

Abstract
The DsrMKJOP transmembrane complex has a most important function in dissimilatory sulfur metabolism and consists of cytoplasmic, periplasmic, and membrane integral proteins carrying FeS centers and b- and c-type cytochromes as cofactors. In this study, the complex was isolated from the purple sulfur bacterium Allochromatium vinosum and individual components were characterized as recombinant proteins. The two integral membrane proteins DsrM and DsrP were successfully produced in Escherichia coli C43(DE3) and C41(DE3), respectively. DsrM was identified as a diheme cytochrome b, and the two hemes were found to be in low-spin state. Their midpoint redox potentials were determined to be +60 and +110 mV. Although no hemes were predicted for DsrP, it was also clearly identified as a b-type cytochrome. To the best of our knowledge, this is the first time that heme binding has been experimentally proven for a member of the NrfD protein family. Both cytochromes were partly reduced after addition of a menaquinol analogue, suggesting interaction with quinones in vivo. DsrO and DsrK were both experimentally proven to be FeS-containing proteins. In addition, DsrK was shown to be membrane associated, and we propose a monotopic membrane anchoring for this protein. Coelution assays provide support for the proposed interaction of DsrK with the soluble cytoplasmic protein DsrC, which might be its substrate. A model for the function of DsrMKJOP in the purple sulfur bacterium A. vinosum is presented.
The purple sulfur bacterium Allochromatium vinosum uses reduced sulfur compounds as electron donors for anoxygenic photosynthesis. During sulfur oxidation, sulfur globules are formed as obligatory intermediates. Their further oxidation is strictly dependent on the proteins encoded in the dsr operon (7). The dsr genes are found not only in sulfur-oxidizing bacteria but also in sulfate- and sulfite-reducing bacteria and archaea. While the occurrence of some of the Dsr proteins differs between sulfate-reducing organisms and sulfur oxidizers (42), all prokaryotic genomes that contain genes coding for the dissimilatory sulfite reductase (DsrAB) inevitably contain genes coding for the DsrMKJOP transmembrane complex, and it is proposed that these two components interact with each other (7, 14, 34, 36).
The genes coding for the DsrMKJOP complex are apparently strictly conserved in sulfate-reducing organisms, indicating a vital role in sulfate reduction (35). In addition, we have previously shown for the sulfur oxidizer A. vinosum that the degradation of sulfur globules is strictly dependent on the presence of the DsrMKJOP proteins (42). Based on amino acid sequence analysis, DsrM is proposed to be an integral membrane protein with five transmembrane helices. It is related to the heme b-containing subunits NarI of bacterial nitrate reductases and HdrE of heterodisulfide reductases (HDRs) from methanogenic archaea. DsrK is proposed to be a cytoplasmic iron sulfur protein, and it has been suggested that it is the catalytic subunit of the DsrMKJOP complex, since it is related to the catalytic subunit of heterodisulfide reductase of methanogenic archaea (36, 37). DsrO and DsrP are related to the noncatalytic subunits of complex iron-sulfur molybdoenzymes (CISM) (41). DsrP is an integral membrane protein with 10 predicted transmembrane helices. It belongs to the NrfD/PsrC protein family, which provides the membrane-spanning, quinone-interacting subunits to a number of CISM. DsrO is a ferredoxin-like protein, and it is related to the electron transfer subunits of respiratory enzymes. The protein contains a typical Tat signal peptide for translocation across the membrane, and four [4Fe-4S] clusters are predicted by sequence analysis. DsrJ covalently binds three hemes c, one of which is characterized by an unusual His/Cys ligation (10).
While we have recently described the production and characterization of A. vinosum DsrJ in Escherichia coli (10), we provide a detailed report on the production and characterization of the remaining components of the DsrMKJOP transmembrane complex in the present work. A study of the individual components permits a more straightforward analysis of the redox cofactors, without interference from the other components, as happens in the whole complex. This individual analysis provided novel insights into the A. vinosum DsrMKJOP proteins.
MATERIALS AND METHODS
Bacterial strains, media, and growth conditions.
The bacterial strains and plasmids used are described in Table 1. E. coli strains were cultivated in LB or 2× YT medium (29). E. coli DH5α was used for molecular cloning. A. vinosum was grown as described earlier (6). Antibiotics were used at the indicated concentrations (in μg ml−1): for E. coli, ampicillin, 100; kanamycin, 50; chloramphenicol, 25; for A. vinosum, kanamycin 10; rifampin, 50.
TABLE 1.
Bacterial strains and plasmids used in this study
| Bacterial strain or plasmid | Genotype or phenotype | Reference or source |
|---|---|---|
| Strains | ||
| Escherichia coli | ||
| DH5α | F− φ80dlacZΔM15 Δ(lacZYA-argF)U169 recA1 endA1 hsdR17(rK− mK+) supE44 λ−thi-1 gyrA relA1 | 13 |
| BL21(DE3) | F−ompT hsdSB(rB− mB−) gal dcm met (DE3) | Novagen |
| C41(DE3) | Derived from BL21(DE3); at least one uncharacterized mutation | 32 |
| C43(DE3) | Derived from C41(DE3); at least two uncharacterized mutations | 32 |
| Allochromatium vinosum | ||
| Rif 50 | Rifr, spontaneous rifampin-resistant mutant of A. vinosum DSMZ 180T | 25 |
| ΔdsrM mutant | Rifr, in-frame deletion in dsrM | 42 |
| ΔdsrK mutant | Rifr, in-frame deletion in dsrK | 42 |
| ΔdsrP mutant | Rifr, in-frame deletion in dsrP | 42 |
| ΔdsrJ+Jhis mutant | Rifr, Kanr, ΔdsrJ mutant carrying plasmid pBBRJHis | 10 |
| Plasmids | ||
| pET11a | Apr, T7 promoter | Novagen |
| pET22b | Apr, His tag, T7 promoter | Novagen |
| pPR-IBA-1 | Apr, Strep tag, T7 promoter | IBA |
| pASK-IBA-3 | Apr, Strep tag, Tet promoter | IBA |
| pMexC | Apr, His tag, NdeI-BamHI fragment of amplified dsrM in pET11a | This work |
| pPexC | Apr, His tag, NdeI-EcoRI fragment of amplified dsrP in pET22b | This work |
| IBAdsrK | Apr, Strep tag, EcoRI-Eco47III fragment of amplified dsrK in pPR-IBA-1 | This work |
| IBAdsrO | Apr, Strep tag, EcoRI-XhoI fragment of amplified dsrO in pASK-IBA-3 | This work |
Recombinant DNA techniques.
Chromosomal DNA of A. vinosum was obtained by standard methods as described in reference 1. Restriction enzymes, T4 ligase, and Pfu DNA polymerase were obtained from Fermentas (St. Leon-Rot, Germany) and used according to the manufacturer's instructions. Oligonucleotides for cloning were obtained from Eurofins MWG (Ebersberg, Germany).
Construction of expression plasmids.
For the amplification of dsrM, primers M1f (GGTGCGGACATATGGCGTTTCTGAC) and Mhis2rev (GGAGGGGATCCTCAGTGATGATGATGATGATGTGACTGCTCGAGC) were used. While M1f introduced an NdeI restriction site, primer Mhis2rev fused a His tag coding sequence followed by a BamHI restriction site to the dsrM gene. NdeI and BamHI were used to clone the DNA fragment into pET11a (Merck Novagen, Darmstadt, Germany).
For amplification of dsrK, primers DsrKEcoRI-f (GAGGTGAATTCATGGCCAAGGC) and DsrKEco47III-r (GGTCAAGCGCTCTCGTCTGTTTC) were used, introducing restriction sites for EcoRI and Eco47III, respectively. The resulting DNA fragment was digested with EcoRI and Eco47III and ligated into pPR-IBA-1 (IBA BioTagnology, Göttingen, Germany), which was digested with the same restriction enzymes.
dsrO was amplified without the signal peptide coding sequence by the use of DsrOEcoRI-f2 (CACAGGAATTCGGCCGCG) and DsrOXho-r (CTCGCTCGAGAAGGCTGAA), introducing EcoRI and XhoI restriction sites, respectively. The amplified product was cloned into pASK-IBA-3 (IBA Tagnology, Göttingen, Germany) with the appropriate restriction enzymes.
Amplification of dsrP with the primers DsrPNde-f2 (TATCCATATGAAACGAGTCGTCTATC) and DsrPEco-r (CATCTGAATTCGGCTTGGCG) leads to an introduction of NdeI and EcoRI restriction sites that were used to clone the DNA fragment into pET22b (Merck Novagen, Darmstadt, Germany).
Production and purification of recombinant proteins from E. coli.
The production of DsrM and DsrP was carried out in E. coli C43(DE3) and C41(DE3), respectively. An overnight culture was used to inoculate 500 ml of 2× YT medium that was supplied with ampicillin, and the cells were grown at 37°C and 180 rpm until an optical density at 600 nm (OD600) of 0.5 was reached. IPTG (isopropyl-β-d-thiogalactopyranoside) was added in a final concentration of 1 mM. For DsrM the medium was additionally supplied with 0.4 mM aminolevulinic acid. Cells were further grown at 25°C and 180 rpm overnight and harvested by centrifugation. Cells were washed in buffer A (50 mM NaH2PO4, 300 mM NaCl) and stored at −20°C until used. For protein purification, cells were thawed, resuspended in buffer A, and broken by sonication. Insoluble material was removed, and the membrane fraction was prepared by ultracentrifugation. The resulting pellet was resuspended in buffer A, and 1% (wt/vol) dodecyl maltoside (DDM) was added. Solubilization was carried out with gentle stirring for 1 to 3 h on ice, followed by another ultracentrifugation. This step was repeated twice. Supernatants containing solubilized protein were combined and purified via nickel-chelate affinity chromatography according to the manufacturer's instructions (Qiagen, Hilden, Germany). All buffers contained 0.1% DDM.
DsrK was produced in E. coli C41(DE3) harboring plasmid pPRIBAdsrK. An overnight culture was used to inoculate 400 ml of LB medium supplied with ampicillin. The cells were grown at 37°C and 180 rpm until an OD600 of 0.6 was reached. Expression was induced by the addition of 0.1 mM IPTG. After 2 h under the same conditions, the cells were harvested by centrifugation. The pellet was resuspended in buffer W (100 mM Tris-HCl, 150 mM NaCl, pH 7.5), and the cells were broken by sonication. Preparation of the membrane fraction and solubilization were carried out as described for DsrM with the exception that Triton X-100 was used as detergent. DsrK was purified by Strep-Tactin affinity chromatography according to the manufacturer's instructions (IBA Tagnology, Göttingen, Germany). All buffers contained 0.1% Triton X-100. For in vitro reconstitution of FeS clusters, a 50-fold molar excess of dithiothreitol (DTT) was added to the purified protein and 7-fold molar excesses of FeCl2 and Na2S were added dropwise under anoxic conditions. The mixture was incubated overnight with gentle stirring at 6°C. Insoluble material was removed by centrifugation, and the protein was applied to a HiTrap desalting column to remove excess iron and sulfide.
For the production of DsrO, an overnight culture of E. coli BL21(DE3) harboring IBAdsrO was used to inoculate 400 ml LB medium supplied with ampicillin. Cells were grown until an OD600 of 0.6 was reached, and expression was induced by the addition of anhydrotetracycline in a final concentration of 0.2 μg/ml. After 3 h the cells were harvested by centrifugation. Cells were resuspended in buffer W and lysed by sonication. After insoluble cell material was removed by centrifugation, DsrO was purified by Strep-Tactin affinity chromatography according to the manufacturer's instructions (IBA Tagnology, Göttingen, Germany). DsrC was overproduced and purified as described in reference 7.
Protein techniques.
Tricine-SDS-PAGE was carried out by the method of Schagger (43). Immunoblot analysis was done as described earlier (7). Heme staining in acrylamide gels was done by the method of Thomas et al. (47). When b-type cytochromes were applied to heme staining, lithium dodecyl sulfate (LDS) was used instead of SDS, electrophoresis was carried out at 4°C, and the samples were not boiled prior to the electrophoresis. Heme quantification was done by the method of Berry and Trumpower (2). Protein concentration of purified protein was determined with the bicinchoninic acid (BCA) kit from Pierce (Rockford, IL).
Other analytical methods.
Nonheme iron was determined by the method of Massey et al. (30), and acid-labile sulfur was analyzed by the method of King and Morris (20).
Spectroscopic methods.
UV/Vis spectra were recorded in an analytic Jena Specord 210 spectrophotometer. Electron paramagnetic resonance (EPR) spectra were recorded using a Brucker EMX spectrometer equipped with an ESR 900 continuous-flow helium cryostat from Oxford Instruments.
Redox titrations.
Redox titrations were performed as recently described (10) with the exception that dichlorophenolindophenol and trimethylhydroquinone were used as additional redox mediators.
Production and purification of DsrMKJOP from A. vinosum.
For the purification of DsrMKJOP from A. vinosum, the strain A. vinosum ΔdsrJ+Jhis was used (10). This strain carries an in-frame deletion of the chromosomal dsrJ gene and produces a plasmid-encoded His-tagged DsrJ. Purification was carried out as described recently (10). The preparation of DsrMKJOP from A. vinosum ΔdsrJ+Jhis was carried out from 9 g of cells that were derived from 2 liters of a 5-day-old culture.
Quinone interaction assays.
All experimental steps were carried out inside an anaerobic chamber. An 0.1 mM concentration of the water-soluble menaquinone analogue menadione was first reduced by the use of a 2-fold molar excess of sodium borohydride. Complete reduction was verified by UV/Vis spectroscopy. The resulting menadiol was used for the desired reduction assays. As a control experiment, a 50-fold molar excess of sodium borohydride was added to the protein solution. Sodium dithionite was used to achieve full reduction of the hemes.
Coelution assays.
To investigate protein-protein interaction between DsrK and DsrC, coelution assays were carried out by taking advantage of the Strep tag fused to the C terminus of DsrK. DsrC (10 pmol) and DsrK (8.1 pmol) in a final volume of 2 ml were incubated at room temperature for 30 min with gentle agitation. The mixture was then applied to a Strep-Tactin superflow column (IBA Tagnology, Göttingen, Germany) with a column volume of 0.5 ml, followed by purification according to the manufacturer's instructions. A control experiment was done under the same conditions with the exception that DsrK was omitted in the sample that was applied to the Strep-Tactin column.
Bioinformatical methods.
Sequence alignments were carried out using the Clustal W algorithm. The helical wheel projection applet of the University of Virginia (Charlottesville, VA), available at http://cti.itc.virginia.edu/∼cmg/Demo/wheel/wheelApp.html, was used for the prediction of amphipathic helices. Transmembrane helices were predicted using MEMSAT-SVM (33).
RESULTS AND DISCUSSION
Purification of DsrMKJOP from A. vinosum.
First trials for purification of DsrMKJOP from A. vinosum membranes were carried out via classical chromatographical methods following the enrichment of DsrK (7). However, the complex obtained appeared to be unstable under the conditions used since no cytochromes were detected by UV/Vis spectroscopy or heme staining. While substoichiometric amounts of DsrJ could be detected by the use of a specific antibody, the presence of DsrM and DsrP in the preparation could not be verified (7). Instead, large amounts of dissimilatory sulfite reductase (DsrAB) were copurified. The presence of this siroheme-containing protein further prevented spectroscopic characterization of the membrane-associated Dsr components.
Here, we applied a different strategy and expressed dsrJ that was engineered with a His tag coding sequence in a ΔdsrJ A. vinosum host. Thus, we were in the position to successfully enrich the DsrMKJOP complex by nickel-chelate affinity chromatography. The visible spectrum of the sample indicates the presence of the cofactors predicted for the DsrMKJOP complex: hemes b and c and FeS clusters (Fig. 1). The spectrum in oxidized state is dominated by the intense Soret peak at 410 nm that shifts to 419 nm upon reduction. In the reduced state α and β peaks were observed at 554 and 522 nm, respectively. The α peak is very broad, and the β peak displays a shoulder at 530 nm, indicating that hemes b and c were simultaneously present in the preparation. This was confirmed by the pyridine hemochrome method whereby the heme b/heme c ratio was determined to be 1:1.26, indicating almost-stoichiometric amounts of the two types of hemes within the DsrMKJOP complex. When menadiol was used for reduction, the α peak had a lower intensity and displayed a maximum at 559 nm, indicating that only the hemes b can be partially reduced by the soluble menaquinol analogue (Fig. 1). The broad absorbance in the 400- to 500-nm region indicates the presence of FeS clusters. The spectrum showed no characteristic signals for the siroheme sulfite reductase, revealing that the DsrMKJOP complex was successfully enriched from A. vinosum without copurification of DsrAB, as previously observed (7). The weak peaks at 485 and 590 nm are probably due to a contamination by carotenoids, which are ubiquitous in the membranes of purple sulfur bacteria. These peaks were also observed in the UV/Vis spectrum of A. vinosum wild-type (wt) membrane extracts that were subjected to nickel-chelate affinity chromatography in a control experiment. No heme compounds were detected in the UV/Vis spectrum of the latter preparation. This showed that, although the DsrMKJOP preparation was not completely homogeneous, the cytochromes of the complex fully accounted for the heme-specific features of the spectrum. Identification of the Dsr proteins in Coomassie blue-stained Tricine-SDS-PAGE gels was not possible since the dominant bands were obviously contaminating proteins that were also present when A. vinosum wild-type extracts were purified via affinity chromatography under identical conditions (Fig. 2). Nevertheless, DsrM, DsrK, DsrJ, and DsrO were readily detected in this preparation by Western blotting, but the presence of DsrP could not be ascertained as a specific antibody was not available (Fig. 2). The theoretical mass of DsrM is 27.9 kDa; however, it appears to be smaller upon SDS-PAGE analysis. Anomalous migration of DsrM has also been observed for the corresponding protein from Archaeoglobus fulgidus (27) and is generally observed in membrane proteins (38). A faint second band at ∼40 kDa was observed with the anti-DsrM antibody; it most likely represents the dimeric form of the protein. A band of the same size was observed for the recombinant protein purified from E. coli (not shown). DsrK was detected above the 55-kDa band of the molecular mass marker, while DsrO ran slightly below the 26-kDa band (Fig. 2). This is in accordance with the calculated value of the processed protein, while the nonprocessed protein would have a molecular mass of 28.9 kDa. It may be concluded that the signal peptide of the protein is cleaved off in A. vinosum as previously suggested (7), although we did not achieve final proof for this proposal. In contrast to this, the signal peptide of DsrJ is not cleaved off and serves as membrane anchor for the protein as recently shown (10). The molecular mass of DsrJ is calculated to be 18.16 kDa, including the signal peptide and three hemes, while the processed form has a calculated molecular mass of 15.6 kDa. DsrJ was observed in the monomeric and in the dimeric form (Fig. 2) (10). Heme staining of Tricine-SDS-PAGE gels identified DsrJ as the sole c-type cytochrome in this preparation (Fig. 2). When Tricine-LDS-PAGE gels run at 4°C were subjected to heme staining, the strong signals of DsrJ multimers and the resulting background prevented the detection of b-type cytochromes (Fig. 2). Furthermore, the smear on top of the resolving gel gave a strong signal, indicating that some amount of cytochromes did not enter the gel (Fig. 2). Most probably, the hydrophobic proteins DsrM and DsrP did not enter the gel to some extent in this preparation.
FIG. 1.

Visible spectra of enriched DsrMKJOP from A. vinosum in the oxidized state (dotted line) and in the dithionite-reduced state (solid line). The inset shows the 500- to 600-nm region in menadiol reduction assays: oxidized (dotted line), after addition of menadiol (dashed line), and after addition of dithionite (solid line).
FIG. 2.

Tricine-SDS-PAGE analysis of enriched DsrMKJOP from A. vinosum ΔdsrJ+Jhis. MM, molecular mass marker. (Left) Western blot analysis using anti-DsrM (M), anti-DsrK (K), anti-DsrJ (J), and anti-DsrO (O) antisera. (Middle) Heme staining in Tricine-SDS-PAGE (SDS) and in Tricine-LDS-PAGE (LDS). (Right) Coomassie blue-stained Tricine-SDS-PAGE of the fraction enriched in DsrMKJOP purified from A. vinosum ΔdsrJ+Jhis (Jhis) and fractions obtained from A. vinosum wild type under identical conditions (wt).
To investigate the individual components of the A. vinosum DsrMKJOP complex, we produced them in E. coli as the heterologous host. While we have recently reported the production and characterization of the triheme cytochrome c DsrJ (10), we report here the characterization of the remaining components of the DsrMKJOP complex.
DsrM is a membrane protein containing two low-spin hemes b.
DsrM was produced in E. coli C43. The prepared membranes revealed a red color in contrast to control cells. Therefore, we were in the position to perform EPR spectroscopy on membranes containing DsrM. Four hundred fifty micrograms of DsrM was obtained from 1 liter of expression culture. The protein was identified by the use of a specific anti-DsrM antibody, and a positive signal was observed in a heme staining assay after Tricine-LDS-PAGE (not shown). Heme quantification revealed a 1.4 heme/protein monomer, suggesting that some protein did not fully incorporate the 2 hemes b predicted from the sequence. The UV/Vis spectrum of DsrM (Fig. 3) exhibited values typical of hemes b, with a Soret peak at 413 nm that shifted to 426 nm upon reduction and α and β peaks at 559 and 530 nm, respectively. The broad α peak could be better resolved at 77 K, revealing a split alpha band with maxima of 554 and 559 nm (Fig. 3A). Similar spectra have been observed for heterodisulfide reductases from Methanosarcina barkeri (16, 17) and Methanosarcina thermophila (46) and also for HmeCD purified from Archaeoglobus profundus (28). These enzymes are membrane bound via the subunits HdrE and HmeC, respectively, which are related to DsrM. The almost similar intensities of the two peaks in the low-temperature spectra suggest that two distinct types of hemes are present in equal amounts in DsrM, as has been proposed for HdrE from M. thermophila (46). DsrM was shown to be partly reduced after addition of menadiol (Fig. 3B), and the reduction was found to be specific, as adding a 50-fold molar excess of the much stronger reductant sodium borohydride had no effect on the spectrum of DsrM. This indicates that the protein interacts with quinones in vivo. A redox titration monitoring the absorbance changes of the Soret band at 426 nm by visible spectroscopy was performed to estimate the midpoint redox potential of the hemes in DsrM. The experimental points are best fitted by adding two Nernst equations with redox potentials of +60 mV and +110 mV in a 1:1 ratio with an error of ±10 mV (Fig. 4). HdrE, HmeC, and DsrM belong to the nitrate reductase gamma subunit superfamily (cl00959), the most prominent member of which is NarI from E. coli. The nitrate reductase NarGHI from E. coli is a well-characterized enzyme that catalyzes the oxidation of quinol by nitrate, which results in the generation of a proton gradient across the membrane (see references 4 and 41 for reviews). Like DsrM, NarI contains five transmembrane helices and binds two hemes b, where each heme is axially ligated by one histidine from helix II and helix V, respectively. One heme is located toward the periplasmic side of the membrane bilayer and is referred to as the distal heme (heme bD), whereas the other is located toward the cytoplasmic side and is referred to as the proximal heme (heme bP). The proximal heme has a relatively low redox potential of +20 mV, and the distal heme has a relatively high redox potential of +110 mV (39). Therefore, the two hemes are also often referred to as heme bL and heme bH, respectively (26, 39). The hemes function as a conduit for electron flow from a quinone binding site that has been identified on the periplasmic side of NarI in close proximity to heme bD, and the electrons are then transferred via the FeS clusters in NarH to the catalytic subunit NarG (3, 22, 26, 40).
FIG. 3.
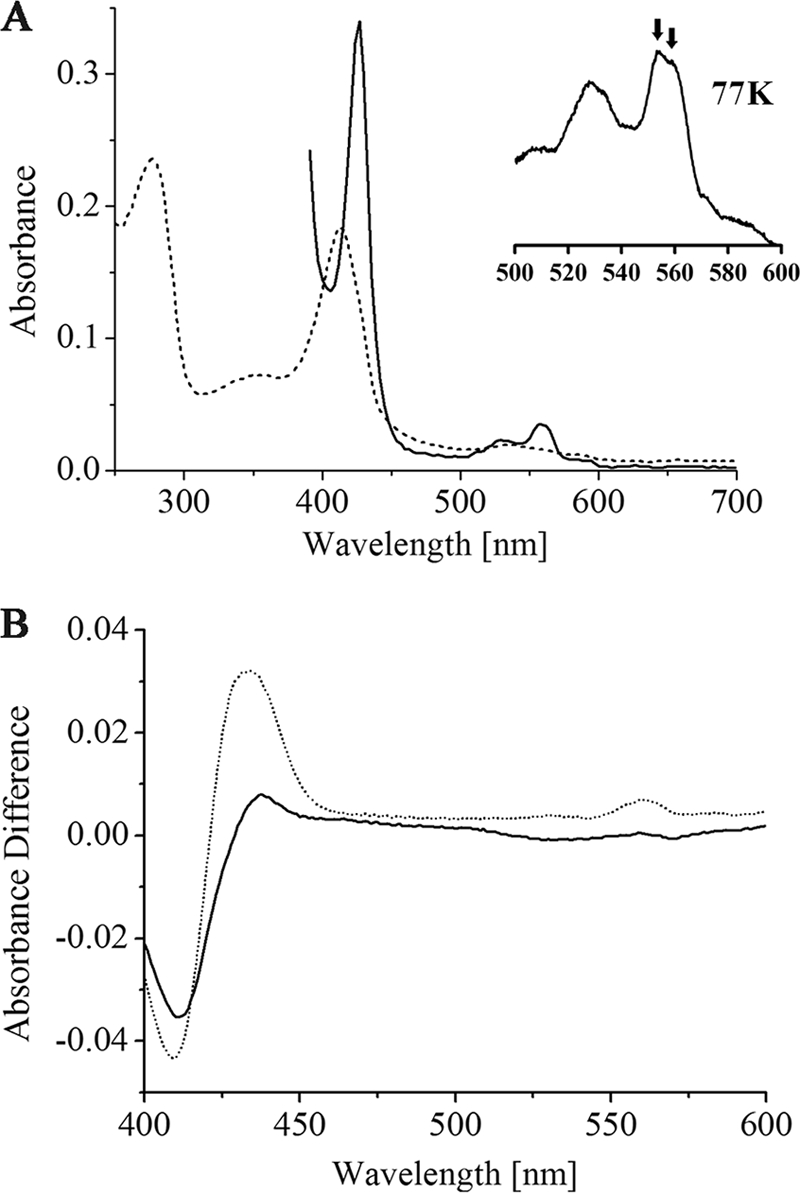
(A) UV/Vis spectra of recombinant DsrM in the oxidized state (dotted line) and in the dithionite-reduced state (solid line); the inset in spectrum A is a close-up of the 500- to 600-nm region recorded at 77 K; the arrows indicate the two maxima of the split α band. (B) Reduced minus oxidized difference spectra of menadiol reduction assays of DsrM, after addition of menadiol (solid line) and after addition of dithionite (dotted line).
FIG. 4.

Redox titration of DsrM followed by UV/visible spectroscopy at 426 nm; the solid line corresponds to a simulation obtained by adding two Nernst equations with redox potentials of +60 and +110 mV.
The localization of the hemes in DsrM is most likely similar to that in NarI because the histidines that serve as axial heme ligands are conserved in DsrM and are also located within the same transmembrane helices (37). Furthermore, with the determined midpoint potentials of +60 and +110 mV for the hemes in DsrM and the quinol reactivity of the protein, an electron flow similar to the situation in NarI, namely, from the quinol via heme bL and bH to the adjacent subunit, can be proposed for DsrM. Similar electron flow has also been proposed for HmeC from Archaeoglobus species and for HdrE from Methanosarcina species (27, 28). The electron donor of the latter is the more negative methanophenazine, and in accordance with that, the midpoint redox potentials of the hemes in M. thermophila HdrE were determined to be −23 and −180 mV (46).
The EPR spectrum of E. coli membranes containing DsrM revealed a signal with a gmax of 2.97 and a gmed of 2.25, while negative-control membranes revealed negligible features in this area (see Fig. S1 in the supplemental material). Other features that can be observed in both types of membranes are the g value of 6.0 that can be attributed to high-spin heme compounds, the signal with a g value of 4.3 that is due to nonspecific iron, and other signals with g values lower than 2. Since the intensities of the peaks are very similar in the control membranes, it can be ruled out that any of the signals could be attributed to a paramagnetic species in DsrM. This includes the peak for high-spin heme compounds, and it can therefore be concluded that both hemes in DsrM are in a low-spin state. This is also consistent with the DsrMKJOP preparation from Desulfovibrio desulfuricans, where no high-spin heme was observed (36).
DsrP is a membrane-bound b-type cytochrome.
DsrP was purified from E. coli C41, and despite the fact that no hemes were expected from sequence analysis, the protein that eluted from the Strep-Tactin column revealed a reddish color. The UV/Vis spectrum of as-isolated DsrP is typical for b-type cytochromes, and it revealed that the protein was partly reduced, indicating a high redox potential for the heme(s). Ferricyanide was used to oxidize the cytochrome, and the Soret peak was observed at 412 nm. After addition of dithionite, the Soret peak shifted to 425 nm and α and β peaks were observed at 559 and 528 nm, respectively (Fig. 5A). Therefore, DsrP was identified as a b-type cytochrome. Even after optimizing expression and purification conditions, we did not achieve sufficient amounts of protein for UV/Vis redox titrations or EPR spectroscopy, as only 50 μg protein was purified from 1 liter expression culture. When UV/Vis spectroscopy was used to compare E. coli control membranes and membranes containing DsrP, significant differences were not observed and DsrP could not yet be identified as a b-type cytochrome. This was possible only with the pure recombinant protein obtained after affinity chromatography. DsrP is a member of the NrfD/PsrC family and bears highest similarity with the PsrC subunit of polysulfide reductases. The identification of DsrP as a b-type cytochrome is highly remarkable since, to the best of our knowledge, DsrP provides the first experimental evidence for heme binding in the NrfD/PsrC family. The first structure of a protein from this family, PsrC from Thermus thermophilus, shows that it does not contain heme (19). The only NrfD/PsrC family member for which heme binding has been debated on the basis of sequence analysis is HybB, the membrane subunit of hydrogenase 2 from E. coli (9, 31). Previous comparisons of DsrP sequences did not identify possible conserved heme-ligating residues, leading to the conclusion that these proteins generally do not contain heme (7, 36). However, when the comparison is restricted to sequences from sulfur-oxidizing prokaryotes, three conserved amino acids that may serve as axial heme ligands become apparent: histidine 42, methionine 47, and histidine 57 (A. vinosum numbering) (see Fig. S3 in the supplemental material). The histidines especially are likely candidates, since they are located within or close to predicted transmembrane helices (see Fig. S2). These conserved residues are present only in proteobacterial sulfur-oxidizing bacteria, and they are absent in sulfate-reducing bacteria and in the Chlorobiaceae (36). Since no hemes b were detected in DsrP from D. desulfuricans (29), it is possible that only DsrP proteins from proteobacterial sulfur-oxidizing bacteria bind heme.
FIG. 5.

(A) UV/Vis spectra of recombinant DsrP in the oxidized state (dotted line) and in the dithionite-reduced state (solid line). (B) PAGE analysis of recombinant DsrP: Tricine-SDS-PAGE with Coomassie blue stain (Coo) and after Western blotting using anti-His antibody (blot) and Tricine-LDS-PAGE after heme staining (heme). MM, molecular mass marker. (C) Reduced minus oxidized difference spectra of menadiol reduction assays of DsrP, after addition of menadiol (solid line) and after addition of dithionite (dotted line).
DsrP was identified in Western blot analysis by using an anti-His tag antibody as well as in a Coomassie blue-stained Tricine-SDS gel and in heme staining (Fig. 5B). Two signals, which most likely represent the monomeric and the dimeric forms of the protein, were observed in each case. The detection of the protein in Tricine-SDS-PAGE gels is quite remarkable, since the related subunits of several CISM holoenzymes were not visible upon denaturing polyacrylamide gel electrophoresis due to their highly hydrophobic character (8, 18, 21, 23, 45). Indeed, DsrP and its homologue HmeB were not detected in the preparations from D. desulfuricans and Archaeoglobus fulgidus, respectively (27, 36).
The majority of amino acids that have been found to participate in quinone binding in T. thermophilus PsrC are conserved in DsrP (see Fig. S2A in the supplemental material), and these residues are located on the periplasmic side of the membrane as evident from a topology model of DsrP (see Fig. S2B). Indeed, DsrP was shown to be partially reduced after addition of menadiol (Fig. 5C). While DsrM may work as a quinol oxidase donating electrons to DsrK, DsrP could act as a quinone reductase, just as the related HybB does.
DsrK is a monotopic membrane protein and interacts with DsrC.
DsrK was most efficiently produced in E. coli C41, a strain well suited for the production of membrane proteins (32). Two hundred micrograms of protein was obtained per liter expression culture. DsrK was exclusively found in the membrane fraction and could not be washed off the membranes. The protein could be solubilized with Triton X-100 and precipitated in the absence of the detergent. After in vitro reconstitution of FeS clusters, the protein showed a broad absorbance between 300 and 500 nm in the UV/Vis spectrum (see Fig. S4A in the supplemental material). Based upon sequence analysis, DsrK contains two classical CX2CX2CX3C binding sites for [4Fe-4S] clusters and one C-terminal CCG domain with the sequence CX43CCGX40CX2C that may bind an additional putatively catalytic [FeS] cluster (36). Although some amount of recombinant DsrK could be purified from E. coli membranes and in vitro reconstitution revealed the presence of FeS clusters, the total protein yield was too low for EPR spectroscopy. Therefore, we were not in the position to elucidate if a [4Fe-4S]3+ cluster—expected to be ligated by the CCG domain—is present in DsrK. Such a [4Fe-4S]3+ cluster has been detected in heterodisulfide reductase (HDR) upon addition of HS-CoM, and it is believed to be an intermediate during enzymatic turnover of HDR (15). A similar paramagnetic species has been observed in DsrMJKOP from D. desulfuricans and in Hme from Archaeoglobus fulgidus; however, it was in the as-isolated forms without addition of a putative substrate (27, 36). DsrK contains only one of the CCG domains, while the members of the CCG domain superfamily contain two. However, it has more recently been shown that only the C-terminal domain—the one that is conserved in DsrK—binds the catalytic FeS cluster, whereas the other domain provides ligands to a Zn site (11, 12).
Previously, we reported that DsrK is associated with the membrane fraction of A. vinosum since it can be purified from the membrane fraction (7). Since no transmembrane helices are predicted for DsrK and the hydropathy profile indicates that it is mainly hydrophilic, we suggested earlier that DsrM or DsrP serves as a membrane anchor for the protein (7). However, this was not experimentally verified and the results with the recombinant protein indicate that it binds to the membrane on its own. To clarify this point, we prepared membrane fractions of A. vinosum wild type (wt) and of strains carrying in-frame deletions in dsrM and dsrP (42), respectively, and analyzed them for the presence of DsrK by Western blot analysis after Tricine-SDS-PAGE. DsrK was present not only in membranes isolated from A. vinosum wt but also in membranes of the ΔdsrM and ΔdsrP strains (Fig. 6). Furthermore, DsrK was detected neither in the soluble fraction of the wild type nor in those of the mutants. We can therefore confidently rule out that DsrK is attached to the membrane via DsrM or DsrP. In accordance, sequence analysis using the helical wheel projection applet revealed the presence of an amphipathic α-helix at the N terminus of DsrK, suggesting monotopic membrane anchoring of the protein. Similar membrane anchoring has been reported for SdhE (formerly SdhC) from Acidianus ambivalens (24), which is closely related to HdrB.
FIG. 6.

Western blot analysis of A. vinosum wt and deletion mutant membrane fractions by using anti-DsrK antibody; MM, molecular mass marker; ΔK, ΔdsrK mutant; wt, wild type; ΔM, ΔdsrM mutant; ΔP, ΔdsrP mutant. The arrow indicates the signal that corresponds to DsrK.
It has been suggested that DsrK is the catalytic subunit of the DsrMKJOP complex, that it acts as a (hetero)disulfide reductase, and that the protein DsrC might be its substrate (37). This protein has two strictly conserved cysteine residues at its carboxy terminus, and these have been shown to form specific disulfide bridges (5) that could be acted upon by DsrK. We therefore analyzed the interaction of recombinant DsrK and DsrC by coelution assays (Fig. 7). When subjected to Strep-Tactin affinity chromatography, DsrC did not bind to the affinity matrix and eluted during the first wash steps. However, when DsrC was incubated with DsrK carrying a Strep tag before being applied to the Strep-Tactin matrix, hardly any DsrC was detected in the wash fractions and the majority of DsrC eluted specifically together with DsrK (Fig. 7). These coelution assays show that DsrK and DsrC interact with each other.
FIG. 7.

DsrK-DsrC coelution assays: detection of DsrC using anti-DsrC antibody in the negative control without DsrK (A) and in the assay with DsrC plus DsrK (B) and detection of DsrK using anti-DsrK antibody (C). W1 to W5, washing fractions 1 to 5, respectively; E1 and E2, elution fractions 1 and 2, respectively.
DsrO binds FeS clusters.
Approximately 3 mg DsrO was purified from 1 liter expression culture. The resulting protein solution revealed a brownish color, and the UV/Vis spectrum indicated the presence of FeS clusters. The spectrum shows maxima at 325 and 417 nm with a shoulder at 460 nm (see Fig. S4B in the supplemental material), which indicates the presence of [2Fe-2S] clusters rather than [4Fe-4S] clusters. A very similar spectrum was observed for IscA, which binds a [2Fe-2S] cluster (48). Indeed iron and sulfur quantification revealed only 4.0 (±0.1) mol iron and 3.9 (±0.2) mol sulfur per mol protein instead of the 16 mol that was expected for a protein that binds four [4Fe-4S] clusters. The DsrMKJOP complex from D. desulfuricans and the Hme complex from Archaeoglobus fulgidus do not contain [2Fe-2S] clusters, suggesting that the wrong cluster form has been inserted in the recombinant protein by the heterologous host. The insertion of wrong cluster forms has also been reported for other FeS proteins that were produced in E. coli (11, 12).
Proposed function of the DsrMKJOP transmembrane complex.
The DsrMKJOP complex has been purified and characterized from D. desulfuricans, but it could not be solved whether DsrJ is an electron entry or exit point (36). However, a more recent model for the function of the complex proposes electron transfer from the periplasm into the cytoplasm (34). The structure of the Desulfovibrio vulgaris dissimilatory sulfite reductase has been solved, with DsrC bound in a cleft between DsrA and DsrB, and a mechanism for the process of sulfite reduction, including DsrAB, DsrC, and DsrMKJOP, in which DsrAB persulfurates DsrC at one of its two conserved cysteine residues has been proposed (34). DsrC then dissociates from the sulfite reductase followed by the reduction of the persulfide by the second conserved cysteine. This results in release of H2S and the formation of an intramolecular disulfide in DsrC. For regeneration the disulfide may be reduced by the DsrMKJOP transmembrane complex.
The two redox active cysteines in DsrC also play a central role in the model proposed by Cort et al. for sulfur oxidation in A. vinosum (5). In this model DsrC is involved in sulfur transfer reactions together with DsrEFH, a protein that is found in sulfur-oxidizing but not in sulfate-reducing organisms. DsrC is suggested to act as a substrate-donating protein for sulfite reductase. In sulfur oxidizers this enzyme is thought to operate in the opposite direction from sulfate reducers (44). In one of the suggested models for sulfur oxidation in A. vinosum, sulfide is reductively released as a substrate for sulfite reductase and an intramolecular disulfide is proposed to be formed in DsrC, similar to the situation suggested for Desulfovibrio species (5). This disulfide has to be reduced to regenerate free thiol groups to restart the cycle. By coelution assays, we have shown here that DsrK indeed interacts with DsrC, and we propose reduction of the DsrC disulfide by DsrK in close analogy to the model for Desulfovibrio. The sulfur metabolisms of D. vulgaris and A. vinosum are the reverse of each other, and it is believed that the dissimilatory sulfite reductase also acts in opposite directions in the two organisms (44). This may lead to the assumption that electron flow through the A. vinosum DsrMKJOP must be exactly the reverse of the electron flow proposed for Desulfovibrio species. However, our results strongly suggest electron flow from the periplasm into the cytoplasm also in A. vinosum. We have recently reported that D. vulgaris DsrJ can perfectly replace its homologue in A. vinosum, indicating that the two can perform the same function in the different organisms (10). Here, we have furthermore shown that DsrM is a quinone-reactive diheme cytochrome b and that the hemes have midpoint potentials of 60 and 110 mV, respectively. The significant similarity to NarI and HdrE suggests electron flow to DsrK (related to HdrD). Moreover, since in DsrMKJOP only the hemes b but not the hemes c are reducible by menadiol, electron flow from DsrM to DsrJ—possibly via DsrO—is most unlikely. The same has also been observed for the purified Dsr complex from D. desulfuricans (36), and it is also in agreement with the difference in the determined midpoint redox potentials of the recombinant A. vinosum proteins: the potentials of the hemes in DsrJ have been determined to be −20, −200, and −220 mV (10), i.e., significantly more negative than those of the hemes in DsrM.
Figure 8 summarizes the proposed model for the function of the DsrMKJOP transmembrane complex. DsrJ may be involved in the oxidation of a putative sulfur substrate in the periplasm as recently proposed (10), and the released electrons would be transported across the membrane via the other components of the DsrMKJOP complex. The fact that DsrO is a periplasmic protein and the similarity of DsrOP to electron-transporting subunits of a number of respiratory enzyme complexes suggest that electrons may be further transported via these proteins. Since DsrP and DsrM are both quinone-interacting proteins, these two could be connected via the quinone pool. Alternatively the heme b that was found in DsrP could be involved in electron transfer from DsrP to DsrM. DsrM would then donate electrons to DsrK—the catalytic subunit of the complex. This would reduce the disulfide formed by the conserved cysteines in DsrC to generate free thiols. This would enable DsrC to restart a cycle of sulfur transfer from DsrEFH to DsrAB, as suggested by Cort et al. (5).
FIG. 8.

Proposed model for the function of the DsrMKJOP complex; the question mark represents a putative sulfur substrate or an alternative electron donor; hemes and FeS clusters in the complex are indicated by circles and cubes, respectively; possibly catalytic prosthetic groups are depicted in black. See text for further explanation.
Supplementary Material
Acknowledgments
This work was supported by grants DA 351/3-4 and 3-5 from the Deutsche Forschungsgemeinschaft, by PTDC/QUI/68368/2006 funded by Fundação para a Ciência e Tecnologia (FCT, MCES, Portugal) and the FEDER program, and by a Luso-German Joint Action funded by the German Academic Exchange Service (DAAD) and Conselho de Reitores das Universidades Portuguesas.
Footnotes
Published ahead of print on 15 October 2010.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Ausubel, F. M., R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, and K. Struhl. 1995. Current protocols in molecular biology. Greene Publishing Associates and Wiley-Interscience, New York, NY.
- 2.Berry, E. A., and B. L. Trumpower. 1987. Simultaneous determination of hemes a, b, and c from pyridine hemochrome spectra. Anal. Biochem. 161:1-15. [DOI] [PubMed] [Google Scholar]
- 3.Bertero, M. G., R. A. Rothery, N. Boroumand, M. Palak, F. Blasco, N. Ginet, J. H. Weiner, and N. C. J. Strynadka. 2005. Structural and biochemical characterization of a quinol binding site of Escherichia coli nitrate reductase A. J. Biol. Chem. 280:14836-14843. [DOI] [PubMed] [Google Scholar]
- 4.Blasco, F., B. Guigliarelli, A. Magalon, M. Asso, G. Giordano, and R. A. Rothery. 2001. The coordination and function of the redox centres of the membrane-bound nitrate reductases. Cell. Mol. Life Sci. 58:179-193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cort, J. R., U. Selan, A. Schulte, F. Grimm, M. A. Kennedy, and C. Dahl. 2008. Allochromatium vinosum DsrC: solution-state NMR structure, redox properties, and interaction with DsrEFH, a protein essential for purple sulfur bacterial sulfur oxidation. J. Mol. Biol. 382:692-707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dahl, C. 1996. Insertional gene inactivation in a phototrophic sulphur bacterium: APS-reductase-deficient mutants of Chromatium vinosum. Microbiology 142:3363-3372. [DOI] [PubMed] [Google Scholar]
- 7.Dahl, C., S. Engels, A. S. Pott-Sperling, A. Schulte, J. Sander, Y. Lübbe, O. Deuster, and D. C. Brune. 2005. Novel genes of the dsr gene cluster and evidence for close interaction of Dsr proteins during sulfur oxidation in the phototrophic sulfur bacterium Allochromatium vinosum. J. Bacteriol. 187:1392-1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dietrich, W., and O. Klimmek. 2002. The function of methyl-menaquinone-6 and polysulfide reductase membrane anchor (PsrC) in polysulfide respiration of Wolinella succinogenes. Eur. J. Biochem. 269:1086-1095. [DOI] [PubMed] [Google Scholar]
- 9.Dubini, A., R. L. Pye, R. L. Jack, T. Palmer, and F. Sargent. 2002. How bacteria get energy from hydrogen: a genetic analysis of periplasmic hydrogen oxidation in Escherichia coli. Int. J. Hydrogen Energy 27:1413-1420. [Google Scholar]
- 10.Grein, F., S. S. Venceslau, L. Schneider, S. Todorovic, P. Hildebrandt, I. A. C. Pereira, and C. Dahl. 2010. DsrJ, an essential part of the DsrMKJOP transmembrane complex in the purple sulfur bacterium Allochromatium vinosum, is an unusual triheme cytochrome c. Biochemistry 49:8290-8299. [DOI] [PubMed] [Google Scholar]
- 11.Hamann, N., E. Bill, J. E. Shokes, R. A. Scott, M. Bennati, and R. Hedderich. 2009. The CCG-domain-containing subunit SdhE of succinate:quinone oxidoreductase from Sulfolobus solfataricus P2 binds a [4Fe-4S] cluster. J. Biol. Inorg. Chem. 14:457-470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hamann, N., G. J. Mander, J. E. Shokes, R. A. Scott, M. Bennati, and R. Hedderich. 2007. A cysteine-rich CCG domain contains a novel [4Fe-4S] cluster binding motif as deduced from studies with subunit B of heterodisulfide reductase from Methanothermobacter marburgensis. Biochemistry 46:12875-12885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hanahan, D. 1983. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 166:557-580. [DOI] [PubMed] [Google Scholar]
- 14.Haveman, S. A., E. A. Greene, C. P. Stilwell, J. K. Voordouw, and G. Voordouw. 2004. Physiological and gene expression analysis of inhibition of Desulfovibrio vulgaris Hildenborough by nitrite. J. Bacteriol. 186:7944-7950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hedderich, R., N. Hamann, and M. Bennati. 2005. Heterodisulfide reductase from methanogenic archaea: a new catalytic role for an iron-sulfur cluster. Biol. Chem. 386:961-970. [DOI] [PubMed] [Google Scholar]
- 16.Heiden, S., R. Hedderich, E. Setzke, and R. K. Thauer. 1993. Purification of a cytochrome b containing H2:heterodisulfide oxidoreductase complex from membranes of Methanosarcina barkeri. Eur. J. Biochem. 213:529-535. [DOI] [PubMed] [Google Scholar]
- 17.Heiden, S., R. Hedderich, E. Setzke, and R. K. Thauer. 1994. Purification of a two-subunit cytochrome-b-containing heterodisulfide reductase from methanol-grown Methanosarcina barkeri. Eur. J. Biochem. 221:855-861. [DOI] [PubMed] [Google Scholar]
- 18.Hussain, H., J. Grove, L. Griffiths, S. Busby, and J. Cole. 1994. A seven-gene operon essential for formate-dependent nitrite reduction to ammonia by enteric bacteria. Mol. Microbiol. 12:153-163. [DOI] [PubMed] [Google Scholar]
- 19.Jormakka, M., K. Yokoyama, T. Yano, M. Tamakoshi, S. Akimoto, T. Shimamura, P. Curmi, and S. Iwata. 2008. Molecular mechanism of energy conservation in polysulfide respiration. Nat. Struct. Mol. Biol. 15:730-737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.King, T. E., and R. O. Morris. 1967. Determination of acid-labile sulfide and sulfhydryl groups. Methods Enzymol. 10:634-641. [Google Scholar]
- 21.Krafft, T., R. Gross, and A. Kröger. 1995. The function of Wolinella succinogenes psr genes in electron transport with polysulphide as the terminal electron acceptor. Eur. J. Biochem. 230:601-606. [DOI] [PubMed] [Google Scholar]
- 22.Lanciano, P., A. Magalon, P. Bertrand, B. Guigliarelli, and S. Grimaldi. 2007. High-stability semiquinone intermediate in nitrate reductase A (NarGHI) from Escherichia coli is located in a quinol oxidation site close to heme bD. Biochemistry 46:5323-5329. [DOI] [PubMed] [Google Scholar]
- 23.Laska, S., F. Lottspeich, and A. Kletzin. 2003. Membrane-bound hydrogenase and sulfur reductase of the hyperthermophilic and acidophilic archaeon Acidianus ambivalens. Microbiology 149:2357-2371. [DOI] [PubMed] [Google Scholar]
- 24.Lemos, R. S., C. M. Gomes, and M. Teixeira. 2001. Acidianus ambivalens complex II typifies a novel family of succinate dehydrogenases. Biochem. Biophys. Res. Commun. 281:141-150. [DOI] [PubMed] [Google Scholar]
- 25.Lübbe, Y. J., H.-S. Youn, R. Timkovich, and C. Dahl. 2006. Siro(haem)amide in Allochromatium vinosum and relevance of DsrL and DsrN, a homolog of cobyrinic acid a,c-diamide synthase, for sulphur oxidation. FEMS Microbiol. Lett. 261:194-202. [DOI] [PubMed] [Google Scholar]
- 26.Magalon, A., D. Lemesle-Meunier, R. A. Rothery, C. Frixon, J. H. Weiner, and F. Blasco. 1997. Heme axial ligation by the highly conserved his residues in helix II of cytochrome b (NarI) of Escherichia coli nitrate reductase A (NarGHI). J. Biol. Chem. 272:25652-25658. [DOI] [PubMed] [Google Scholar]
- 27.Mander, G. J., E. C. Duin, D. Linder, K. O. Stetter, and R. Hedderich. 2002. Purification and characterization of a membrane-bound enzyme complex from the sulfate-reducing archaeon Archaeoglobus fulgidus related to heterodisulfide reductase from methanogenic archaea. Eur. J. Biochem. 269:1895-1904. [DOI] [PubMed] [Google Scholar]
- 28.Mander, G. J., A. J. Pierik, H. Huber, and R. Hedderich. 2004. Two distinct heterodisulfide reductase-like enzymes in the sulfate-reducing archaeon Archaeoglobus profundus. Eur. J. Biochem. 271:1106-1116. [DOI] [PubMed] [Google Scholar]
- 29.Maniatis, T., E. F. Fritsch, and J. Sambrook. 1982. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY.
- 30.Massey, V. 1957. Studies on succinic dehydrogenase. VII. Valency state of the iron in beef heart succinic dehydrogenase. J. Biol. Chem. 229:763-770. [PubMed] [Google Scholar]
- 31.Menon, N. K., C. Y. Chatelus, M. Dervartanian, J. C. Wendt, K. T. Shanmugam, H. D. Peck, Jr., and A. E. Przybyla. 1994. Cloning, sequencing, and mutational analysis of the hyb operon encoding Escherichia coli hydrogenase 2. J. Bacteriol. 176:4416-4423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miroux, B., and J. E. Walker. 1996. Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J. Mol. Biol. 260:289-298. [DOI] [PubMed] [Google Scholar]
- 33.Nugent, T., and D. T. Jones. 2009. Transmembrane protein topology prediction using support vector machines. BMC Bioinformatics 10:159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oliveira, T. F., C. Vonrhein, P. M. Matias, S. S. Venceslau, I. A. C. Pereira, and M. Archer. 2008. The crystal structure of Desulfovibrio vulgaris dissimilatory sulfite reductase bound to DsrC provides novel insights into the mechanism of sulfate respiration. J. Biol. Chem. 283:34141-34149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pereira, I. 2007. Respiratory membrane complexes of Desulfovibrio, p. 24-35. In C. Dahl and C. G. Friedrich (ed.), Microbial sulfur metabolism. Springer, Berlin, Germany.
- 36.Pires, R. H., S. S. Venceslau, F. Morais, M. Teixeira, A. V. Xavier, and I. A. C. Pereira. 2006. Characterization of the Desulfovibrio desulfuricans ATCC 27774 DsrMKJOP complex—a membrane-bound redox complex involved in the sulfate respiratory pathway. Biochemistry 45:249-262. [DOI] [PubMed] [Google Scholar]
- 37.Pott, A. S., and C. Dahl. 1998. Sirohaem sulfite reductase and other proteins encoded by genes at the dsr locus of Chromatium vinosum are involved in the oxidation of intracellular sulfur. Microbiology 144:1881-1894. [DOI] [PubMed] [Google Scholar]
- 38.Rath, A., M. Glibowicka, V. G. Nadeau, G. Chen, and C. M. Deber. 2009. Detergent binding explains anomalous SDS-PAGE migration of membrane proteins. Proc. Natl. Acad. Sci. U. S. A. 106:1760-1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rothery, R. A., F. Blasco, A. Magalon, M. Asso, and J. H. Weiner. 1999. The hemes of Escherichia coli nitrate reductase A (NarGHI): potentiometric effects of inhibitor binding to NarI. Biochemistry 38:12747-12757. [DOI] [PubMed] [Google Scholar]
- 40.Rothery, R. A., F. Blasco, A. Magalon, and J. H. Weiner. 2001. The diheme cytochrome b subunit (Narl) of Escherichia coli nitrate reductase A (NarGHI): structure, function, and interaction with quinols. J. Mol. Microbiol. Biotechnol. 3:273-283. [PubMed] [Google Scholar]
- 41.Rothery, R. A., G. J. Workun, and J. H. Weiner. 2008. The prokaryotic complex iron-sulfur molybdoenzyme family. Biochim. Biophys. Acta 1778:1897-1929. [DOI] [PubMed] [Google Scholar]
- 42.Sander, J., S. Engels-Schwarzlose, and C. Dahl. 2006. Importance of the DsrMKJOP complex for sulfur oxidation in Allochromatium vinosum and phylogenetic analysis of related complexes in other prokaryotes. Arch. Microbiol. 186:357-366. [DOI] [PubMed] [Google Scholar]
- 43.Schägger, H. 2006. Tricine-SDS-PAGE. Nat. Protoc. 1:16-22. [DOI] [PubMed] [Google Scholar]
- 44.Schedel, M., M. Vanselow, and H. G. Trüper. 1979. Siroheme sulfite reductase isolated from Chromatium vinosum. Purification and investigation of some of its molecular and catalytic properties. Arch. Microbiol. 121:29-36. [Google Scholar]
- 45.Schröder, I., A. Kröger, and J. M. Macy. 1988. Isolation of the sulphur reductase and reconstitution of the sulphur respiration of Wolinella succinogenes. Arch. Microbiol. 149:572-579. [Google Scholar]
- 46.Simianu, M., E. Murakami, J. M. Brewer, and S. W. Ragsdale. 1998. Purification and properties of the heme- and iron-sulfur-containing heterodisulfide reductase from Methanosarcina thermophila. Biochemistry 37:10027-10039. [DOI] [PubMed] [Google Scholar]
- 47.Thomas, P. E., D. Ryan, and W. Levin. 1976. An improved staining procedure for the detection of the peroxidase activity of cytochrome P-450 on sodium dodecyl sulfate polyacrylamide gels. Anal. Biochem. 75:168-176. [DOI] [PubMed] [Google Scholar]
- 48.Wollenberg, M., C. Berndt, E. Bill, J. D. Schwenn, and A. Seidler. 2003. A dimer of the FeS cluster biosynthesis protein IscA from cyanobacteria binds a [2Fe2S] cluster between two protomers and transfers it to [2Fe2S] and [4Fe4S] apo proteins. Eur. J. Biochem. 270:1662-1671. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.