

Abstract
Activation of the cholinergic anti-inflammatory pathway through direct activation of nicotinic acetylcholine receptors on immune cells can inhibit pro-inflammatory chemokine and cytokine release and thereby protect in a variety of inflammatory diseases. The aim of this study was to investigate whether nicotine treatment protected against acute lung inflammation. Mice challenged with intratracheal lipopolysaccharide (LPS, 50 μg) were treated with nicotine (0.2 or 0.4 mg/kg, sc). After 24 h, bronchoalveolar lavage fluid (BALF) was obtained to measure leukocyte infiltration, lung edema, and pro-inflammatory chemokine (MIP-1α, MIP-2, and eotaxin) and cytokine (IL-1, IL-6, and TNF-α) levels. Nicotine treatment reduced the LPS-mediated infiltration of leukocytes and edema as evidenced by decreased BALF inflammatory cells, myeloperoxidase, and protein. Nicotine also downregulated lung production of pro-inflammatory chemokines and cytokines. These data support the proposal that activation of the cholinergic anti-inflammatory pathway may represent a useful addition to the therapy of acute respiratory distress syndrome.
Keywords: inflammation, nicotine, acetylcholine, lung, chemokine
INTRODUCTION
Acute respiratory distress syndrome (ARDS) is a major complication of conditions such as sepsis, trauma, and severe pneumonia. ARDS is characterized by an extensive lung inflammation involving the local recruitment and activation of polymorphonuclear neutrophils [1–3] and the release of pro-inflammatory mediators such as chemokines and cytokines [4–6] and both reactive oxygen and nitrogen species [7]. This inflammation causes alveolocapillary damage, high permeability pulmonary edema, altered lung mechanics, and severe gas exchange abnormalities [1, 2]. Currently, treatment remains largely supportive as despite significant advances in understanding the underlying pathogenic mechanisms of ARDS, no satisfying therapeutic approach has emerged. To investigate prospective clinical therapeutic strategies to combat ARDS, several animal models have been developed, in particular the in vivo administration of lipopolysaccharide (LPS), a component of the cell wall of Gram-negative bacteria, which has gained wide acceptance as a clinically relevant model of severe lung inflammation [8].
The cholinergic anti-inflammatory pathway has been identified as a major neuromodulator of immune cell function [9] with stimulation of the vagus nerve protecting against endotoxic shock by directly affecting immune cells [10–12]. Nicotine also attenuated inflammation in human subjects exposed to endotoxin [12]. Nicotinic acetylcholine receptor alpha7 has been identified on the surface of immune cells which, following activation, mediates cholinergic regulation of inflammation [13] via the activation of jak2 and STAT 3 [14], resulting in a decrease in pro-inflammatory cytokine production particularly TNF-α [11, 13, 15]. The involvement of a nicotinic receptor supported the previously observed anti-inflammatory effect of nicotine in colitis both in animal models [16] and in human patients [17]. Nicotine treatment also reduced the incidence of type I diabetes in two animal models [18], again an effect observed in the human population [19]. Nicotine has also proved to be anti-inflammatory in animal models of CNS inflammation [20], arthritis [21], allergy/asthma [22], intestinal inflammation [23], peritonitis [11], and ricin toxicity [24].
This study was undertaken to evaluate the effects of activating the cholinergic anti-inflammatory pathway with exogenous nicotine on acute lung inflammation induced in mice by the intratracheal instillation of LPS in vivo.
METHODS
In vivo studies were performed in accordance with the National Institutes of Health guidelines and with the approval of the local institutional animal care and use committee.
Intratracheal LPS Administration
Male BALB/c mice, 8–10 weeks old, were anesthetized with a mixture of ketamine (80 mg/kg) and xylazine (30 mg/kg) intraperitoneally (ip). A 1-cm midline cervical incision was made, exposing the trachea. Intratracheal administration of LPS (Escherichia coli, O127:B8) or vehicle (phosphate-buffered saline (PBS), pH 7.4) was performed with a bent 27G tuberculin syringe (in a volume of 100 μl), as described previously. The cervical incision was closed with 5.0 silk suture and the mice returned to their cage. The animals recovered rapidly after surgery.
Bronchoalveolar Lavage
Twenty-fourhours after the surgery, the mice were reanesthetized with ketamine/xylazine, ip. The animals were bled by transection of the inferior vena cava to reduce hemorrhage into the lungs. Bronchoalveolar lavage was performed by the intratracheal instillation of 1 ml PBS (pH 7.4) into the exposed lungs (maintained within the thoracic cavity). The lavage fluid was infused a total of three times into the lungs before final collection. The bronchoalveolar lavage fluid (BALF) was then centrifuged at 5,000 rpm for 10 min and the cell-free supernatant frozen at −70°C until further analysis. The volume of cell-free supernatant was measured for each animal.
Treatment Groups
Animals challenged with intratracheal LPS were treated with nicotine, 0.2 or 0.4 mg/kg total dose given over 24 h (n=14), or vehicle (isotonic saline, 0.2 ml, n= 14, control group), administered i.p. BID at 1 and 12 h after surgery. In addition, there were two groups of sham-operated mice (n=10) challenged with intratracheal PBS instead of LPS and treated either with saline or nicotine (0.4 mg/kg).
Measurements
Protein Assay
The amount of proteins in the BALF was assayed using the Bradford assay [25]. Proteins are expressed in micrograms protein per milliliter BALF
Myeloperoxidase Activity
The activity of myeloperoxidase (MPO), an indicator of neutrophil accumulation, was directly measured in the BALF. An aliquot of BALF (20 μl) was mixed with 1.6 mM tetra-methyl-benzidine and 1 mM hydrogen peroxide. Myeloperoxidase activity was then measured as the change in absorbance at 650 nm at 37°C using a Spectramax microplate reader (Molecular Devices, Sunnyvale, CA, USA) [26]. Results are expressed as milliunits of MPO activity per milliliter BALF.
Cytokines and Chemokines
The amount of the pro-inflammatory cytokines IL-1, IL-6, and TNF-α and chemokines MIP-1α, MIP-2, and eotaxin were determined in the BALF using commercially available ELISAs following the protocol provided by the manufacturer (R&D Systems, Minneapolis, MN). For the measurement of pro-inflammatory cytokines and chemokines in mice challenged with LPS, BALF was diluted 1:2 to 1:5. BALF was assayed undiluted in sham animals.
Lung Histology and Immunohistochemistry
Histopathological changes induced by LPS were evaluated in five mice treated with nicotine treated with 0.4 mg/kg BID at 1 and 12 h and five mice treated with vehicle. Twenty-fourhours after surgery, the animals were anesthetized, killed by exsanguination, and the lungs were inflated fixed with 4% paraformaldehyde. Paraffin-embedded lungs were sectioned at 3 μm and stained with hematoxylin and eosin for morphological analysis [27, 28]. Photomicrographs were taken with a Zeiss Axiovert 25 microscope equipped with a digital camera and show representative images.
Data Analysis
All the data were statistically evaluated using analysis of variance. When the relevant F values were significant at the 5% level, further pairwise comparisons were made using Tukey post hoc test. Statistical significance was assigned to p<0.05.
RESULTS
Nicotine Reduces Lung Leukocyte Accumulation and Formation of High Permeability Edema After Intratracheal Instillation of LPS
Treatment with nicotine (0.4 mg/kg) alone had no effect on lung leukocyte accumulation, as evidenced by BALF cell numbers and MPO activity being not significantly different to those found in sham animals (Fig. 1a, b). Administration of LPS elicited a massive inflammation of the lung as demonstrated by a marked increase in BALF cell count (Fig. 1a) and MPO activity (Fig. 1b), indicating the presence of a significant proportion of polymorphonuclear cells. Nicotine treatment, at both 0.2 and 0.4 mg/kg, significantly suppressed the accumulation of leukocytes in the alveolar spaces, as indicated by a significant reduction BALF cell numbers (Fig. 1a) and MPO activity from 618± 78 mU/ml to 211±27 and 116±19 mU/ml, respectively (Fig. 1b).
Fig. 1.

Protective effect of nicotine against leukocyte infiltration (a), increased MPO activity (b), and edema (c) in an acute model of lung inflammation. Male BALBc mice received an intratracheal instillation of lipopolysaccharide (LPS from E. coli serotype 0127:B8, 50 μg) and were treated with nicotine at 0.2 or 0.4 mg/kg divided into two doses at 1 and 12 h after surgery. Control mice are sham-treated, receiving vehicle instead of LPS intratracheally. Inflammatory cell number, myeloperoxidase activity, and protein levels in the bronchoalveolar lavage fluid (BALF) were determined 24 h after the LPS administration. Data are expressed as mean ± SEM from 8–12 animals. * p<0.05, **p<0.01 vs. control mice; † p<0.05 vs. LPS-treated mice.
A hallmark of ARDS is the development of high permeability edema, characterized by a high protein content of the edema fluid. Such abnormalities were present in the lungs of mice exposed to LPS where the protein concentration in the BALF increased more than fivefold from that in sham animals (Fig. 1c), an effect attenuated by both 0.2 and 0.4 mg/kg nicotine (Fig. 1c). Nicotine alone had no effect on BALF protein level as compared to the vehicle-treated sham mice (Fig. 1c).
Effects of Nicotine on LPS-Mediated Increases in BALF Levels of Chemokines and Inflammatory Cytokines
The BALF from sham animals treated with vehicle or 0.4 mg/kg nicotine had virtually undetectable levels of the inflammatory chemokines MIP-1α, MIP-2, and eotaxin (Fig. 2a). Administration of LPS dramatically increased BALF levels of MIP-1α, MIP-2, and eotaxin (Fig. 2a), with nicotine at both 0.2 and 0.4 mg/kg significantly attenuating the increase in both MIP-1α and MIP-2 (Fig. 2a) while returning the eotaxin levels to that observed in sham-treated animals (Fig. 2a). LPS administration also significantly increased BALF levels of the inflammatory cytokines IL-1, IL-6, and TNF-α (Fig. 2b, c), with nicotine treatment (0.2 and 0.4 mg/kg) significantly reducing the LPS-mediated increase in inflammatory cytokine levels (Fig. 2b, c).
Fig. 2.

Nicotine reduces bronchoalveolar lavage fluid chemokine (a) and cytokine (b, c) levels in an acute model of lung inflammation. LPS (50 μg) induced a significant increase in BALF levels of chemokines MIP-1α, MIP-2 (a) and eotaxin (a), and the pro-inflammatory cytokines IL-1 (b), IL-6 (b), and TNF-α (c). The increased levels of chemokines and cytokines in the BALF were significantly suppressed by nicotine at both 0.2 and 0.4 mg/kg. ata are expressed as mean ± SEM from 8–12 animals. *p<0.05, **p<0.01 vs. control mice; †p<0.05 vs. LPS-treated mice.
Nicotine Reduces Morphologic Damage in Lungs Exposed to Lipopolysaccharide
As illustrated in Fig. 3, the lungs of mice exposed to LPS showed marked inflammatory alterations characterized by a thickening of the alveolocapillary membrane, the presence of alveolar hemorrhage, and massive extravasation of both mono- and polymorphonuclear leukocytes into the alveolar space as compared to sham mice. In contrast, the histological lung damage was less pronounced in mice treated with nicotine, with both erythrocytes and leukocyte infiltration into the alveolar spaces being reduced. Morphological analysis showed no difference between mice treated with nicotine (0.4 mg/kg) alone and sham mice (data not shown).
Fig. 3.
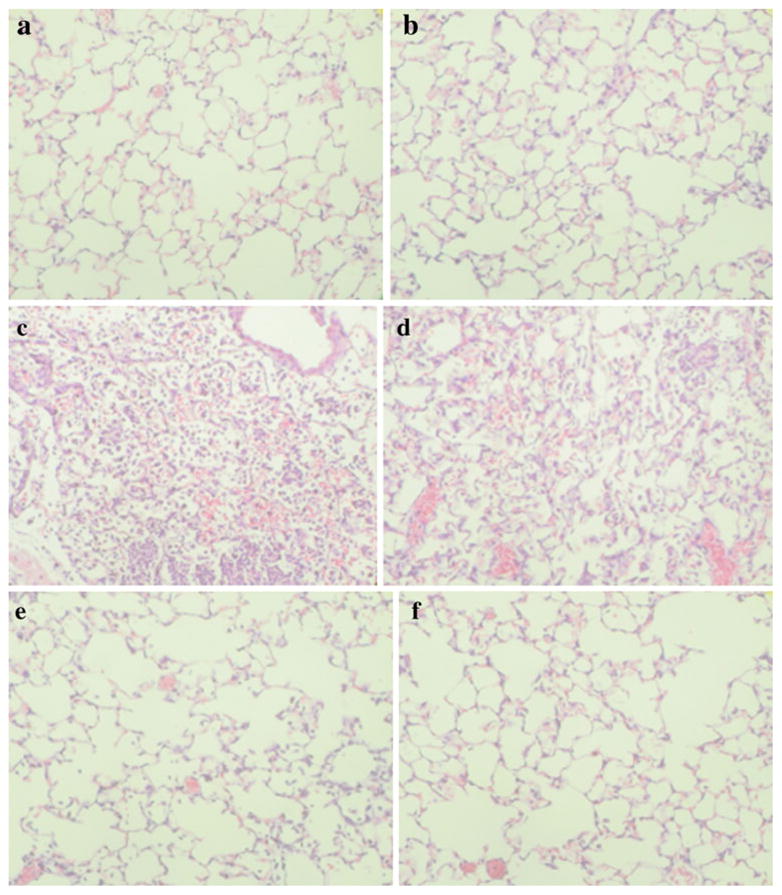
Lung morphology in mice challenged with lipopolysaccharide (LPS). Representative histological sections of lungs from two separate mice harvested 24 h in sham mice (a, b), LPS-treated mice (c, d), and mice treated with LPS and nicotine 0.4 mg/kg (e, f) starting at 1 and 12 h. The morphologic alterations induced by LPS (infiltration of the alveolar spaces with mono- and polymorphonuclear cells, presence of alveolar hemorrhages and exudates, thickening of the alveolar septa) were reduced by nicotine treatment. Pictures are representative of n=5 mice/treatment group, magnification shown at ×320.
DISCUSSION
The data presented demonstrate that nicotine treatment reduces LPS-induced lung inflammation in a murine model of ARDS. The protective effect of nicotine is likely to be associated with its anti-inflammatory effect, suggesting a possible therapeutic strategy of activating the cholinergic anti-inflammatory pathway in ARDS.
Activation of the anti-inflammatory cholinergic pathway, either through vagal stimulation [10, 29] or direct activation of nicotinic acetylcholine receptors by nicotine administration [18, 30, 31], has been demonstrated to be anti-inflammatory in a wide variety of inflammatory disease states [9]. More recently, the cholinergic anti-inflammatory pathway has been activated following pharmacologic cholinesterase inhibition [32, 33] or application of the acetylcholine precursor choline [34].
Specific receptors for nicotine have been shown to be present on lymphocytes from human, rat, and mouse [9]. Nicotine has been shown to have T cell-dependent and -independent effects, with the anti-inflammatory effects of nicotine receptor activation having been identified as mediated through the activation of the alpha7 nicotinic acetylcholine receptor (alpha7nAChR) located on immune cells [13, 34]. The subsequent alpha7nAChR-meduiated activation of jak2 and STAT 3 [14] results in a decrease in pro-inflammatory cytokine production, particularly TNF-α [34, 35], through an inhibitory effect on NF-κB activation [36]. Nicotine suppresses the production of inflammatory mediators such as IL-1, IL-8, and PGE2 from human macrophages [36] and IL-2 and TNF-α from human mononuclear cells [34], an effect again mediated through the modulation of NF-κB activation [36]. Recently, administration of the cholinesterase inhibitor physostigmine in an experimental model of sepsis was shown to reduce the activation of NF-κB and suppress circulating pro-inflammatory cytokines (TNF-α, IL-1, and IL-6) and neutrophil infiltration [33].
ARDS is associated with the development of interconnected inflammatory cascades, with pro-inflammatory cytokines playing a central role in the initiation and propagation of the inflammatory response leading to lung injury. Of these cytokines, TNF-α, IL-1, and IL-6 are considered pivotal, with high levels of these cytokines observed in non-surviving ARDS patients as compared to surviving patients [5, 6]. Pro-inflammatory cytokines including TNF-α, IL-1, and IL-6 have been implicated in the destruction of alveolar epithelium and leakage of capillaries leading to pulmonary edema, a hallmark of ARDS [37]. Nicotine had previously proved effective in reducing lung inflammation in asthma [22], pneumonitis [38], and acid-induced injury [31]. In this model of ARDS, nicotine again proved very effective at reducing the inflammation, decreasing the levels of TNF-α, IL-1, and IL-6 and hence the lung damaging effects of these cytokines reducing lung edema. Nicotine has been shown to decrease inflammatory cytokine production by macrophages and other mononuclear cells, and this may account for the protective effect in ARDS; however, it may also be possible that nicotine initially affects chemokine production, thus reducing migration of inflammatory cells such as leukocytes to the site of inflammation and damage.
The presence of infiltrating leukocytes is the hallmark of pulmonary inflammation associated with acute lung injury [3, 39], with chemokines [4] such as MIP-1α and MIP-2 regulating the recruitment to and activation of these leukocytes at sites of inflammation; lung levels of both increase following LPS treatment in vivo [27]. Another chemokine, eotaxin, had previously been thought to increase in asthma and other allergic reactions [40], but recent human data have indicated that this chemokine also goes up in ARDS [41], and here, we have shown for the first time in an animal model of ARDS that LPS treatment in vivo also increases lung levels of eotaxin, though not to the same extent as MIP-1α and MIP-2. The reduction in leukocyte infiltration is an important mechanism underlying the beneficial affects on edema as neutrophils and the pro-inflammatory environment they produce are considered the primary effectors of alveolocapillary damage in ARDS [2, 3].
The protective effects of nicotine in ARDS may well be a direct effect reducing both immune cell infiltration via decreased chemokine levels and reducing inflammatory cellular damage via decreased pro-inflammatory cytokine levels through inhibitory second messenger systems on NF-κB activation. However, the two are obviously linked with reduced immune cell infiltration also reducing inflammatory cytokine production at the site on inflammation and vice versa, with a reduction in inflammatory cytokine-mediated cellular damage reducing immune cell infiltration. Therefore, an overall effect following activation of the cholinergic anti-inflammatory pathway may be to alter the development of chemotactic gradients, thereby acting as a powerful downregulator of leukocyte trafficking in inflammatory conditions.
This study indicates that activation of the cholinergic anti-inflammatory pathway produces marked anti-inflammatory effects in a clinically relevant animal model of ARDS. Nicotine may not be suitable for therapeutic application in the clinical setting for ARDS; however, studies have indicated that other methods of cholinergic activation exist, including direct activation of the vagus nerve [10] or administration of either a cholinesterase inhibitor [32] or acetylcholine precursor choline [34], all of which reduce inflammation. Therefore, future development of therapeutic strategies to activate the cholinergic anti-inflammatory pathway may be useful in treating ARDS and other inflammatory conditions.
References
- 1.Artigas A, Bernard GR, Carlet J, et al. The American-European Consensus Conference on ARDS, part 2. Ventilatory, pharmacologic, supportive therapy, study design strategies and issues related to recovery and remodeling. Intensive Care Medicine. 1998;24:378–398. doi: 10.1007/s001340050585. [DOI] [PubMed] [Google Scholar]
- 2.Kollef MH, Schuster DP. The acute respiratory distress syndrome. The New England Journal of Medicine. 1995;332:27–37. doi: 10.1056/NEJM199501053320106. [DOI] [PubMed] [Google Scholar]
- 3.Abraham E, Carmody A, Shenkar R, Arcaroli J. Neutrophils as early immunologic effectors in hemorrhage- or endotoxemia-induced acute lung injury. American Journal of Physiology Lung Cellular and Molecular Physiology. 2000;279:L1137–L1145. doi: 10.1152/ajplung.2000.279.6.L1137. [DOI] [PubMed] [Google Scholar]
- 4.Strieter RM, Kunkel SL, Keane MP, Standiford TJ. Chemokines in lung injury: Thomas A. Neff Lecture. Chest. 1999;116:103S–110S. doi: 10.1378/chest.116.suppl_1.103s. [DOI] [PubMed] [Google Scholar]
- 5.Meduri GU, Headley S, Kohler G, et al. Persistent elevation of inflammatory cytokines predicts a poor outcome in ARDS. Plasma IL-1 beta and IL-6 levels are consistent and efficient predictors of outcome over time. Chest. 1995;107:1062–1073. doi: 10.1378/chest.107.4.1062. [DOI] [PubMed] [Google Scholar]
- 6.Takala A, Jousela I, Takkunen O, et al. A prospective study of inflammation markers in patients at risk of indirect acute lung injury. Shock. 2002;17:252–257. doi: 10.1097/00024382-200204000-00002. [DOI] [PubMed] [Google Scholar]
- 7.Peng X, Abdulnour RE, Sammani S, et al. Inducible nitric oxide synthase contributes to ventilator-induced lung injury. American Journal of Respiratory and Critical Care Medicine. 2005;172:470–479. doi: 10.1164/rccm.200411-1547OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Borron P, McIntosh JC, Korfhagen TR, Whitsett JA, Taylor J, Wright JR. Surfactant-associated protein A inhibits LPS-induced cytokine and nitric oxide production in vivo. American Journal of Physiology Lung Cellular and Molecular Physiology. 2000;278:L840–L847. doi: 10.1152/ajplung.2000.278.4.L840. [DOI] [PubMed] [Google Scholar]
- 9.Tracey KJ. Physiology and immunology of the cholinergic antiinflammatory pathway. The Journal of Clinical Investigation. 2007;117:289–296. doi: 10.1172/JCI30555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Borovikova LV, Ivanova S, Zhang M, et al. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature. 2000;405:458–462. doi: 10.1038/35013070. [DOI] [PubMed] [Google Scholar]
- 11.van Westerloo DJ, I, Giebelen A, Florquin S, et al. The cholinergic anti-inflammatory pathway regulates the host response during septic peritonitis. The Journal of Infectious Diseases. 2005;191:2138–2148. doi: 10.1086/430323. [DOI] [PubMed] [Google Scholar]
- 12.Wittebole X, Hahm S, Coyle SM, Kumar A, Calvano SE, Lowry SF. Nicotine exposure alters in vivo human responses to endotoxin. Clinical and Experimental Immunology. 2007;147:28–34. doi: 10.1111/j.1365-2249.2006.03248.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang H, Yu M, Ochani M, et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature. 2003;421:384–388. doi: 10.1038/nature01339. [DOI] [PubMed] [Google Scholar]
- 14.de Jonge WJ, van der Zanden EP, The FO, et al. Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2-STAT3 signaling pathway. Nature Immunology. 2005;6:844–851. doi: 10.1038/ni1229. [DOI] [PubMed] [Google Scholar]
- 15.de Jonge WJ, Ulloa L. The alpha7 nicotinic acetylcholine receptor as a pharmacological target for inflammation. British Journal of Pharmacology. 2007;151:915–929. doi: 10.1038/sj.bjp.0707264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eliakim R, Karmeli F, Rachmilewitz D, Cohen P, Fich A. Effect of chronic nicotine administration on trinitrobenzene sulphonic acid-induced colitis. European Journal of Gastroenterology & Hepatology. 1998;10:1013–1019. [PubMed] [Google Scholar]
- 17.Karban A, Eliakim R. Effect of smoking on inflammatory bowel disease: Is it disease or organ specific? World Journal of Gastroenterology. 2007;13:2150–2152. doi: 10.3748/wjg.v13.i15.2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mabley JG, Pacher P, Southan GJ, Salzman AL, Szabo C. Nicotine reduces the incidence of type I diabetes in mice. The Journal of Pharmacology and Experimental Therapeutics. 2002;300:876–881. doi: 10.1124/jpet.300.3.876. [DOI] [PubMed] [Google Scholar]
- 19.Carlsson S, Midthjell K, Grill V. Smoking is associated with an increased risk of type 2 diabetes but a decreased risk of autoimmune diabetes in adults: an 11-year follow-up of incidence of diabetes in the Nord-Trondelag Study. Diabetologia. 2004;47:1953–1956. doi: 10.1007/s00125-004-1554-9. [DOI] [PubMed] [Google Scholar]
- 20.Shi FD, Piao WH, Kuo YP, Campagnolo DI, Vollmer TL, Lukas RJ. Nicotinic attenuation of central nervous system inflammation and autoimmunity. Journal of Immunology. 2009;182:1730–1739. doi: 10.4049/jimmunol.182.3.1730. [DOI] [PubMed] [Google Scholar]
- 21.van Maanen MA, Lebre MC, van der Poll T, et al. Stimulation of nicotinic acetylcholine receptors attenuates collagen-induced arthritis in mice. Arthritis and Rheumatism. 2009;60:114–122. doi: 10.1002/art.24177. [DOI] [PubMed] [Google Scholar]
- 22.Mishra NC, Rir-Sima-Ah J, Langley RJ, et al. Nicotine primarily suppresses lung Th2 but not goblet cell and muscle cell responses to allergens. Journal of Immunology. 2008;180:7655–7663. doi: 10.4049/jimmunol.180.11.7655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.The FO, Boeckxstaens GE, Snoek SA, et al. Activation of the cholinergic anti-inflammatory pathway ameliorates postoperative ileus in mice. Gastroenterology. 2007;133:1219–1228. doi: 10.1053/j.gastro.2007.07.022. [DOI] [PubMed] [Google Scholar]
- 24.Mabley JG, Pacher P, Szabo C. Activation of the cholinergic antiinflammatory pathway reduces ricin-induced mortality and organ failure in mice. Molecular Medicine. 2009;15:166–172. doi: 10.2119/molmed.2008.00105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bradford MM. A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemistry. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 26.Mabley JG, Pacher P, Liaudet L, et al. Inosine reduces inflammation and improves survival in a murine model of colitis. American Journal of Physiology Gastrointestinal and Liver Physiology. 2003;284:G138–G144. doi: 10.1152/ajpgi.00060.2002. [DOI] [PubMed] [Google Scholar]
- 27.Liaudet L, Mabley JG, Pacher P, et al. Inosine exerts a broad range of antiinflammatory effects in a murine model of acute lung injury. Annals of Surgery. 2002;235:568–578. doi: 10.1097/00000658-200204000-00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liaudet L, Pacher P, Mabley JG, et al. Activation of poly (ADP-ribose) polymerase-1 is a central mechanism of lipopolysaccharide-induced acute lung inflammation. American Journal of Respiratory and Critical Care Medicine. 2002;165:372–377. doi: 10.1164/ajrccm.165.3.2106050. [DOI] [PubMed] [Google Scholar]
- 29.Rosas-Ballina M, Ochani M, Parrish WR, et al. Splenic nerve is required for cholinergic antiinflammatory pathway control of TNF in endotoxemia. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:11008–11013. doi: 10.1073/pnas.0803237105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gwilt CR, Donnelly LE, Rogers DF. The non-neuronal cholinergic system in the airways: An unappreciated regulatory role in pulmonary inflammation? Pharmacology & Therapeutics. 2007;115:208–222. doi: 10.1016/j.pharmthera.2007.05.007. [DOI] [PubMed] [Google Scholar]
- 31.Su X, Lee JW, Matthay ZA, et al. Activation of the {alpha}7 nAChR reduces acid-induced acute lung injury in mice and rats. American Journal of Respiratory Cell and Molecular Biology. 2007;37:186–192. doi: 10.1165/rcmb.2006-0240OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pavlov VA, Parrish WR, Rosas-Ballina M, et al. Brain acetylcholinesterase activity controls systemic cytokine levels through the cholinergic anti-inflammatory pathway. Brain, Behavior, and Immunity. 2009;23:41–45. doi: 10.1016/j.bbi.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hofer S, Eisenbach C, Lukic IK, et al. Pharmacologic cholinesterase inhibition improves survival in experimental sepsis. Critical Care Medicine. 2008;36:404–408. doi: 10.1097/01.CCM.0B013E31816208B3. [DOI] [PubMed] [Google Scholar]
- 34.Parrish WR, Rosas-Ballina M, Gallowitsch-Puerta M, et al. Modulation of TNF release by choline requires alpha7 subunit nicotinic acetylcholine receptor-mediated signaling. Molecular Medicine. 2008;14:567–574. doi: 10.2119/2008-00079.Parrish. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yoshikawa H, Kurokawa M, Ozaki N, et al. Nicotine inhibits the production of proinflammatory mediators in human monocytes by suppression of I-kappaB phosphorylation and nuclear factor-kappaB transcriptional activity through nicotinic acetylcholine receptor alpha7. Clinical and Experimental Immunology. 2006;146:116–123. doi: 10.1111/j.1365-2249.2006.03169.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sugano N, Shimada K, Ito K, Murai S. Nicotine inhibits the production of inflammatory mediators in U937 cells through modulation of nuclear factor-kappaB activation. Biochemical and Biophysical Research Communications. 1998;252:25–28. doi: 10.1006/bbrc.1998.9599. [DOI] [PubMed] [Google Scholar]
- 37.Ware LB. Pathophysiology of acute lung injury and the acute respiratory distress syndrome. Seminars in Respiratory and Critical Care Medicine. 2006;27:337–349. doi: 10.1055/s-2006-948288. [DOI] [PubMed] [Google Scholar]
- 38.Blanchet MR, Israel-Assayag E, Cormier Y. Inhibitory effect of nicotine on experimental hypersensitivity pneumonitis in vivo and in vitro. American Journal of Respiratory and Critical Care Medicine. 2004;169:903–909. doi: 10.1164/rccm.200210-1154OC. [DOI] [PubMed] [Google Scholar]
- 39.Abraham E, Arcaroli J, Shenkar R. Activation of extracellular signal-regulated kinases, NF-kappa B, and cyclic adenosine 5′-monophosphate response element-binding protein in lung neutrophils occurs by differing mechanisms after hemorrhage or endotoxemia. Journal of Immunology. 2001;166:522–530. doi: 10.4049/jimmunol.166.1.522. [DOI] [PubMed] [Google Scholar]
- 40.Sugita A, Ogawa H, Azuma M, et al. Antiallergic and anti-inflammatory effects of a novel I kappaB kinase beta inhibitor, IMD-0354, in a mouse model of allergic inflammation. International Archives of Allergy and Immunology. 2009;148:186–198. doi: 10.1159/000161579. [DOI] [PubMed] [Google Scholar]
- 41.Miyazaki E, Nureki S, Ono E, et al. Circulating thymus-and activation-regulated chemokine/CCL17 is a useful biomarker for discriminating acute eosinophilic pneumonia from other causes of acute lung injury. Chest. 2007;131:1726–1734. doi: 10.1378/chest.06-2596. [DOI] [PubMed] [Google Scholar]