different point mutants of the endonuclease. Three typical results are observed at various positions in the substitution matrix (Supplementary Figure S3). I-Ssp6803I tolerates base substitutions at several positions (such as basepair −11) very well, with little or no difference between F(wt) and F(i,j). At many positions (most significantly, at basepairs +/−5), incorporation of mismatches is poorly tolerated, leading to F(i,j) ≫F(wt) (indicating that the altered DNA sequences fail to compete with the wild-type target). Finally, I-Ssp6803I occasionally binds more strongly to sequences harboring specific basepair substitutions (for example, at position −3), resulting in F(i,j)<F(wt).
The raw data was converted to a matrix of relative binding affinities to individual DNA targets harboring single-base substitutions (rKa(i,j)) as described in methods. The relative binding affinities are plotted in Figure 4b and listed in Supplementary Table S2. The binding specificity profile of I-Ssp6803I resembles its cleavage profile, but is not as tightly constrained to the wild-type target sequence. Significant binding specificity again extends over a 17 basepair sequence (positions −8 to +8), and is strongest at positions +/− 4, 5 and 6. Similar to the cleavage specificity of the enzyme, basepair substitutions at positions +/− 5 uniformly cause the largest decrease in binding affinity. The estimated overall binding specificity is somewhat lower than for cleavage, corresponding to near wild-type affinity for approximately 1 in 2×10−4 random DNA sequences. This estimate corresponds to the number of possible target site variants that could contain one or more substitutions that individually display greater than 50% of the wild-type Ka (3 × 106), again divided by the total number of possible unique 17 basepair sequences (417 = 1.7 × 1010).
Almost half of the seventeen positions that display significant binding specificity also exhibit at least one basepair substitution that increases protein binding affinity. Of these, one substitution (−3C to T) gives the most significant increase in binding affinity; this is the only such sequence change that also increases cleavability of the DNA target. This substitution converts the basepair at position −3 to the same identity as its counterpart in the right half-site.
DISCUSSION
DNA cleavage by catalytically active I-Ssp6803I
This manuscript describes the production of catalytically active I-Ssp6803I, as a prerequisite for determination of the enzyme’s cleavage specificity, using a method where the active site of the enzyme is initially blocked by expression of a fusion construct consisting of a large multi-domain protein (chitin binding protein and a self-cleaving intein) at the N-terminal end of the endonuclease. Subsequent release of the fusion partner after initial purification results in liberation of free active endonuclease. The solution behavior and thermal stability of the final construct was compared to previous mutants (including E11Q and K51M described in this study) by dynamic light scattering, size exclusion chromatography and circular dichroism thermal denaturation and found to be unchanged (data not shown). The active enzyme was used in initial digests with wild-type DNA target (in a plasmid backbone) in order to confirm its activity, and to determine reaction conditions appropriate for subsequent measurements of cleavage specificity (corresponding to approximately 50% cleavage with a concentration of enzyme at least 10-fold greater than the expected Kd for the wild-type substrate).
The enzyme displays substantial generation of transient nicked DNA intermediates when digests are conducted at a concentration (7 nM nM enzyme tetramer) that is near or below the expected KD for the protein-DNA interaction; this condition corresponds to an approximately molar equivalence of the protein relative to the plasmid substrate concentration (10 nM). At higher enzyme concentrations, this behavior is significantly reduced at the time points monitored in our digests (although it is possible that at elevated enzyme concentrations, the same nicked species are transiently produced at earlier time points).
There are two possible explanations for this behavior: either the enzyme’s two active sites (each located in an individual protein subunit) display asymmetric catalytic behavior in the bound DNA complex (which might be promoted by the asymmetry of the DNA target site across its central three basepairs) or the conformation, quaternary structure or solution behavior of the enzyme is destabilized or otherwise perturbed at low enzyme concentrations.
Specificity of binding recognition versus specificity of cleavage
The overall profiles of binding and cleavage specificity displayed by I-Ssp6803I are well correlated, with 11 out of 17 positions in the target site, and 42 out of 51 possible DNA basepair substitutions, displaying similar effects on both activities (Figure 4). In particular, six positions across the target site (−2, +/− 4, +/−5 and 0) exhibit substantial and uniform decreases in the substrate’s binding affinity and its cleavability as a result of all possible basepair substitutions. This indicates that all mutations at these positions have a significant effect on ground state binding energy that leads directly to poor substrate cleavage. In addition, five additional positions in the target site (−8, −6, +3, +6 and +7) display less severe, but still well correlated, reductions in binding affinity and cleavage activity as a result of at least one basepair subsitution at those positions.
At the remaining six positions in the target site (−7, −3, −1, 1, 2 and +8) at least one basepair substitution is observed to cause a substantial reduction in cleavage, without a significant decrease in binding affinity (Figure 4; these basepair substitutions are denoted with arrows). This observation, combined with the correlation between binding affinity and cleavage at other positions, leads to two conclusions. First, high affinity binding is necessary, but not completely sufficient for catalysis of DNA hydrolysis. Second, an additional component of the overall specificity of the enzyme, realized at several basepair positions during the actual hydrolysis event, corresponds to the enzyme’s reduced ability to stabilize the transition state.
Base-pair substitutions within the enzyme’s cognate DNA target (for example, +2A to C) that uniquely increase the free energy of the enzyme-bound transition state could be caused by at least three factors: (1) by increasing the energy required to distort the DNA backbone around the scissile phosphate groups into the transition state conformation (a state which corresponds to a highly polarized, pentacoordinate geometry for both scissile phosphates required for hydrolysis); (2) by causing a misalignment of accompanying reactive groups (side chains, metals and/or solvent) at the reaction center; or (3) by causing a significant unfavorable change in the overall entropy of the system at the reaction transition state (corresponding to a negative value for ΔΔS‡). In addition, substitutions that uniquely stabilize the ground state only, without a corresponding reduction in the transition state energy, would also result in a decrease in the catalytic efficiency of the enzyme reaction, in that case with a corresponding increase in substrate binding affinity.
In I-Ssp6803I, as in all the structures of homing endonucleases in complex with DNA that have been visualized to date, the phosphate backbone and DNA duplex are significantly distorted near the sites of cleavage 16; 17. Given the amount of bending of the DNA substrate induced by these enzymes in order to produce a catalytically complex, it is not surprising that the formation of an activated reaction complex at the moment of DNA strand cleavage is a measurable component of sequence discrimination by the enzyme. As well, examination of the effect of target site substitutions on cleavage versus binding (Figure 4) indicates that the majority of mutations that decrease only the cleavability of the substrate (indicated by arrows in that figure) actually increase target site binding affinity, in agreement with the general principle of enzymatic catalysis that transition state stabilization, coupled with ground state destabilization, is essential for overall catalytic efficiency.
The position-specific base preferences of I-Ssp6803I
A position-specific probability matrix was calculated from positions −8 to +8 of the target site, based on the binding and cleavage specificities of I-Ssp6803I, in order to generate a more intuitive visualization of base preference at each position and their relationship to the corresponding bases of the tRNAfMet product (Figure 4c). Overall, from positions +/− 3 to 8 in each half-site of the DNA target (corresponding to the base-paired region of the tRNA anticodon stem-loop), I-Ssp6803I displays a palindromic recognition preference, even at positions where double or triple degeneracy are tolerated. This is especially pronounced at position −3, where I-Ssp6803I seems to strongly disfavor the wild type base at position −3.
The central five basepairs of the target site, which correspond to the unpaired bases of the tRNA anticodon loop, display more asymmetric and promiscuous recognition patterns of binding and cleavage. Positions −2 and 0 strongly favor the wild type base identity both in binding and in cleavage, while the enzyme binds efficiently to all three possible mismatches at position −1 (and cleaves one, −1C to T, with almost wild-type efficiency). In contrast, at positions +1 and +2 the enzyme can bind strongly to at least one alternate basepair, albeit with reduced cleavage activity. The reduced symmetry of target site specificity at these central positions agrees with the crystal structure of the endonuclease/DNA complex. Unlike the rest of the the target site (basepairs +/− 3 to 8, which are recognized by symmetrical protein elements through interactions within the major groove; Figure 1c), the central five basepairs are contacted by non-specific contacts of protein main chain atoms from long surface loops, that are arranged asymmetrically at the dimer interface, and protein side chains that make additional contacts within the DNA minor groove.
Correlation of the recognition specificity profile of I-Ssp6803I with constraints from its host target gene
The tRNAfMet gene is the most conserved tRNA gene from all three biological kingdoms. Comparison of 30 bacterial genomes indicates that 58% of the basepairs are absolutely conserved in tRNAfMet gene, while elongator tRNA genes display much less conservation (35% identity across the elongator tRNAMetgene, and 22% across the elongator tRNAIlegene) (Figure 5a).
Figure 5. Conservation of anticodon coding regions in bacterial tRNAs.
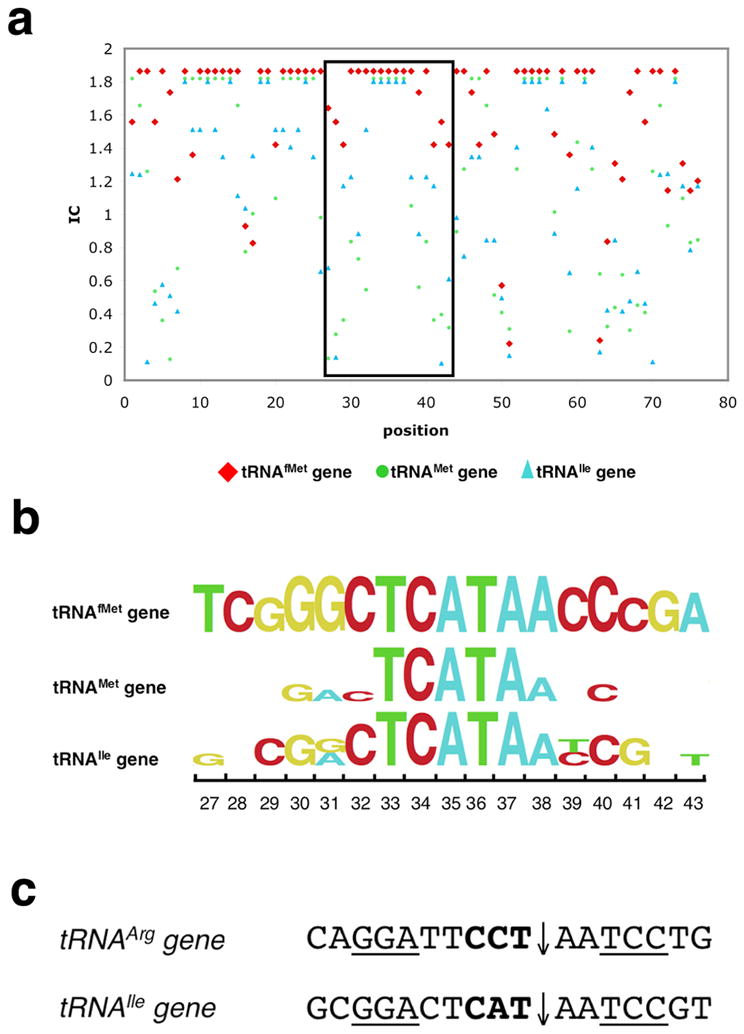
(a) The conservation of tRNA genes from 30 bacterial genomes. X-axis is the position along the tRNA gene and Y-axis is the corresponding level of sequence conservation expressed as ‘information content’ (IC) at that position. IC is generated by program enoLOGOS (http://biodev.hgen.pitt.edu/cgi-bin/enologos/enologos.cgi).
(b) The detail of the conservation of base 27 to 43 (boxed area in panel a) for tRNAfMet, tRNAMet and tRNAIle genes are shown in a logo plot. Besides the center highly conserved region (corresponding to the tRNA anticodon loop), the flanking sequence of tRNAfMet shows higher degree of conservation compared to the other two tRNA genes (indicated by positions with no labeled base preference, indicating that there is no conservation of RNA base identify at those positions.
(c) Fragments (base 27 to 43) of tRNAArg genes (from α-purple bacterium Agrobacterium tumefaciens, Agrobacterium halopraeferens; Agrobacterium marginale) and tRNAIle gene(Azoarcus sp. BH72) that contain group I introns. Intron insertion sites are indicated by arrows and anticodon region are shown in bold. The regions encoding anti-codon stem are underlined. Cleavage site and sequence characteristics are similar with those of target site of I-Ssp6803I, which might be responsible for the accidental breakage and intron insertion by a homolog of I-Ssp6803. The homing endonuclease genes were subsequently lost after fixation the intron genes in the population.
The target site of I-Ssp6803I is the most conserved regions of the tRNAfMet gene, corresponding to bases 27 to 43, which corresponds to the entire anticodon stem-loop of tRNAfMet (Figure 1a). The unpaired anticodon loop, containing base 33 to 37, including the actual anticodon sequence 5′-CAT-3′ (bases 34–36) is absolutely conserved in all genomes (Figure 5b). Bases 29 to 31(GGG, occasionally AGG), paired with bases 39 to 41 (CCC or CCT) form the lower part of the anticodon stem (Figure 1). These three basepairs are directly involved in tRNAfMet binding to the ribosomal P site and initiating transcription 18–21. Since this binding event is unique for initiator tRNA, it is used as one of most unique criterion to distinguish the tRNAfMet gene from other elongator tRNA genes, including the very similar tRNAMet and the tRNAIle genes (which also uses an anticodon CAT sequence to read the Ile codon ATA in bacteria) 22; 23 (Figure 5b).
Comparisons between the sequence conservation of the host target gene and the specificity profile of I-Ssp6803I at each base pair indicates a strong correlation between specificity at individual positions in the target site and constraints on the sequence of the tRNAfMet gene. The most specifically recognized position within the I-Ssp6803I recognition site, as observed both in the binding and the cleavage profiles, is at basepairs +/−5 and the immediately adjacent basepairs +/− 4 and 6) (Figure 4); basepair substitutions at these positions are uniformly not well tolerated. This region coincides with the strongly conserved anti-codon stem region (position 29–31 and 39–41) in target on tRNAfMet gene described above. Such an adaptation of I-Ssp6803I towards regions under strong host gene constraints increases its chance of genetic persistence in its immediate host and of mobilization into additional organisms (because the host gene is less likely to escape recognition by random mutational drift), and also reduces its toxicity by preventing accidental cleavage on closely related tRNA sequences (i.e. on tRNAMet gene and on tRNAIle gene).
However, I-Ssp6803I is not fully optimized against its host target site. At position −3, a “T” is more favored over the wild type “C”, which is one of the highly conserved bases (position 32) in on tRNAfMet gene. This clearly shows that although I-Ssp6803I is evolved under selective pressure to acquire and maintain recognition of a highly conserved sequence in a ubiquitous and essential host gene, its specificity profile is simultaneously constrained by its highly symmetrical tetrameric structure in a manner that is not fully optimal for recognition of the biological target site. The quaternary structure of I-Ssp6803I, while limiting the ability of the enzyme to completely optimize recognition of its target site, is a necessary structural requirement in order for the type II PD-(D/E)XK fold to recognize a long DNA target sequence with the relaxed fidelity required of a homing endonuclease.
Evolution of the restriction fold for use as a homing endonuclease
The ‘restriction fold’ has successfully evolved into many DNA cleavage enzyme systems, including non-specific λ endonuclease, DNA repairing enzymes (Mut H and Vsr endonuclease) and Archaeal Holliday junction resolvases(Hjc). The discovery of restriction conserved common fold in homing endonuclease I-Ssp6803I further broadens the substrate diversity for this fold. However, compared to other protein folds utilized by homing endonucleases, the PD-(D/E)XK motif is surrounded by a relatively bulky protein domain. This is a potential drawback for a homing endonuclease, which generally are under pressure to limit the length of their genes in order to avoid interfering with the folding and splicing of their host introns. To use protein residues economically while still recognizing a long DNA target site, I-Ssp6803I employs a tetrameric quaternary structure (Figure 1c), which positions two DNA binding subunits at distant ends of the target site and creates a elongated DNA binding surface, while the other two subunits stabilize such arrangement13.
There are very few examples of group I introns within tRNA genes; most of those that have been discovered are found to interrupt tRNALeu genes (338 out of 349). It is likely that these introns (or at least a large fraction of them) are descended from a common ancient origin, not through horizontal gene transfer24–27. The remainder of group I introns that interrupt tRNA genes are from bacterial genomes, including tRNAfMet genes (7), tRNAArg genes (3) and tRNAIle genes (1) (Comparative RNA Web site, http://www.rna.ccbb.utexas.edu/, and 10; 24–28). Among these introns, the intron in tRNAfMet gene of cyanobacterial Synechocystis is the only one carrying a homing endonuclease ORF, which is responsible for the mobility and persistence of same intron in a few closely related Synechocystis strains. However, the intron insertion position and intron targeting sequences of the tRNAArg genes and tRNAIle genes both display similarity to the cleavage pattern and recognition profile of I-Ssp6803I (Figure 5c). It is possible that a close homologue of I-Ssp6803I may be responsible for the horizontal intron transfer in these genes, but was subsequently lost after intron fixation in those host genes.
The primary evolutionary advantage for the PD-(D/E)XK nuclease superfamily is its stable and multifunctional core fold, which orients catalytic residues in optimal geometry for a phosphoryl transfer reaction, while providing a highly adaptable surrounding scaffold that can easily accommodate additional structural elaborations. This fold also facilitates a variety of quaternary assemblages, which can be used to create variable cleavage patterns (5′ sticky ends, 3′ sticky ends, or blunt ends), variable types of cooperative DNA binding and cleavage behaviors, and multiple types of DNA binding surfaces capable of recognizing highly diverse DNA substrates. These targets include short DNA targets for restriction endonucleases and mismatch repair enzymes, such as MutH and Vsr, long DNA target for homing endonucleases, and specific tertiary DNA structures for Holliday junction resolvases.
A growing body of evidence indicates that restriction-modification enzyme (R-M) systems have arisen within bacterial genomes via invasive horizontal transfer events. At least one study has demonstrated a restriction endonuclease gene (encoding EcoRI) displays homing-type mobility when placed into the appropriate sequence context 29. Upon residence in a bacterial genome, R-M gene systems act as selfish elements and promote their own survival through two mechanisms: participation in cellular defense (which provides an advantage to the host) and post-segregational host killing (in which loss of the R-M system results in cell death as unmethylated sites in newly replicated DNA are cleaved by residual levels of restriction endonuclease) 30–32.
Given that restriction endonucleases display many of the characteristics of invasive genes and selfish DNA elements, it seems plausible that such gene systems might share distant evolutionary relationships with modern-day microbial invasive elements, such as homing endonucleases, and may even have initially descended from such genes. If this hypothesis is true, examples of catalytic protein folds that transcend the boundaries of modern homing and restriction endonuclease families should be observed. This prediction has been fulfilled in multiple systems. Two separate folds typically associated with homing endonucleases have been predicted in canonical restriction endonucleases 33,34; a recent review of restriction endonuclease genes indicates that up to 40% of putatitive REases contain these catalytic motifs35. Similarly, the homing endonuclease I-Ssp6803I is a tetrameric homing endonuclease built around the PD-(D/E)-XK motif and surrounding restriction fold 13 but with a DNA recognition profile that is unique to the biological and genetic requirements of a mobile intron making its way amongst bacterial genomes.
Materials and Methods
Overview
A catalytically active I-Ssp6803I construct was used to study its cleavage mechanism and corresponding specificity profile. The latter experiments used a panel of 69 substrate variants (each harboring one of three possible basepair substitution at one position in the target site). Subsequently, two separate catalytically inactive point mutants of the enzyme were used to measure the enzyme’s specificity profile of DNA binding, using a corresponding matrix of synthetic double stranded DNA oligos in a high-throughput competitive binding assay in 96-well plate format.
Expression and purification of catalytically active I-Ssp6803I
As described previously 13, the wild-type I-Ssp6803I enzyme is extremely toxic to E. coli and cannot be produced either as an untagged protein or with a short N- or C-terminal affinity tag. In order to produce active enzyme in quantities sufficient for determination of its cleavage specificity, the gene encoding the endonuclease was subcloned into a commercial pTYB expression vector (New England Biolabs) between restriction site SapI and PstI. The resulting vector encodes a 454 amino acid polypeptide chain, with an autocleaving intein domain and chitin-binding protein fused to the N-terminus of the endonuclease. Because the N-terminal end of I-Ssp6803I is involved in DNA interactions, the N-terminal fusion to the intein domain blocks DNA binding, and the protein is relatively inactive until the free endonuclease is liberated in vitro by treatment with DTT.
The protein contains two mutations (F99L and F55K), both quite distant from the DNA-binding surface and the active site, each of which appear to improve the folding and solubility of the protein. The generation of the F55K mutation was described in our earlier crystallographic study13. The position of the F99L mutation is shown in Supplementary Figure S1.
The protein was expressed in the E. coli strain BL21 (DE3) RIL+ at 37 °C in 2 liters LB medium containing 50 μg/mL ampicillin until OD600 reached approximately 0.6. Expression was induced for 16 hours at 16 °C by addition of 0.5mM isopropyl-thio-β-D-galactosidase (IPTG). Cells were harvested by centrifugation and re-suspended with a 100ml lysis/storage buffer (20mM Tris 7.5, 800mM NaCl and 10% glycerol) and lysed by sonication. Cell debris was removed by centrifugation at 17,000 rpm for 30 minutes at 4 °C, then forced through a 0.2 um syringe filter and applied to a 3ml chitin affinity column at a flow rate of 1 ml/minute. The column was washed with 50 ml of lysis/storage buffer, followed by exchange with of 10mL lysis/storage buffer containing 50mM DTT(1,4-Dithiothreitol). The protein was eluted with 5ml lysis/storage buffer after 20 hours on-column autolytic cleavage at room temperature. Protein was then concentrated to 0.5 mg/ml and frozen by liquid nitrogen for long term storage.
Expression and purification of mutant proteins for binding specificity studies
Because our method for determination of the binding specificity of I-Ssp6803I requires his-tagged protein (which precludes the use of wild-type protein), two catalytically inactive endonuclease point mutants (E11Q and K51M) were generated using the commercial QuickChange mutagenesis kit and protocol (Stratagene). Both mutations were sequenced to verify the protein constructs used for binding studies and binding specificity determinations.
The mutant protein constructs were cloned into the pET-28b vector (Novagen) between its NcoI and XhoI restriction sites, in order to express each protein with a C-terminal his-tag. Both proteins were expressed and purified from Escherichia coli strain BL21(DE3)RIL+ using an affinity purification protocol developed previously for crystallographic studies 13. Expression was induced at 16 °C for 18 hours. Cells were harvested by centrifugation and lysed using a microfluidizer in 400 mM NaCl, 50 mM Tris pH 7.5, and 10% glycerol. Cell debris was removed by centrifugation, then forced through a 0.2 um syringe filter and applied to a heparin affinity column. Eluted protein was concentrated and dialyzed against storage buffer (600mM NaCl, 50 mM Tris pH 7.5 and 10% glycerol). Size exclusion chromatography using a Superdex-200 column equilibrated against the same buffer was then performed, and the protein was concentrated to 3.5 mg/ml.
Quantitative binding assays using isothermal titration calorimetry were conducted as previously described 13 in the presence of 10 mM calcium chloride.
DNA cleavage activity assays
An oligonucleotide containing the physiological substrate of I-Ssp6803I (5′-TCGTCGGGCTCATAACCCGAAGG-3′) was synthesized with six consecutive “A” flanking on each end (Integrated DNA technology, 100-nmole scale, salt-free). This DNA strand was then annealed to its corresponding synthetic complementary DNA strand and cloned into pTOPO-2.1 (Stratagene) to create plasmid substrates for endonuclease activity assays. All plasmid clones putatively harboring target site inserts were amplified and sequenced to confirm the desired substrate sequence.
The relative activity of the enzyme on the resulting plasmid substrate was measured as a function of two parameters: the enzyme concentration and the timecourse of the reaction. The digest conditions for the reactions were 10 nM DNA, 100 mM NaCl, 50 mM Tris-Cl pH 7.9, 10 mM MgCl2 and 1 mM DTT, with variable concentrations of enzyme, incubated at 37°C for variable time points, as described in the Results section and shown in accompanying figures. Products of the digests were visualized and quantitated by electrophoretic analyses on 1.2% TAE agarose gels and subsequent densitometry using program ImageJ (NIH).
Measurement of I-Ssp6803I cleavage specificity profile
A matrix of oligonucleotides, each containing a single base substitution within the wild type target site (Supplementary Table 1), was synthesized and each target site was cloned into the same plasmid vector described above, and verified by sequencing. This DNA target site plasmid matrix thus contains 70 unique DNA target sites (one wild- type and 69 single basepair variants). The individual target sites in the matrix are denoted ‘ssp(i,j)’, where i varies from −11 to +11 (corresponding to the position of the substitution) and j = A, G, C or T (corresponding to the unique basepair substitution at that position). The complementary strands were arrayed and annealed in parallel by mixing equal amounts of top strand and bottom strand in a 96-well PCR plate and incubating for 10 min at 95°C followed by slow cooling for six hours in a PCR thermal cycler.
All plasmid-encoded substrates, as well at the wild-type site in the same backbone, were then individually incubated with the enzyme under conditions that generate approximately 50% cleavage of the wild-type target, at concentrations of protein that exceed the dissociation constant for the wild-type target site by at least ten-fold (700 nM protein and 10 nM DNA in 100 mM NaCl, 50 mM Tris-Cl pH 7.9, 10 mM MgCl2 and 1 mM DTT, incubated at 37°C for 30 minutes). The reactions were quenched with 100 mM EDTA and 2.5% SDS, and products were separated by electrophoresis using 1.2% TAE gels. Substrate and product bands were quantitated using program ImageJ36 and converted to relative cleavability.
Measurement of DNA binding specificity profiles
We developed a high throughput fluorescence-based competition binding assay for determination of the enzyme’s DNA binding specificity based on previous approaches 37; 38. In this assay, the relative binding affinities of the homing endonuclease to each individual variant of its cognate target site are measured by determining the ability of each DNA sequence to compete for binding against a fluorescently labeled wild type sequence, using homing endonucleases immobilized on micro-plates (Figure 2). The extent to which the fluorescently labeled DNA duplex is retained in each test well is proportional to the difference in binding affinity relative to the wild-type target.
The synthetic matrix of annealed DNA oligonucleotides described above was used as the actual competitors for binding against the labeled wild-type DNA sequence. A fluorescently labeled wild-type top strand oligonucleotide, modified with 5′ Cy3™, was synthesized separately and annealed with a complementary unlabeled bottom strand. In addition, an unrelated DNA oligonucleotide sequence (5′-ATCGATCATCGTCGCATGATCAT-3′) (ssp(random)) and its complement were also synthesized and annealed as non-specific binding control.
His-tagged I-Ssp6803I was immobilized onto nickel-coated 96 well plates (Ni-NTA HisSorb plates) by incubating 200 μl of 100 nM I-Ssp6803I in TBS/BSA buffer (50mM Tris pH=7.5, 150mM NaCl, 0.2%BSA) in wells for 2 hours at room temperature. The plates were washed four times with TBS/Tween-20 (50mM Tris pH=7.5, 150mM NaCl, 0.05%Tween-20) to remove unbound protein prior to addition of DNA. The immobilized I-Ssp6803I in each well was incubated for two hours against a mixture of 100 nM labeled wild type DNA duplex and 3 μM (a 30-fold excess) of unlabeled competitor duplex in 200 microliters of binding buffer (50 mM Tris pH=7.5, 150 mM NaCl, 0.02 mg/mL poly dI-dC, 10mM CaCl2).
Each individual competitive binding assay (referred to below as ‘tests’) consists of one of three unlabeled oligonucletides versus the fluorescently labeled wild-type target: (1) ssp(wt) (the unlabeled wild type DNA site competing against itself); (2) ssp(random) (a completely randomized sequence used as a negative control to account for competition by a nonspecific DNA sequence); or (3) ssp(i,j) (one of the target sequences in the substrate matrix, containing a single basepair mismatch). The plates were washed four times with TBS (50mM Tris pH=7.5, 150mM NaCl). The fluorescent signal retained from each test well was counted using a SpectraMax® M5/M5e micro-plate reader (Molecular Devices) (excitation: 510nm, emission: 565nm, cutoff: 550 nm). All measurements were performed in triplicate. Additional negative control experiments performed in the absence of protein indicated that no significant detectable fluorescent signal was retained after the protocol described above was completed. The raw fluorescence data for the entire matrix is shown in Supplementary Figure S2.
The measurements of the retained fluorescent signal for each mismatch sequence variant (F(i,j)) were then converted to relative binding affinities as compared to the wild-type target site using the relationship . This formula gives a close approximation of the binding affinity of each sequence variant.
The relative basepair preference of I-Ssp6803I at each position was then calculated using the relationship: . An example of the full calculation of Ka (i,j) and BP (i,j) from raw fluorescence intensity measurements is given in Supplementary Figure S3.
Additional validating quantitative analyses of individual binding affinities using either isothermal titration calorimetry and competition titrations with variable amounts of unlabeled competitor indicated the calculated binding affinity generates values within close agreement (+/− 5%) with that measured by the high-throughput binding assay (Supplementary Figure S4).
Sequence alignment and information content
The bacterial tRNA sequences described in this manuscript were extracted from 30 bacterial genomes and aligned as described in an earlier study23. The degree of conservation at each position in the tRNA gene was observed by calculating IC by enoLOGOS(http://biodev.hgen.pitt.edu/cgi-bin/enologos/enologos.cgi)39.
Supplementary Material
Acknowledgments
This work was funded by a pair of grants from the NIH to BLS (R01 GM49857 and RL1 CA133833) and generous support from the FHCRC President’s Circle fund to LZ.
References
- 1.Belfort M, Reaban ME, Coetzee T, Dalgaard JZ. Prokaryotic introns and inteins: a panoply of form and function. J Bacteriol. 1995;177:3897–903. doi: 10.1128/jb.177.14.3897-3903.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Belfort M, Perlman PS. Mechanisms of intron mobility. J Biol Chem. 1995;270:30237–40. doi: 10.1074/jbc.270.51.30237. [DOI] [PubMed] [Google Scholar]
- 3.Dujon B. Group I introns as mobile genetic elements: facts and mechanistic speculations--a review. Gene. 1989;82:91–114. doi: 10.1016/0378-1119(89)90034-6. [DOI] [PubMed] [Google Scholar]
- 4.Stoddard BL. Homing endonuclease structure and function. Q Rev Biophys. 2005;38:49–95. doi: 10.1017/S0033583505004063. [DOI] [PubMed] [Google Scholar]
- 5.Guhan N, Muniyappa K. Structural and functional characteristics of homing endonucleases. Crit Rev Biochem Mol Biol. 2003;38:199–248. doi: 10.1080/713609235. [DOI] [PubMed] [Google Scholar]
- 6.Chevalier B, Monnat RJJ, Stoddard BL. The LAGLIDADG homing endonuclease family. In: MBe, et al., editors. Homing endonucleases and inteins. Vol. 16. Springer Verlag; Berlin: 2005. pp. 34–47. [Google Scholar]
- 7.Shen BW, Landthaler M, Shub DA, Stoddard BL. DNA binding and cleavage by the HNH homing endonuclease I-HmuI. J Mol Biol. 2004;342:43–56. doi: 10.1016/j.jmb.2004.07.032. [DOI] [PubMed] [Google Scholar]
- 8.Van Roey P, Meehan L, Kowalski JC, Belfort M, Derbyshire V. Catalytic domain structure and hypothesis for function of GIY-YIG intron endonuclease I-TevI. Nat Struct Biol. 2002;9:806–11. doi: 10.1038/nsb853. [DOI] [PubMed] [Google Scholar]
- 9.Flick KE, Jurica MS, Monnat RJ, Jr, Stoddard BL. DNA binding and cleavage by the nuclear intron-encoded homing endonuclease I-PpoI. Nature. 1998;394:96–101. doi: 10.1038/27952. [DOI] [PubMed] [Google Scholar]
- 10.Biniszkiewicz D, Cesnaviciene E, Shub DA. Self-splicing group I intron in cyanobacterial initiator methionine tRNA: evidence for lateral transfer of introns in bacteria. EMBO J. 1994;13:4629–35. doi: 10.1002/j.1460-2075.1994.tb06785.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bonocora RP, Shub DA. A novel group I intron-encoded endonuclease specific for the anticodon region of tRNA(fMet) genes. Mol Microbiol. 2001;39:1299–306. doi: 10.1111/j.1365-2958.2001.02318.x. [DOI] [PubMed] [Google Scholar]
- 12.Orlowski J, Boniecki M, Bujnicki JM. I-Ssp6803I: the first homing endonuclease from the PD-(D/E)XK superfamily exhibits an unusual mode of DNA recognition. Nucleic Acids Res. 2007 doi: 10.1093/bioinformatics/btm007. [DOI] [PubMed] [Google Scholar]
- 13.Zhao L, Bonocora RP, Shub DA, Stoddard BL. The restriction fold turns to the dark side: a bacterial homing endonuclease with a PD-(D/E)-XK motif. EMBO J. 2007;26:2432–42. doi: 10.1038/sj.emboj.7601672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pingoud A, Fuxreiter M, Pingoud V, Wende W. Type II restriction endonucleases: structure and mechanism. Cell Mol Life Sci. 2005;62:685–707. doi: 10.1007/s00018-004-4513-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Niv MY, Ripoll DR, Vila JA, Liwo A, Vanamee ES, Aggarwal AK, Weinstein H, Scheraga HA. Topology of Type II REases revisited; structural classes and the common conserved core. Nucleic Acids Res. 2007 doi: 10.1093/nar/gkm045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stoddard BL. Homing endonuclease structure and function. Quarterly Reviews of Biophysics. 2005;38:49 – 95. doi: 10.1017/S0033583505004063. [DOI] [PubMed] [Google Scholar]
- 17.Zhao L, Bonocora RP, Shub DA, Stoddard BL. The restriction fold turns to the dark side: a bacterial homing endonuclease with a PD-(D/E)-XK motif. Embo J. 2007;26:2432–42. doi: 10.1038/sj.emboj.7601672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kapp LD, Kolitz SE, Lorsch JR. Yeast initiator tRNA identity elements cooperate to influence multiple steps of translation initiation. RNA. 2006;12:751–64. doi: 10.1261/rna.2263906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guillon JM, Mechulam Y, Blanquet S, Fayat G. Importance of formylability and anticodon stem sequence to give a tRNA(Met) an initiator identity in Escherichia coli. J Bacteriol. 1993;175:4507–14. doi: 10.1128/jb.175.14.4507-4514.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Senger B, Fasiolo F. Yeast tRNA(Met) recognition by methionyl-tRNA synthetase requires determinants from the primary, secondary and tertiary structure: a review. Biochimie. 1996;78:597–604. doi: 10.1016/s0300-9084(96)80006-x. [DOI] [PubMed] [Google Scholar]
- 21.Mandal N, Mangroo D, Dalluge JJ, McCloskey JA, Rajbhandary UL. Role of the three consecutive G:C base pairs conserved in the anticodon stem of initiator tRNAs in initiation of protein synthesis in Escherichia coli. RNA. 1996;2:473–82. [PMC free article] [PubMed] [Google Scholar]
- 22.RajBhandary UL, Chow CM. tRNA: Structure, biosynthesis, and function. ASM press; Washington D.C: 1995. Initiator tRNAs and initiation of protein synthesis; pp. 511–528. [Google Scholar]
- 23.Marck C, Grosjean H. tRNomics: analysis of tRNA genes from 50 genomes of Eukarya, Archaea, and Bacteria reveals anticodon-sparing strategies and domain-specific features. RNA. 2002;8:1189–232. doi: 10.1017/s1355838202022021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rudi K, Fossheim T, Jakobsen KS. Nested evolution of a tRNA(Leu)(UAA) group I intron by both horizontal intron transfer and recombination of the entire tRNA locus. J Bacteriol. 2002;184:666–71. doi: 10.1128/JB.184.3.666-671.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rudi K, Jakobsen KS. Complex evolutionary patterns of tRNA Leu(UAA) group I introns in the cyanobacterial radiation [corrected] J Bacteriol. 1999;181:3445–51. doi: 10.1128/jb.181.11.3445-3451.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rudi K, Jakobsen KS. Cyanobacterial tRNA(Leu)(UAA) group I introns have polyphyletic origin. FEMS Microbiol Lett. 1998;156:293–8. doi: 10.1016/s0378-1097(97)00446-1. [DOI] [PubMed] [Google Scholar]
- 27.Paquin B, Kathe SD, Nierzwicki-Bauer SA, Shub DA. Origin and evolution of group I introns in cyanobacterial tRNA genes. J Bacteriol. 1997;179:6798–806. doi: 10.1128/jb.179.21.6798-6806.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nord D, Torrents E, Sjoberg BM. A functional homing endonuclease in the Bacillus anthracis nrdE group I intron. J Bacteriol. 2007;189:5293–301. doi: 10.1128/JB.00234-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eddy SR, Gold L. The phage T4 nrdB intron: a deletion mutant of a version found in the wild. Genes Dev. 1991;5:1032–1041. doi: 10.1101/gad.5.6.1032. [DOI] [PubMed] [Google Scholar]
- 30.Chinen A, Naito Y, Handa N, Kobayashi I. Evolution of sequence recognition by restriction-modification enzymes: selective pressure for specificity decrease. Mol Biol Evol. 2000;17:1610 – 1619. doi: 10.1093/oxfordjournals.molbev.a026260. [DOI] [PubMed] [Google Scholar]
- 31.Handa N, Ichige A, Kusano K, Kobayashi I. Cellular responses to postsegregational killing by restriction-modification genes. J Bacteriology. 2000;182:2218 –2229. doi: 10.1128/jb.182.8.2218-2229.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Handa N, Kobayashi I. Post-segregational killing by restriction modification gene complexes: observations of individual cell deaths. Biochimie. 1999;81:931 – 938. doi: 10.1016/s0300-9084(99)00201-1. [DOI] [PubMed] [Google Scholar]
- 33.Saravanan M, Bujnicki JM, Cymerman IA, Rao DN, Nagaraja V. Type II restriction endonuclease R.KpnI is a member of the HNH nuclease superfamily. Nucleic Acids Res. 2004;32:6129 – 6135. doi: 10.1093/nar/gkh951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ibryashkina EM, Zakharova MV, Baskunov vB, Bogdanova ES, Nagornykh MO, Denmukhamedov MM, Melnik BS, Kolinski A, Gront D, Feder M, et al. Type II restriction endonuclease R.Eco29kI is a member of the GIY-YIG nuclease superfamily. BMC Struct Biol. 2007;7:48 – 56. doi: 10.1186/1472-6807-7-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Orlowski J, Bujnicki JM. Structural and evolutionary classification of Type II restriction enzymes based on theoretical and experimental analyses. Nucleic Acids Res. 2008;36:1 – 13. doi: 10.1093/nar/gkn175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Girish V, Vijayalaskshima A. Affordable image analysis using NIH Image/ImageJ. Indian J Cancer. 2004;1:47 – 56. [PubMed] [Google Scholar]
- 37.Zhang ZR, Palfrey D, Nagel DA, Lambert PA, Jessop RA, Santos AF, Hine AV. Fluorescent microplate-based analysis of protein-DNA interactions. I: Immobilized protein. BioTechniques. 2003;35:980–2. 984–986. doi: 10.2144/03355st06. [DOI] [PubMed] [Google Scholar]
- 38.Hallikas O, Taipale J. High-throughput assay for determining specificity and affinity of protein-DNA binding interactions. Nature protocols. 2006;1:215–22. doi: 10.1038/nprot.2006.33. [DOI] [PubMed] [Google Scholar]
- 39.Workman CT, Yin Y, Corcoran DL, Ideker T, Stormo GD, Benos PV. enoLOGOS: a versatile web tool for energy normalized sequence logos. Nucleic Acids Res. 2005;33:W389–92. doi: 10.1093/nar/gki439. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.