

Abstract
Fibronectin fragments (FN-f), including the 110-kDa fragment that binds the α5β1 integrin, stimulate collagenase-3 (MMP-13) production and cartilage destruction. In the present study, treatment of chondrocytes with the 110-kDa FN-f or an activating antibody to the α5β1 integrin was found to increase tyrosine autophosphorylation (Tyr-402) of the proline-rich tyrosine kinase-2 (PYK2) without significant change in autophosphorylation (Tyr-397) of focal adhesion kinase (FAK). The tyrosine kinase inhibitor tyrphostin A9, shown previously to block a PYK2-dependent pathway, blocked the FN-f-stimulated increase in MMP-13, whereas tyrphostin A25 did not. FN-f-stimulated PYK2 phosphorylation and MMP-13 production was also blocked by reducing intracellular calcium levels. Adenovirally mediated overexpression of wild type but not mutant PYK2 resulted in increased MMP-13 production. The protein kinase C (PKC) activator phorbol 12-myristate 13-acetate stimulated PYK2 phosphorylation and MMP-13 production. MMP-13 expression stimulated by either phorbol 12-myristate 13-acetate or FN-f was blocked by PKC inhibitors including the PKCδ inhibitor rottlerin. Furthermore, PKCδ translocation from cytosol to membrane was noted within 5 min of stimulation with FN-f. Immortalized human chondrocytes, transiently transfected with MMP-13 promoter-luciferase reporter constructs, showed increased promoter activity after FN-f treatment that was inhibited by co-transfection with either of two dominant negative mutants of PYK2 (Y402F and K457A). No inhibition was seen after co-transfection with wild type PYK2, a dominant negative of FAK (FRNK) or empty vector plasmid. FN-f-stimulated MMP-13 promoter activity was also inhibited by chemical inhibitors of ERK, JNK, and p38 mitogen-activated protein (MAP) kinases or by co-transfection of dominant negative MAP kinase mutant constructs. These studies have identified a novel pathway for the MAP kinase regulation of MMP-13 production which involves FN-f stimulation of the α5β1 integrin and activation of the nonreceptor tyrosine kinase PYK2 by PKC, most likely PKCδ.
Matrix metalloproteinases (MMPs)1 are expressed by a number of different cell types and play a key role in diverse processes ranging from morphogenesis to tumor invasion to tissue remodeling (reviewed in Ref. 1). Because of their potent ability to degrade the extracellular matrix and the undesirable consequences of excess matrix degradation, the production and activation of MMPs must be tightly regulated. In articular cartilage, excess production of MMPs results in destruction of the cartilage matrix (2-4). MMP-13 (collagenase-3) is a very potent degrader of type II collagen (5), the major collagen type in cartilage, and overexpression of MMP-13 in a transgenic mouse model was found to reproduce the joint changes characteristic of osteoarthritis (6). A better understanding of the cellular mechanisms responsible for regulating MMP expression could lead to the development of novel therapies aimed at controlling excess matrix degradation.
Signals generated from the extracellular matrix and mediated through the integrin family of matrix receptors can regulate MMP expression. Treatment of cultured synovial fibroblasts with the 120-kDa fibronectin fragment (FN-f), which binds the α5β1 integrin, or with antibodies to the α5β1 integrin was shown to stimulate expression of collagenase-1 (MMP-1) and stromelysin (MMP-3) (7). Similar FN-fs have been identified in arthritic cartilage and synovial fluid (8, 9) and have been found to stimulate chondrocyte-mediated cartilage matrix degradation (10, 11). We recently demonstrated that the 120-kDa FN-f stimulated human articular chondrocytes to express high levels of MMP-13 (12). The increase in MMP-13 was accompanied by an increase in expression of IL-1β, although IL-1β was not required for the initial FN-f stimulation of MMP-13 expression. These studies suggest that FN-f stimulation of integrin signaling could play an important role in mediating cartilage matrix degradation through stimulation of MMP and cytokine production.
The intracellular signaling pathways responsible for regulating MMP expression are incompletely understood. Integrin-mediated signals that stimulated MMP-1 expression by synovial fibroblasts required Rac activation and the generation of reactive oxygen species (13). MAP kinase activation has been linked to MMP-13 expression in response to IL-1β in chondrocytes (14) and in response to fibroblast culture in collagen gels (15). Both the N-terminal 29-kDa FN-f (16) and the 120-kDa FN-f (12) were found to stimulate chondrocyte MAP kinase phosphorylation, and inhibition of either MEK, JNK, or p38 inhibited the FN-f-stimulated MMP-13 production (12). Importantly, the upstream signals that mediate MAP kinase activation and subsequent MMP-13 expression have not been identified.
Depending on the cell type and the stimulus, both focal adhesion kinase (FAK) and the closely related proline-rich tyrosine kinase-2 (PYK2) have been shown to mediate signals from integrins that can lead to MAP kinase activation (17-21). PYK2 was identified as a calcium-dependent tyrosine kinase (20) and has also been called RAFTK (22) or CAKβ (23). Although a role for FAK has been demonstrated for MMP secretion in response to concanavalin A (24) and hyaluronan (25), to our knowledge the possibility that activation of either FAK or PYK2 might be required for integrin signaling which regulates MMP expression has not been studied.
The aim of the present study was to determine whether activation of FAK and/or PYK2 was required for the stimulation of chondrocyte MMP-13 expression in response to treatment with FN-f. Because PYK2 can be activated by increases in intracellular calcium and activation of protein kinase C (PKC) (18, 20), inhibitors or activators of these pathways were tested. Experiments were performed using primary human articular chondrocytes and, for transfection experiments, an immortalized human chondrocyte cell line C-28I2. We have shown previously that C-28I2 cells demonstrate chondrocyte phenotypic features including expression of type II collagen and expression of the α5β1 integrin at levels similar to primary chondrocytes (26). The present results demonstrate a role for PYK2 in mediating FN-f-induced signals that stimulate MMP-13 expression. Both increases in intracellular calcium and activation of PKC are required.
EXPERIMENTAL PROCEDURES
Reagents and Plasmids
The α5β1 integrin activating antibody (JBS5) and the 120-kDa fibronectin fragment were from Chemicon (Temecula, CA). Anti-phospho-PYK2 (Tyr-402) was from BIOSOURCE (Camarillo, CA). The 110-kDa fibronectin fragment, anti-phospho-FAK-(Tyr-397), and anti-FAK were from Upstate Biotechnology, Inc. (Lake Placid, NY). Anti-PYK2(CADTK) was a gift from Dr. Shelley Earp (University of North Carolina, Chapel Hill). The PKC antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). The MEK inhibitor PD98059, PKC inhibitors bisindolylmaleimide I (BIM) and rottlerin, tyrphostins A9 (AG 17) and A25 (AG 82), and calcium inhibitors nifedipine and BAPTA-AM were purchased from Calbiochem. The p38 inhibitor SB203580 was purchased from Sigma, and the JNK inhibitor SP600125 was a gift from Signal Pharmaceuticals (San Diego, CA). IL-1β was from R&D Systems (Minneapolis, MN). Sheep polyclonal MMP-13 antibody L2916 was generously provided by Dr. Gillian Murphy (Norwich, UK).
The PYK2 expression plasmids (wild type and dominant negative mutants Y402F and K457A) used in transient transfection experiments were provided by Drs. Archana Sanjay and Roland Baron (Yale University School of Medicine, New Haven, CT). The cDNAs encoding wild type and the same dominant negative mutant forms of PYK2 used for construction of replication-defective adenoviruses (described below) were kindly provided by Dr. Tom Parsons (University of Virginia, Charlottesville). The dominant negative FAK construct FRNK was provided by Dr. Michael Schaller (University of North Carolina, Chapel Hill); the ERK1(K71R), ERK2(K52R), and JNK(K-R) dominant negative constructs were provided by Dr. Shu Chien (University of California San Diego, La Jolla), and the p38 dominant negative (pcDNA3-dn-p38) was provided by Dr. Francis Berenbaum (Universite Pierre et Marie Curie, Paris, France).
Adenoviral Constructs
cDNAs encoding wild type and dominant negative mutants of PYK2 were subcloned in-frame into pEGFP-C1 vector (Clontech, Palo Alto, CA). The GFP-PYK2 inserts were then sequentially subcloned into pShuttle-CMV plasmid and then pAdeno-X™ viral DNA (Clontech) for the preparation of replication-defective adenoviruses. Linearized pAdeno-X+GFP-PYK2 sequences were introduced into HEK293 cells using a liposome-based transient transfection procedure (SuperFect, Qiagen, Valencia, CA). Resulting wild type and mutant GFP-PYK2 adenoviruses were amplified from cell extracts and purified by double CsCl gradient centrifugation. The multiplicity of viral infection was determined by viral dilution assay in HEK293 cells grown in 96-well clusters. An adenovirus expressing GFP alone (Adv-GFP) was used to control for nonspecific effects of adenoviral infection.
Chondrocyte Culture
Normal human ankle cartilage was obtained from tissue donors through a joint agreement between Rush Medical College and the Gift of Hope Organ and Tissue Network under a protocol approved by the Rush University institutional review board. Each donor specimen was graded for gross degenerative changes based on a modified version of the 5-point scale of Collins (see Ref. 27). Samples used for this study were grade 0 or 1. Chondrocytes were isolated by enzymatic digestion using Pronase followed by overnight digestion with collagenase-P as described previously (12). Isolated cells were resuspended in media at 1 × 106 cells per ml and added to 60-mm plates (6 ml/plate) or 6-well plates at 2 ml/well. Cells were cultured in DMEM/F-12 containing 10% fetal bovine serum and antibiotics (complete media) for 5–7 days before use. Immortalized human chondrocytes (C-28I2 cells), described in detail previously (26), were also cultured in complete media.
Chondrocyte Stimulation and Immunoblotting
Media were changed to serum-free DMEM/F-12 with antibiotics 18 h (overnight) and again 2 h before each experiment. For inhibitor studies, cells were preincubated with inhibitor for 30 min before stimulation. Although preliminary studies were performed with a 120-kDa FN-f, similar results were noted using the 110-kDa FN-f which was used in the experiments reported here because of greater availability. FN-f or α5β1 integrin activating antibody were added directly to the culture media for the indicated periods. In a previous study (12), non-activating integrin antibodies and intact fibronectin were tested as controls and found not to stimulate MAP kinase activation or MMP-13 expression in this system and so were not repeated here. Experiments were terminated with removal of media and cell lysate preparation. The conditioned media was aliquoted and stored at −70 or 4 °C with 0.1% NaN3. Cell lysates were prepared using modified cell lysis RIPA buffer: 20 mm Tris (pH 7.5), 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 1% Nonidet P-40, 0.25% deoxycholate, 2.5 mm sodium pyrophosphate, 1 mm glycerol phosphate, 1 mm Na3VO4, with 2 mm phenylmethylsulfonyl fluoride (Sigma). For PYK2 and FAK immunoblotting experiments, 2 mm diisopropyl fluorophosphate was included as an additional protease inhibitor. Cold RIPA buffer was added to cells; dishes were scraped, and lysates were centrifuged to remove insoluble material. The soluble lysate protein concentration was determined with BCA reagent (Pierce). Lysate samples containing equal amounts of total protein were separated by SDS-PAGE and transferred to nitrocellulose for immunoblotting using the ECL system (Amersham Biosciences). Conditioned media were concentrated (10:1) and analyzed by immunoblotting for MMP-13 as described (12). All immunoblotting experiments were repeated at least three times. Densitometric scanning of blots was analyzed using 1D Image Analysis software (Eastman Kodak Co.).
PKC Analysis
Cytosolic and particulate membrane fractions were isolated from cells stimulated with FN-f in order to determine translocation of specific PKC isoforms as a measure of PKC activation. After stimulation, cells were washed once for 3 min with ice-cold phosphate-buffered saline containing 1 mm diisopropyl fluorophosphate. Cells were scraped and briefly sonicated in a small volume of a hypotonic lysis buffer (1 ml of buffer/1 × 107 cells) containing 50 mm Tris-HCl (pH 7.5), 2 mm 4-(2-aminoethyl)benzenesulfonylfluoride, 1 mm EDTA, 1 mm diisopropyl fluorophosphate, 14 μm E-64, 130 μm bestatin, 1 mm leupeptin, and 0.3 μm aprotinin. Cell extracts were then gently placed on top of a 15% sucrose cushion (4 ml) and centrifuged at 75,000 × g for 45 min. The clear supernatant at the top of the sucrose was collected and used as the cytosolic fraction. After careful removal of the remaining sucrose, the pellet at the bottom of the centrifuge tube was solubilized with brief sonication in the above lysis buffer (0.5 ml buffer/pellet) with the addition of 150 mm NaCl and 1% Triton X-100 (final concentration). This lysate was transferred to a microcentrifuge tube and centrifuged at 14,000 × g for 10 min to remove insoluble cellular debris. The clear supernatant was collected and used as the particulate membrane fraction. Total protein was measured, and the samples were processed for immunoblotting as described above.
Transfection Studies
MMP-13 (SacI/BglII) and MMP-1 (XhoI/Hin-dIII) promoter regions, kindly provided by Drs. Yubo Sun and Herman Cheung (University of Miami School of Medicine, Miami, FL), were subcloned into a firefly luciferase promoterless vector, pGL2-Enhancer (Promega, Madison, WI), generating −1600MMP-13, −736MMP-13, −370MMP-13, −186MMP-13, and −562MMP-1 promoter luciferase reporter constructs. Details of these constructs including their promoter sites are reported elsewhere.2 For transient transfection studies, immortalized human chondrocytes were plated at a density of 1 × 106 cells per well in 6-well plates and incubated for 24 h in complete media. The cells were then transfected with 2 μg of the specific promoter-reporter constructs using FuGENE 6 transfection reagent (Roche Applied Science) following the manufacturer’s instructions. The promoterless vector, pGL2-Enhancer, was used as a control, and a Renilla luciferase construct was included as an internal control for transfection efficiency. For co-transfection of dominant negative constructs, 500 ng of the dominant negative construct was used except for the PYK2 constructs that were used at 150 ng. For controls, the same amount of empty vector, pcDNA3.1 Zeo(−) (Invitrogen), was co-transfected in place of the dominant negative plasmid constructs. The cells were incubated for 24 h in complete media after transfection and then rinsed in phosphate-buffered saline and changed to serum-free conditions for 20–24 h prior to stimulation. Cells were incubated for an additional 24 h, and then cell lysates were prepared. Luciferase activity was assayed using a dual luciferase reporter assay system (Promega). All transfection experiments were repeated three times in duplicate.
For adenovirus infection, primary human chondrocytes were plated at a density of 2 × 106 cells per well in 6-well plates and incubated for 24 h in complete media. The cells were then infected with a replication-defective adenovirus encoding PYK2 wild type and the PYK2 dominant negative constructs kinase dead-PYK2 and Y402F-PYK2 at a multiplicity of infection of 50. Four hours after the transduction, the cells were fed with complete media and incubated for 10 h at 37 °C in a humidified environment containing 5% CO2. The cells were then changed to serum-free conditions and incubated for 18 h prior to removal of the media for MMP-13 analysis and preparation of cell lysates for PYK2 immunoblotting as described above.
RESULTS
The 110-kDa Fibronectin Fragment and Antibodies to the α5β1 Integrin Stimulate PYK2 Phosphorylation
In a previous study we found that treatment of chondrocytes with FN-f stimulated expression of MMP-13 which was associated with MAP kinase activation (12). Integrin signals upstream of MAP kinases can include FAK and/or PYK2 (17-21). When human articular chondrocytes in primary monolayer culture were treated with the 110-kDa FN-f, a time-dependent increase in phosphorylation of PYK2 at the autophosphorylation site (Tyr-402) was noted (Fig. 1, A and C). Phosphorylation returned to base line by 24 h (not shown). A similar increase in PYK2 phosphorylation was seen when chondrocytes were treated with an α5β1 integrin antibody (Fig. 1B). In contrast, there was a constitutive level of FAK phosphorylation at the autophosphorylation site (Tyr-397) that did not appear to change after the addition of FN-f (Fig. 1, A and C) or anti-α5β1 (Fig. 1B). No effect of FN-f was noted on total PYK2 or FAK (data not shown).
Fig. 1. Phosphorylation of PYK2 in chondrocytes treated with fibronectin fragments or an α5β1 integrin antibody.
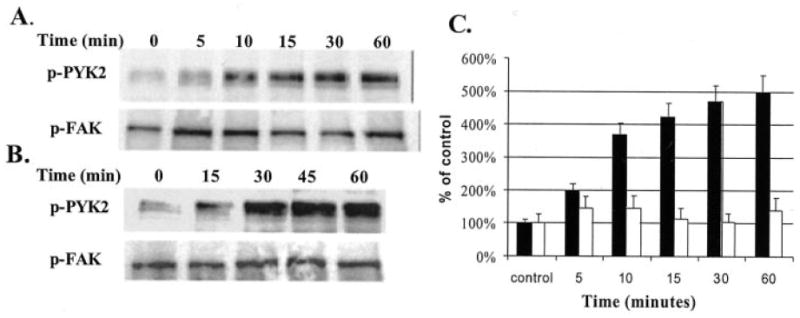
Human articular chondrocyte cultures were made serum free and then stimulated with 1 μm of the 110-kDa FN-f (A) or 25 μg/ml of an activating antibody to the α5β1 integrin (B). Cell lysates were prepared at the indicated time points and used for immunoblotting with antibodies to the autophosphorylation sites of PYK (Tyr-402, p-PYK2) and FAK (Tyr-397, p-FAK). C, mean band intensity from immunoblots of cells treated with FN-f was calculated as % of time 0 controls for p-PYK2 (filled bars) and p-FAK (open bars).
Activation or Inhibition of PYK2 Is Associated with Parallel Changes in Fibronectin Fragment-stimulated MMP-13 Expression
In order to determine further a role for signaling through PYK2 in the fibronectin fragment-mediated expression of MMP-13, we next tested known activators and inhibitors of PYK2 for their ability to affect MMP-13 expression. PYK2 activation has been shown in some systems to be downstream of PKC activation (18, 28, 29). Consistent with a similar pathway in chondrocytes, treatment of chondrocytes with the PKC activator PMA stimulated PYK2 phosphorylation which was similar to the effects of FN-f and IL-1β (Fig. 2, A and C). As expected, both PMA and IL-1β also stimulated an increase in MMP-13 protein production (data not shown). The broad spectrum PKC inhibitor BIM at 10 μm and the PKCδ-specific inhibitor rottlerin at 4 μm both inhibited FN-f-stimulated PYK2 phosphorylation (Fig. 2, A and C). Likewise, both PKC inhibitors blocked FN-f-stimulated MMP-13 production (Fig. 2, B and D).
Fig. 2. PKC inhibition blocks fibronectin fragment stimulation of PYK2 phosphorylation and MMP-13 expression.
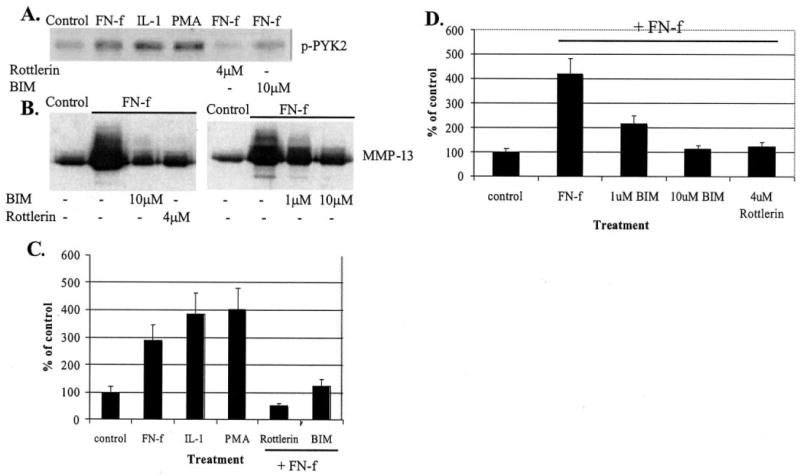
A, human articular chondrocytes were treated for 30 min with control media or media containing 1 μm FN-f, 10 ng/ml IL-β, 50 nm PMA, or FN-f after pretreatment with 4 μm rottlerin or 10 μm BIM. Cell lysates were immunoblotted with antibodies to phosphorylated PYK2 (p-PYK2). B, conditioned media were collected from human articular chondrocytes treated for 24 h with FN-f with or without the indicated concentrations of BIM or rottlerin. Media samples were concentrated and equal amounts used for immunoblotting with antibodies to MMP-13. C and D, mean band intensity results for the experiments shown in A and B, respectively, expressed as % of unstimulated controls.
In order to further confirm a role for PKC in FN-f-stimulated signaling in chondrocytes, activation of specific PKC isoforms was examined by determining translocation from the cytosol to the membrane after FN-f treatment. Whereas PKCα appeared in the membrane fraction 60 min after addition of FN-f, PKCδ appeared much earlier at 5 min (Fig. 3). There was a very low level of PKCβ expression without movement to the membrane as well as no apparent translocation of PKCε. These results taken together with the inhibitory effect of rottlerin suggest that PKCδ is part of the initial signal that activates PYK2 and subsequent MMP-13 expression, whereas activation of PKCα could be part of an autocrine loop acting further downstream from PYK2.
Fig. 3. Chondrocyte PKC activation in response to fibronectin fragments.
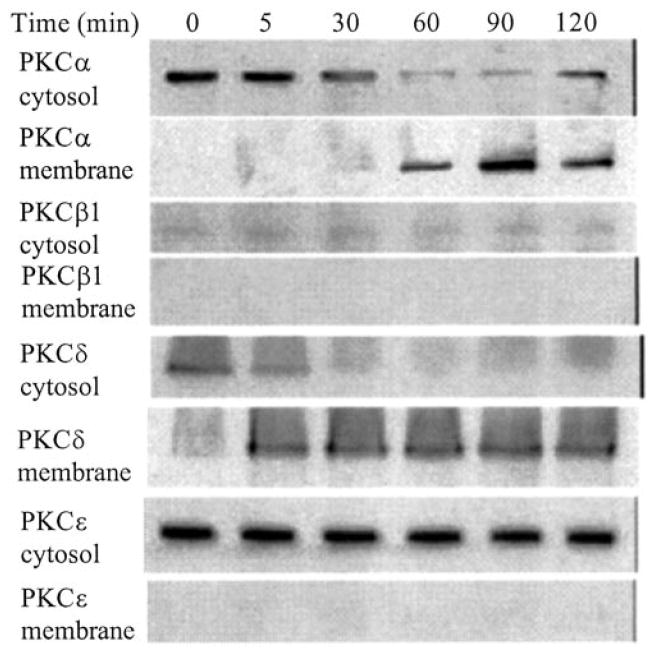
Human articular chondrocytes were stimulated for the indicated periods with 1 μm FN-f. Cytosol and membrane fractions were prepared, and samples equalized for total protein were immunoblotted with antibodies to the specific PKC isoforms.
PYK2 has been shown to be activated by increases in intracellular calcium (18, 20). Treatment with BAPTA-AM to reduce intracellular calcium levels inhibited FN-f-induced PYK2 phosphorylation and likewise inhibited the FN-f-stimulated increase in MMP-13 (Fig. 4). A more modest inhibition of the FN-f stimulation of PYK2 phosphorylation and MMP-13 production was noted when the cells were pretreated with the calcium channel blocker nifedipine suggesting that much of the calcium-dependent activation of PYK2 in this system was from intracellular calcium stores.
Fig. 4. Inhibition of intracellular calcium signaling blocks fibronectin fragment stimulation of PYK2 phosphorylation and MMP-13 expression.
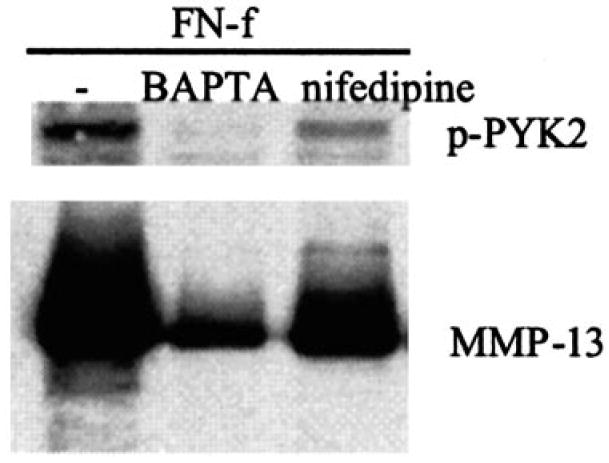
Human articular chondrocytes were pretreated for 30 min with control media or either 10 μm BAPTA-AM or 20 μm nifedipine and then stimulated with 1 μm FN-f. After 30 min, cell lysates were prepared and used for immunoblotting with antibodies to phosphorylated PYK2 (p-PYK2). Parallel cultures were allowed to incubate with FN-f and inhibitors for 24 h, and conditioned media were collected. Samples of equal volume were immunoblotted with antibodies to MMP-13.
In a study of tumor necrosis factor-α-stimulated neutrophils, the tyrosine kinase inhibitor tyrphostin A9 (AG-17) was the most selective of 51 tyrosine kinase inhibitors in blocking the PYK2 signaling pathway (30). When tested with FN-f-stimulated chondrocytes, tyrphostin A9 dose-dependently inhibited MMP-13 production (Fig. 5A). In contrast, tyrphostin A25 (AG-82), which has been shown in some cell types to inhibit FAK activity (31), had no affect on FN-f-stimulated MMP-13 (Fig. 5B). Because these results and the results of our previous study (12) suggested a link between PKC and PYK2 activation, activation of one or more of the MAP kinases, and increased MMP-13 expression, we determined whether inhibition with rottlerin or tyrphostin A9 affected MAP kinase activation. Treatment of chondrocytes with FN-f resulted in phosphorylation of JNK, p38, and ERK as we had reported previously (12). Pretreatment with rottlerin or tyrphostan A9 but not tyrphostan A25 inhibited the FN-f stimulation of JNK phosphorylation (Fig. 6). The FN-f-stimulated phospho-p54(JNK2) band was not visible in samples from cells pretreated with rottlerin or tyrphostan A9, and the lower phospho-JNK1 and -JNK3 bands were reduced by about 65%. Inhibition of ERK or p38 phosphorylation was not observed.
Fig. 5. Tyrosine kinase inhibition with tyrphostin A9 (AG-17) blocks fibronectin fragment stimulation of MMP-13 synthesis.
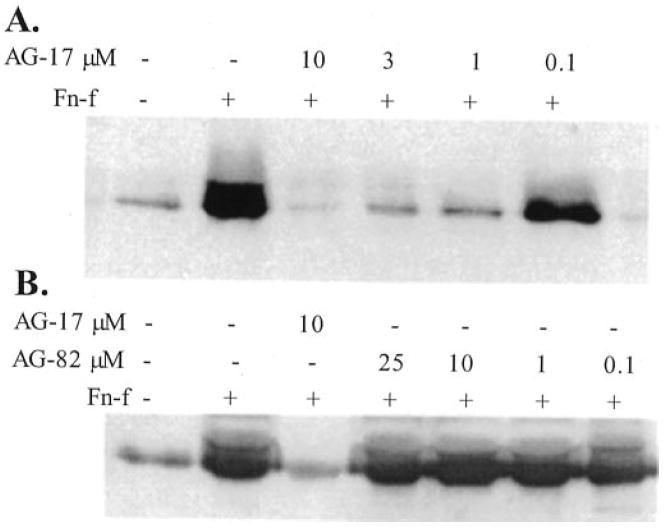
Human articular chondrocytes were pretreated for 30 min with the increasing concentrations of tyrphostin A9 (AG-17) (A) or tyrphostin A25 (AG-82) (B) and then treated overnight with 1 μm FN-f or control media without FN-f. Conditioned media were used for immunoblotting with antibodies to MMP-13.
Fig. 6. Inhibition of fibronectin fragment induced JNK phosphorylation by tyrphostin A9 (AG-17) and rottlerin.
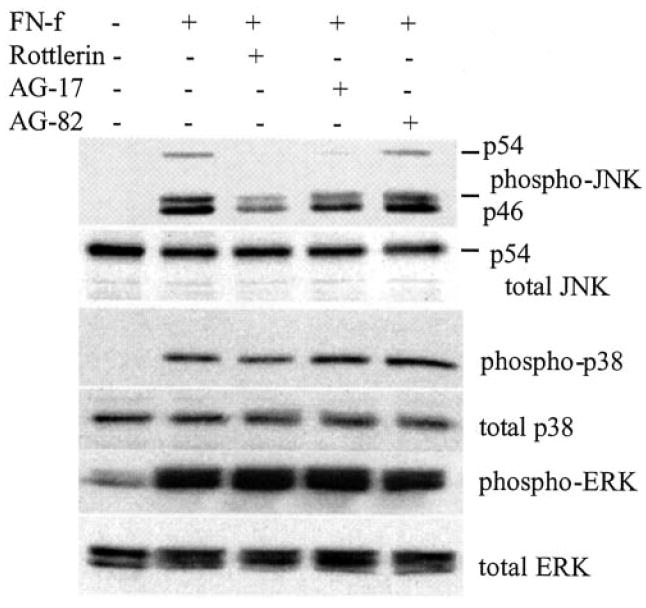
Human articular chondrocytes were pretreated for 30 min with the indicated inhibitors: 4 μm rottlerin, 10 μm tyrphostin A9 (AG-17), or 10 μm tyrphostin A25 (AG-82). The cells were then treated with 1 μm FN-f or control media without FN-f or inhibitors for 30 min at which time cell lysates were prepared. Chondrocyte lysates were immunoblotted with phosphospecific (phospho-) and non-phosphospecific (total) antibodies to JNK, p38, and ERK.
Adenoviral Overexpression of PYK2 Stimulates Chondrocyte MMP-13 Production
In order to provide additional evidence for PYK2 in the stimulation of MMP-13 expression by chondrocytes, primary chondrocytes were treated with adenoviruses expressing GFP-linked wild type PYK2 and two dominant negative PYK2 constructs as well as GFP control virus. Overexpression of wild type PYK2 resulted in increased autophosphorylation of the construct which was not observed with either dominant negative construct or the GFP control (Fig. 7A). Likewise, only overexpression of wild type PYK2 was associated with increased production of MMP-13 (Fig. 7B). We were not able to determine whether overexpression of PYK2 using adenovirus resulted in activation of a specific MAP kinase pathway because all of the constructs, including the GFP control, resulted in some degree of phosphorylation of all three MAP kinases at the time point tested probably as a result of cell stress from the adenoviral infection (data not shown).
Fig. 7. Adenoviral overexpression of PYK2 stimulates chondrocyte MMP-13 production.
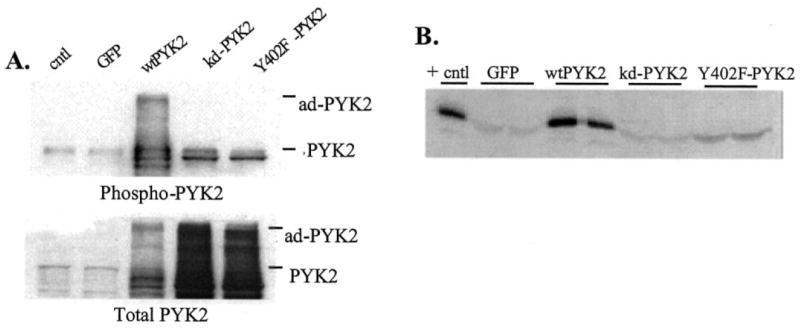
Primary chondrocytes were treated with adenovirus containing a GFP expression construct (GFP) as control or GFP linked to wild type PYK2 (wtPYK2), a kinase dead PYK2 (kd-PYK2), or Y402F-PYK2. A, cell lysates were immunoblotted with phosphospecific and non-phosphospecific (total) antibodies to PYK2. A sample of lysate from an untreated control culture was included (cntl). B, media samples from duplicate wells of adenoviral treated cells were analyzed for MMP-13 by immunoblotting. The positive control (+cntl) was from cells treated with FN-f without adenoviral treatment, whereas none of the adenoviral treated cultures had received FN-f.
MAP Kinase Inhibition Blocks Fibronectin Fragment Stimulation of MMP-13 Promoter Activity
In order to investigate further the signaling pathways regulating FN-f stimulation of MMP-13 expression, transient transfection studies were carried out in the immortalized human chondrocyte line C-28I2. These cells were found to respond to FN-f, PMA, and IL-1β with increased production of MMP-13 protein in a similar manner as the primary cells (data not shown). The cells were transiently transfected with a series of 5′-promoter region deletion constructs that contained various lengths of the human MMP-13 promoter sequence fused to the firefly luciferase reporter gene. Comparisons were made to cells transfected with an MMP-1 promoter-luciferase reporter construct (−562 MMP-1) because previous studies in fibroblasts had demonstrated FN-f regulation of MMP-1 expression (7), and we noted FN-f stimulation of MMP-1 production in chondrocytes as well (data not shown).
Treatment of immortalized chondrocytes with the 110-kDa FN-f resulted in a 3-fold increase in luciferase reporter activity in cells transfected with the −1600 MMP-13 promoter construct or the −736 construct, whereas the −370 promoter showed a 2-fold increase, and the minimal promoter (−186) showed about a 1.5-fold increase (Fig. 8A). The −562 MMP-1 promoter demonstrated an ~2.5-fold increase with FN-f, whereas no change in activity was seen with the promoterless basic vector. Preincubation with any one of the three MAP kinase inhibitors tested (30 μm PD98059 for MEK, 20 μm SP600125 for JNK, or 10 μm SB203580 for p38) blocked the FN-f-stimulated MMP-13 and MMP-1 promoter-reporter activity (Fig. 8A). This inhibition of MMP-13 promoter activity was consistent with our previous finding that FN-f stimulation of MMP-13 protein in conditioned media was blocked using the same inhibitors (12). To support further the chemical inhibitor results, chondrocytes were co-transfected with dominant negative constructs shown previously to inhibit chondrocyte ERK-1, ERK-2, JNK (32), and p38 (33). These experiments showed consistent inhibition of FN-f-induced MMP-13 as well as MMP-1 promoter activity (Fig. 8B). These results suggest that activation of all three major MAP kinase pathways is required for FN-f stimulation of chondrocyte MMP-13 expression.
Fig. 8. MAP kinase inhibition blocks fibronectin fragment stimulation of MMP-13 and MMP-1 promoter activity.
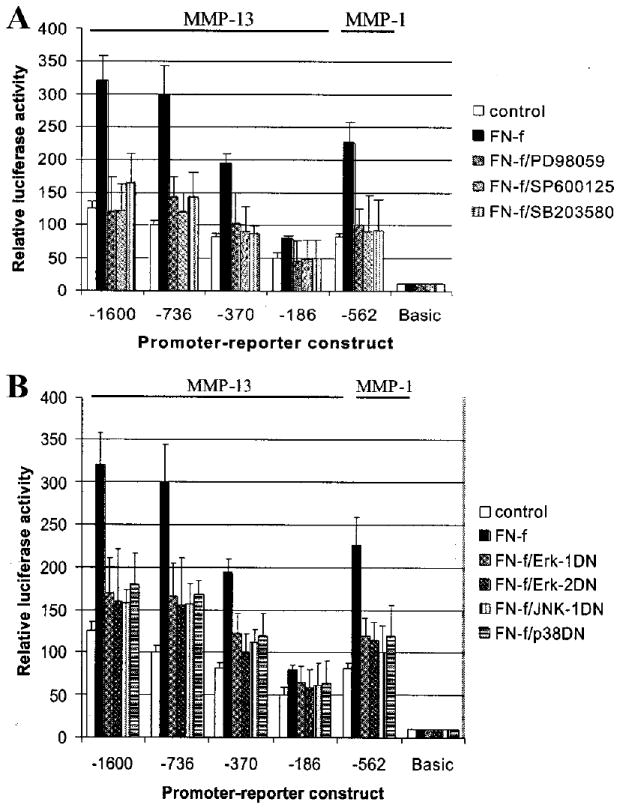
A, immortalized human chondrocytes (C-28I2 cells) were transiently transfected with −1600, −736, −370, −186 MMP-13, and −562 MMP-1 promoter-luciferase reporter constructs or the promoterless luciferase vector (basic) as control. After 24 h in complete media, the cells were changed to serum-free conditions for 20–24 h and then treated with 1 μm FN-f with or without MAP kinase inhibitors (30 μm PD98059, 20 μm SP600125, or 10 μm SB203580). After 24 h of incubation, cell lysates were prepared and used for luciferase assays. B, C-28I2 cells were co-transfected with the MMP promoter-reporter constructs and dominant negative (DN) mutant constructs for ERK-1, ERK-2, JNK, or p38 as indicated. Cells were incubated for 24 h in complete media, made serum-free for 24 h, and then treated with 1 μm FN-f for 24 h followed by analysis of luciferase activity.
Inhibition of PYK2 Blocks Fibronectin Fragment and PMA Stimulation of MMP-13 Promoter Activity
Co-transfection of the MMP-13 and MMP-1 promoter-reporter constructs with a wild type PYK2 expression construct resulted in a modest increase in FN-f-stimulated promoter activity, whereas co-transfection with either of two dominant negative mutants of PYK2 (Y402F and K457A) blocked FN-f-stimulated promoter activity (Fig. 9). Pretreatment with tyrphostin A9 reduced FN-f-stimulated promoter activity below basal levels. Co-transfection with a dominant negative form of FAK (FRNK) or empty vector had no affect on basal MMP-13 or MMP-1 promoter activity and did not inhibit FN-f-stimulated promoter activity.
Fig. 9. Inhibition of PYK2 blocks fibronectin fragment stimulation of MMP-13 and MMP-1 promoter activity.
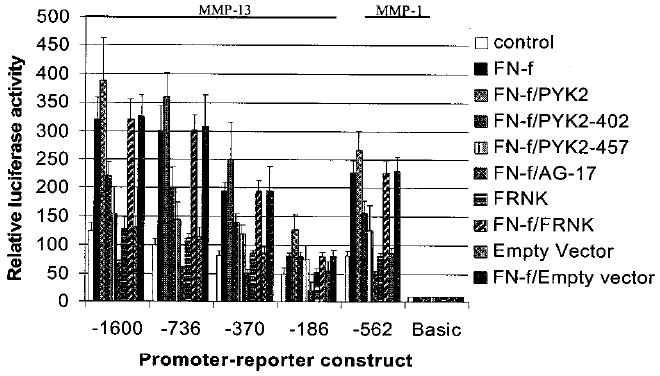
Immortalized human chondrocytes were co-transfected with the MMP promoter-reporter constructs described in FIG. 8 and constructs for wild type PYK2, dominant negative PYK2 mutants (PYK2–402 and -457), dominant negative FAK (FRNK), or an empty control vector. Tyrosine kinase inhibition by 10 μm tyrphostin A9 (AG-17) was also tested. Following 24 h of incubation in complete media and 24 h incubation in serum-free media, the cultures were treated with 1 μm FN-f as indicated. After 24 h of treatment, cell lysates were prepared and analyzed by luciferase assay.
The potential role of PKC in FN-f stimulation of MMP-13 expression was also further explored in chondrocytes transfected with the MMP-13 and MMP-1 promoter-reporter constructs. PMA stimulated a very significant increase in MMP-13 and MMP-1 promoter activity which was completely inhibited by either the PKC inhibitor BIM or the tyrosine kinase inhibitor tyrphostin A9 (Fig. 10A). Co-transfection with either of the two dominant negative PYK2 constructs also blocked PMA-induced MMP promoter-reporter activity, whereas wild type PYK2, the dominant negative FAK, or empty vector control did not. For further evidence of a PKC requirement for FN-f stimulation of MMP-13 expression, immortalized chondrocytes were treated with FN-f in the presence of the PKC inhibitor BIM. Consistent with the MMP-13 protein results (Fig. 2), the PKC inhibitor blocked FN-f induced MMP-13 and MMP-1 promoter activity (Fig. 10B).
Fig. 10. Inhibition of PYK2 blocks PMA-stimulated MMP-13 and MMP-1 promoter activity and PKC inhibition blocks fibronectin fragment stimulated activity.
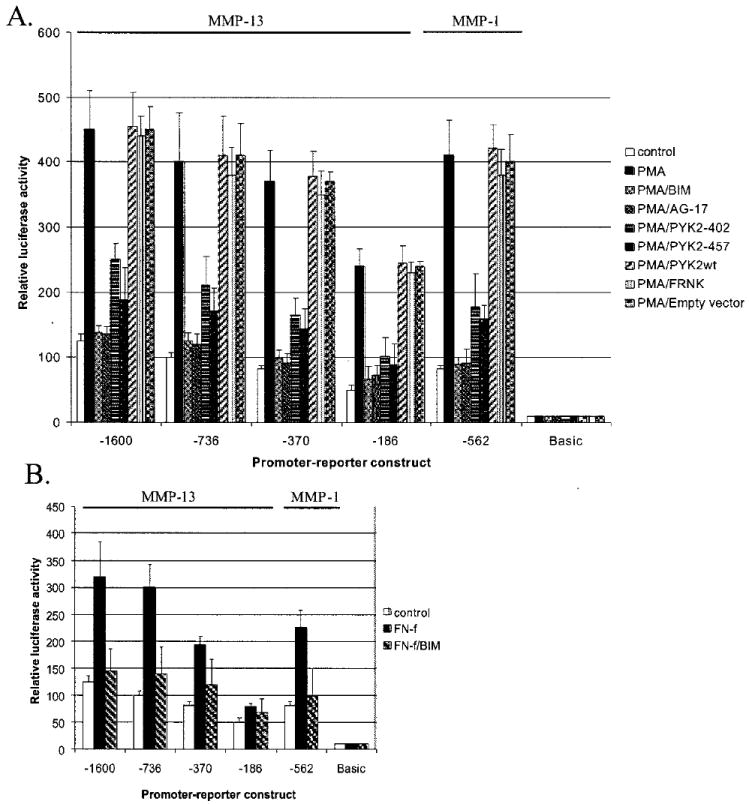
A, immortalized human chondrocytes were transiently transfected with MMP-13 and MMP-1 promoter-reporter constructs and in some cultures co-transfected with the PYK2 and FAK constructs as described in Fig. 9. The cultures were stimulated with 50 nM PMA with or without 5 μm BIM or 10 μm tyrphostin A9 (AG-17). After 24 h, cell lysates were prepared and used to measure luciferase activity. B, chondrocytes transiently transfected with the promoter-reporter constructs were treated with the FN-f in the absence or presence of 5 μm BIM.
DISCUSSION
Recent studies have indicated that in arthritic cartilage overproduction of collagenases, in particular MMP-13 and MMP-1, by chondrocytes plays a central role in collagen degradation (4, 5, 34). Therefore, it has become important to determine the signaling pathways that regulate chondrocyte collagenase expression. These signaling pathways may be activated by cytokines such as IL-1 (35, 36) or by stimulation of chondrocyte integrins such as α5β1 by FN-f (12). The present results provide evidence for a pathway by which FN-f induces MMP-13 expression through activation of PKC, PYK2 and the ERK1/2, JNK, and p38 MAP kinases. The finding that inhibition of any one of these signaling proteins will block FN-f induced MMP-13 expression indicates that activation of each one is required for sufficient stimulation of chondrocyte MMP-13 promoter activity. Although MMP-13 expression was the focus of these experiments because of its potent collagenase activity, similar findings were noted when testing an MMP-1 promoter-reporter construct, suggesting FN-f stimulation of both collagenases utilizes a similar pathway in chondrocytes.
PYK2 was initially identified in neuronal cells and found to be activated through PKC and elevation of intracellular calcium levels and was linked to downstream activation of MAP kinase signaling (18). We found evidence for a similar signaling pathway in articular chondrocytes after stimulation with FN-f. FN-f stimulated early activation of PKCδ (within 5 min) with an apparently delayed activation of PKCα at 60 min. Inhibition of increased intracellular calcium levels by BAPTA-AM blocked chondrocyte PYK2 phosphorylation in response to FN-f as did the general PKC inhibitor BIM and the PKCδ-specific inhibitor rottlerin. Also consistent with a role for PKC in mediating chondrocyte PYK2 activation was the finding that the PKC activator PMA-stimulated chondrocyte PYK2 phosphorylation.
The inhibition of FN-f-stimulated PYK2 phosphorylation by rottlerin and the early translocation of PKCδ to the membrane suggest PKCδ may be responsible for at least the initial PYK2 activation in response to FN-f. Subsequent activation of PKCα may be involved in an autocrine signaling loop. Consistent with our findings with FN-f, activation of PYK2 by PKCδ has recently been demonstrated in vascular smooth muscle cells as a mechanism by which angiotensin II activates Janus kinase 2 (29). Likewise, serotonin-induced MMP-13 expression in uterine smooth muscle cells was found to require PKC activation and similar to our FN-f results was inhibited by the PKCδ inhibitor rottlerin (37).
Changes in MMP-13 expression paralleled the changes in PKC and PYK2 activation, which is important for understanding the function of FN-f-stimulated PKC and PYK2 activation. Treatment of chondrocytes with either FN-f or PMA, both which stimulated PYK2 phosphorylation, resulted in increased MMP-13 expression as did overexpression of wild type PYK2. Similar to our findings, Melendez et al. (38) have shown that adenovirally mediated overexpression of wild type PYK2 markedly increases the amount of PYK2 phosphorylated at Tyr-402. Further evidence for a role for PKC and PYK2 in the FN-f stimulation of MMP-13 expression was that inhibition of PKC or PYK2 activation with BIM, rottlerin, BAPTA-AM, or tyrphostin A9 each inhibited MMP-13 expression. Furthermore, use of dominant negative constructs for PYK2, which were previously found to inhibit PYK2 activity in 293-VnR cells (39), confirmed a requirement for PYK2. Although FAK is closely related to PYK2, we did not observe significant stimulation of FAK phosphorylation in response to FN-f and did not inhibit MMP-13 expression in cells transfected with FRNK which acts as a dominant negative for FAK (40). This is consistent with previous work in other cell systems that express both PYK2 and FAK where differential signaling by FAK and PYK2 has been observed despite the similarities in the two proteins (41, 42).
Our results also demonstrate a critical role for MAP kinase activation in the FN-f stimulation of MMP-13 expression. We had demonstrated previously activation of ERK1/2, JNK, and p38 when chondrocytes were treated with FN-f or anti-α5β1 antibodies and, by using chemical inhibitors, that inhibition of any one of the three would inhibit MMP-13 expression at the protein level (12). In the present study, transfection of dominant negative MAP kinase constructs confirmed the chemical inhibitor studies. A requirement for activation of all three MAP kinases in order to obtain an increase in MMP-13 expression might explain why growth factors such as IGF-I, which also activate chondrocyte ERK1/2 but not JNK or p38 (43), do not stimulate chondrocyte MMP-13 expression. A critical role for JNK in the regulation of chondrocyte MMP-13 expression is consistent with a recent study demonstrating that JNK inhibition could inhibit MMP-13 production and subsequent joint destruction in a mouse model of inflammatory arthritis (44).
MAP kinase activation appeared to be downstream of the PYK2 signaling pathway, as has been reported in other cell types (17-21, 45). The coupling of PYK2 to activation of MAP kinases has been shown to involve the Grb2-Sos complex for ERK activation, whereas p130Cas and Crk connect PYK2 to JNK activation (45). In the present study inhibition of FN-f-stimulated JNK phosphorylation was noted using the PKCδ inhibitor rottlerin or the tyrosine kinase inhibitor tyrphostin A9 suggesting PYK2 activation was connected to JNK phosphorylation. The same inhibitors did not block phosphorylation of ERK or p38. These results suggest that pathways in addition to PKCδ and PYK2 may be activated by FN-f in chondrocytes that are responsible for activation of ERK and p38. Further work will be necessary to define the other components of these complex signaling pathways including the potential requirement for the generation of reactive oxygen species as noted in fibroblast expression of MMP-1 in response to FN-f (13). Preliminary experiments with reactive oxygen species scavengers suggest this to be the case.3
In summary, we have identified a novel signaling pathway that regulates MMP-13 and MMP-1 expression in articular chondrocytes stimulated by the 110-kDa fibronectin fragment. This pathway includes PKC and PYK2 and the ERK1/2, JNK, and p38 MAP kinases. Activation of the MAP kinase pathway would in turn activate transcription factors known to regulate chondrocyte MMP-13 expression such as AP-1 and Runx-2 (14). It will be of interest to determine the mechanism of transcriptional regulation of the MMP-13 gene in response to FN-f induced signaling. The response of the various promoter deletional constructs used in the present study suggests that multiple promoter sites are involved, including distal cis-acting elements present in the full-length promoter, as well as the essential binding sites including AP-1, Runx-2, and Ets present in the −186 construct. The present results indicate that chondrocyte MMP-13 expression is highly regulated within the chondrocyte cell signaling network.
Acknowledgments
We thank Drs. Yubo Sun, Archana Sanjay, Mike Schaller, Shu Chien, Tom Parsons, Francis Berenbaum, Shelly Earp, and Gillian Murphy for providing valuable reagents. We also thank the Gift of Hope Organ and Tissue Network for providing tissue, Dr. Arkady Margulis for assistance in collecting tissue, and Hong Chen for technical assistance.
Footnotes
This work was supported by National Institutes of Health Grants AR49003, P50-AR39239, and HL63711 and the Arthritis Foundation. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The abbreviations used are: MMP, matrix metalloproteinase; FN-f, fibronectin fragment; MAP kinase, mitogen-activated protein kinase; MEK, MAPK/ERK kinase; ERK, extracellular signal-related kinase; JNK, c-Jun N-terminal kinase; FAK, focal adhesion kinase; PYK2, proline-rich tyrosine kinase; PKC, protein kinase C; PMA, phorbol 12-myristate 13-acetate; BIM, bisindolylmaleimide I; IL, interleukin; GFP, green fluorescent protein; BAPTA, 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid; DMEM, Dulbecco’s modified Eagle’s medium.
H.-J. Im, C. Pacione, S. Chubinskya, A. J. van Wijnen, Y. Sun, and R. F. Loeser, submitted for publication.
R. F. Loeser, C. B. Forsyth, and H.-J. Im, unpublished observations.
References
- 1.Sternlicht MD, Werb Z. Annu Rev Cell Dev Biol. 2001;17:463–516. doi: 10.1146/annurev.cellbio.17.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dean DD, Martel-Pelletier J, Pelletier JP, Howell DS, Woessner JF., Jr J Clin Invest. 1989;84:678–685. doi: 10.1172/JCI114215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Billinghurst RC, Dahlberg L, Ionescu M, Reiner A, Bourne R, Rorabeck C, Mitchell P, Hambor J, Diekmann O, Tschesche H, Chen J, Van Wart H, Poole AR. J Clin Invest. 1997;99:1534–1545. doi: 10.1172/JCI119316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shlopov BV, Lie WR, Mainardi CL, Cole AA, Chubinskaya S, Hasty KA. Arthritis Rheum. 1997;40:2065–2074. doi: 10.1002/art.1780401120. [DOI] [PubMed] [Google Scholar]
- 5.Reboul P, Pelletier JP, Tardif G, Cloutier JM, Martel-Pelletier J. J Clin Invest. 1996;97:2011–2019. doi: 10.1172/JCI118636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neuhold LA, Killar L, Zhao W, Sung ML, Warner L, Kulik J, Turner J, Wu W, Billinghurst C, Meijers T, Poole AR, Babij P, De-Gennaro LJ. J Clin Invest. 2001;107:35–44. doi: 10.1172/JCI10564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Werb Z, Tremble PM, Behrendtsen O, Crowley E, Damsky CH. J Cell Biol. 1989;109:877–889. doi: 10.1083/jcb.109.2.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xie DL, Meyers R, Homandberg GA. J Rheumatol. 1992;19:1448–1452. [PubMed] [Google Scholar]
- 9.Homandberg GA, Wen C, Hui F. Osteoarthr Cartil. 1998;6:231–244. doi: 10.1053/joca.1998.0116. [DOI] [PubMed] [Google Scholar]
- 10.Homandberg GA, Meyers R, Xie DL. J Biol Chem. 1992;267:3597–3604. [PubMed] [Google Scholar]
- 11.Homandberg GA. Front Biosci. 1999;4:D713–D730. doi: 10.2741/homandberg. [DOI] [PubMed] [Google Scholar]
- 12.Forsyth CB, Pulai J, Loeser RF. Arthritis Rheum. 2002;46:2368–2376. doi: 10.1002/art.10502. [DOI] [PubMed] [Google Scholar]
- 13.Kheradmand F, Werner E, Tremble P, Symons M, Werb Z. Science. 1998;280:898–902. doi: 10.1126/science.280.5365.898. [DOI] [PubMed] [Google Scholar]
- 14.Mengshol JA, Vincenti MP, Brinckerhoff CE. Nucleic Acids Res. 2001;29:4361–4372. doi: 10.1093/nar/29.21.4361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ravanti L, Heino J, Lopez-Otin C, Kahari VM. J Biol Chem. 1999;274:2446–2455. doi: 10.1074/jbc.274.4.2446. [DOI] [PubMed] [Google Scholar]
- 16.Gemba T, Valbracht J, Alsalameh S, Lotz M. J Biol Chem. 2002;277:907–911. doi: 10.1074/jbc.M109690200. [DOI] [PubMed] [Google Scholar]
- 17.Schlaepfer DD, Hunter T. Trends Cell Biol. 1998;8:151–157. doi: 10.1016/s0962-8924(97)01172-0. [DOI] [PubMed] [Google Scholar]
- 18.Lev S, Moreno H, Martinez R, Canoll P, Peles E, Musacchio JM, Plowman GD, Rudy B, Schlessinger J. Nature. 1995;376:737–745. doi: 10.1038/376737a0. [DOI] [PubMed] [Google Scholar]
- 19.Sorokin A, Kozlowski P, Graves L, Philip A. J Biol Chem. 2001;276:21521–21528. doi: 10.1074/jbc.M008869200. [DOI] [PubMed] [Google Scholar]
- 20.Yu H, Li X, Marchetto GS, Dy R, Hunter D, Calvo B, Dawson TL, Wilm M, Anderegg RJ, Graves LM, Earp HS. J Biol Chem. 1996;271:29993–29998. doi: 10.1074/jbc.271.47.29993. [DOI] [PubMed] [Google Scholar]
- 21.Rocic P, Lucchesi PA. J Biol Chem. 2001;276:21902–21906. doi: 10.1074/jbc.M101684200. [DOI] [PubMed] [Google Scholar]
- 22.Avraham S, London R, Fu Y, Ota S, Hiregowdara D, Li J, Jiang S, Pasztor LM, White RA, Groopman JE, Avraham H. J Biol Chem. 1995;270:27742–27751. doi: 10.1074/jbc.270.46.27742. [DOI] [PubMed] [Google Scholar]
- 23.Sasaki H, Nagura K, Ishino M, Tobioka H, Kotani K, Sasaki T. J Biol Chem. 1995;270:21206–21219. doi: 10.1074/jbc.270.36.21206. [DOI] [PubMed] [Google Scholar]
- 24.Sein TT, Thant AA, Hiraiwa Y, Amin AR, Sohara Y, Liu Y, Matsuda S, Yamamoto T, Hamaguchi M. Oncogene. 2000;19:5539–5542. doi: 10.1038/sj.onc.1203932. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Y, Thant AA, Hiraiwa Y, Naito Y, Sein TT, Sohara Y, Matsuda S, Hamaguchi M. Biochem Biophys Res Commun. 2002;290:1123–1127. doi: 10.1006/bbrc.2001.6321. [DOI] [PubMed] [Google Scholar]
- 26.Loeser RF, Sadiev S, Tan L, Goldring MB. Osteoarthr Cartil. 2000;8:96–105. doi: 10.1053/joca.1999.0277. [DOI] [PubMed] [Google Scholar]
- 27.Muehleman C, Bareither D, Huch K, Cole AA, Kuettner KE. Osteoarthr Cartil. 1997;5:23–37. doi: 10.1016/s1063-4584(97)80029-5. [DOI] [PubMed] [Google Scholar]
- 28.Wu SS, Chiu T, Rozengurt E. Am J Physiol. 2002;282:C1432–C1444. doi: 10.1152/ajpcell.00323.2001. [DOI] [PubMed] [Google Scholar]
- 29.Frank GD, Saito S, Motley ED, Sasaki T, Ohba M, Kuroki T, Inagami T, Eguchi S. Mol Endocrinol. 2002;16:367–377. doi: 10.1210/mend.16.2.0768. [DOI] [PubMed] [Google Scholar]
- 30.Fuortes M, Melchior M, Han H, Lyon GJ, Nathan C. J Clin Invest. 1999;104:327–335. doi: 10.1172/JCI6018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsuda T, Kusui T, Jensen RT. Biochemistry. 1997;36:16328–16337. doi: 10.1021/bi971448o. [DOI] [PubMed] [Google Scholar]
- 32.Jin G, Sah RL, Li YS, Lotz M, Shyy JY, Chien S. J Orthop Res. 2000;18:899–908. doi: 10.1002/jor.1100180608. [DOI] [PubMed] [Google Scholar]
- 33.Thomas B, Thirion S, Humbert L, Tan L, Goldring MB, Bereziat G, Berenbaum F. Biochem J. 2002;362:367–373. doi: 10.1042/0264-6021:3620367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu W, Billinghurst RC, Pidoux I, Antoniou J, Zukor D, Tanzer M, Poole AR. Arthritis Rheum. 2002;46:2087–2094. doi: 10.1002/art.10428. [DOI] [PubMed] [Google Scholar]
- 35.Shlopov BV, Gumanovskaya ML, Hasty KA. Arthritis Rheum. 2000;43:195–205. doi: 10.1002/1529-0131(200001)43:1<195::AID-ANR24>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 36.Mengshol JA, Vincenti MP, Coon CI, Barchowsky A, Brinckerhoff CE. Arthritis Rheum. 2000;43:801–811. doi: 10.1002/1529-0131(200004)43:4<801::AID-ANR10>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 37.Shum JK, Melendez JA, Jeffrey JJ. J Biol Chem. 2002;277:42830–42840. doi: 10.1074/jbc.M205094200. [DOI] [PubMed] [Google Scholar]
- 38.Melendez J, Welch S, Schaefer E, Moravec CS, Avraham S, Avraham H, Sussman MA. J Biol Chem. 2002;277:45203–45210. doi: 10.1074/jbc.M204886200. [DOI] [PubMed] [Google Scholar]
- 39.Sanjay A, Houghton A, Neff L, DiDomenico E, Bardelay C, Antoine E, Levy J, Gailit J, Bowtell D, Horne WC, Baron R. J Cell Biol. 2001;152:181–195. doi: 10.1083/jcb.152.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Richardson A, Parsons T. Nature. 1996;380:538–540. doi: 10.1038/380538a0. [DOI] [PubMed] [Google Scholar]
- 41.Schaller MD, Sasaki T. J Biol Chem. 1997;272:25319–25325. doi: 10.1074/jbc.272.40.25319. [DOI] [PubMed] [Google Scholar]
- 42.Zheng C, Xing Z, Bian ZC, Guo C, Akbay A, Warner L, Guan JL. J Biol Chem. 1998;273:2384–2389. doi: 10.1074/jbc.273.4.2384. [DOI] [PubMed] [Google Scholar]
- 43.Geng Y, Valbracht J, Lotz M. J Clin Invest. 1996;98:2425–2430. doi: 10.1172/JCI119056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Han Z, Boyle DL, Chang L, Bennett B, Karin M, Yang L, Manning AM, Firestein GS. J Clin Invest. 2001;108:73–81. doi: 10.1172/JCI12466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Blaukat A, Ivankovic-Dikic I, Gronroos E, Dolfi F, Tokiwa G, Vuori K, Dikic I. J Biol Chem. 1999;274:14893–14901. doi: 10.1074/jbc.274.21.14893. [DOI] [PubMed] [Google Scholar]