

Abstract
Recent studies indicate that tissue factor (TF) acts in embryogenesis, metastasis, and angiogenesis. Three independent groups showed that targeted disruption of the murine TF (mTF) gene results in 90% lethality of mTF null embryos at embryonic days 9.5–10.5. We have demonstrated that expression of wild-type human TF (hTF) from a minigene rescues the embryonic lethality of mTF null embryos. To investigate the role of TF in embryogenesis, we made mutant hTF minigenes whose products either bound FVII/VIIa at a reduced level or lacked the cytoplasmic domain. Two independent transgenic lines expressing the hTF extracellular domain mutant failed to rescue the embryonic lethality of mTF null embryos, suggesting that FVII/VIIa binding by TF, proteolytic activity by the TF/FVIIa complex, or both were required for embryogenesis. In contrast, two transgenic lines expressing the hTF cytoplasmic domain mutant rescued the embryonic lethality of mTF null embryos, indicating that the cytoplasmic domain of TF was not required for embryogenesis. We propose that TF/FVIIa-dependent extracellular protease activity is required for embryogenesis.
Introduction
Tissue factor (TF) is a 263–amino acid transmembrane glycoprotein that serves as the cellular receptor and cofactor for plasma FVII/VIIa (1, 2). TF is expressed in the adventitia of blood vessels and at epithelial surfaces, where it functions as a hemostatic barrier ready to activate blood coagulation after vessel injury (3, 4). In disease, TF expression within the vasculature contributes to thrombosis in patients with a variety of clinical disorders, including atherosclerosis, sepsis, and cancer (5–7). TF also plays a role in other biologic processes, including metastasis, tumor-associated angiogenesis, and embryogenesis (8–13). However, the precise mechanisms by which TF contributes to these processes has yet to be defined.
Targeted disruption of the murine TF (mTF) gene results in embryonic lethality of approximately 90% of mTF null embryos at embryonic days (E) 9.5–10.5 (14–16). Similarly, 50% of embryos deficient in FV or prothrombin die between E9.5 and E10.5 (17–19). In contrast, 100% of FVII-deficient embryos survive to birth (20). The survival of FVII-deficient embryos was surprising but may be explained by the rescue of FVII-deficient embryos by low levels of maternal FVII. Embryos deficient in TF, FV, and prothrombin all exhibit an abnormal yolk sac vasculature (16, 17, 19), which may be the primary defect that leads to death of these embryos. Vitelline vessels from TF null embryos showed a deficiency in smooth muscle α-actin–expressing mesenchymal cells that maintain the structural integrity of the vessel (16). The survival of fibrinogen-deficient embryos and NF-E2–deficient embryos, which exhibit profound thrombocytopenia, suggest that fibrin deposition, platelets, and hemostasis are not essential for embryonic development (21–23). These studies and the similarity in the phenotypes of TF-, FV-, and prothrombin-deficient embryos suggest a hemostasis-independent role of TF in the maintenance of blood vessels during embryogenesis. TF may exert its effect on blood vessel development by the generation of extracellular proteases, such as thrombin. It is provocative that 50% of protease-activated receptor 1–deficient (PAR-1–deficient) embryos die at approximately E10.5 (24, 25), suggesting that TF-dependent thrombin generation and activation of PAR-1 are required for embryogenesis.
TF may function to localize the serine protease FVIIa to the cell surface in a TF:VIIa complex that subsequently generates various extracellular proteases, such as FXa and thrombin, by initiating the coagulation protease cascade (26). Extracellular protease generation is required for TF-mediated metastasis (10, 27, 28). In addition, FVIIa induction of VEGF expression by human fibroblasts and induction of the urokinase receptor (uPAR) in human pancreatic carcinoma cells requires FVIIa proteolytic activity (29, 30). More recently, FVIIa and FXa were shown to activate the ERK1/2 signaling pathway and induce the Egr-1 gene expression in keratinocytes (31). TF:VIIa-dependent generation of thrombin and subsequent activation of PARs may also modulate many cellular functions (28).
The TF cytoplasmic domain may also play a role in intracellular signaling. The 21–amino acid cytoplasmic domain of TF is highly conserved in five mammalian species (26) and contains serine residues that are phosphorylated when cells are stimulated with protein kinase C activators (32, 33). Deletion of the cytoplasmic domain of TF abolishes prometastatic functions of TF in an in vivo model of melanoma cell metastasis (11, 27). Moreover, a recent study using human melanoma cell lines suggested that the cytoplasmic domain of TF is essential for VEGF production (34). Studies by Ott et al. (35) have demonstrated a role for TF in cell adhesion and migration that is mediated by interaction with actin-binding protein 280. However, the significance of this in vitro data to the process of metastasis or other biologic processes has yet to be defined.
We reported a novel mouse model system in which expression of human TF (hTF) from a transgene rescued the embryonic lethality of mTF null embryos (36). In the present study, we investigated the role of the TF extracellular and cytoplasmic domains in embryogenesis using this transgenic mouse model.
Methods
Generation of transgenic mice.
To construct a minigene expressing hTF lacking the cytoplasmic domain (mutated intracellular domain [mutID]), we mutated the sequence coding for amino acid 244 to a stop codon using site-directed mutagenesis. Briefly, a 2.7-kb XbaI fragment from the wild-type minigene was cloned into pUC18. A 773-bp AgeI/NcoI fragment was removed from the XbaI fragment by digestion and replaced with a corresponding fragment containing a mutation that introduced a stop codon at amino acid 244 (a gift of W. Ruf, The Scripps Research Institute, La Jolla, California, USA). The 2.7-kb fragment containing the mutation was then cloned back into the wild-type minigene construct. The extracellular domain mutant (mutated extracellular domain [mutED]) was constructed in three stages. First, the wild-type 773-bp AgeI/NcoI fragment from the 2.7-kb XbaI fragment was replaced by an AgeI/NcoI fragment containing the F140→A mutation in a manner similar to that just described. Second, site-directed mutagenesis was performed on this template using the QuikChange system (Stratagene Cloning Systems, La Jolla, California, USA) to mutate K20→A. Finally, a second round of mutagenesis was performed to mutate D44→A. The 2.7-kb fragment containing all three mutations was then cloned back into the wild-type minigene construct. All intermediates in the cloning were characterized by DNA sequencing. The mutated minigenes were excised from pBluescript (Stratagene Cloning Systems) on a 6.1-kb KpnI-BstXI DNA fragment. Purified DNA was injected into the pronucleus of fertilized mouse embryos (strain CB6 [C57BL/6 X BALB/c]) by the Scripps Transgenic Core facility. Transgenic animals were identified by hybridizing Southern blots of EcoRI-digested genomic DNA with a 641-bp hTF cDNA probe. Heterozygous F1 and F2 transgenic progeny were obtained by mating the founder animals with C57BL/6 mice.
Experiments to examine rescue of mTF null embryos with the mutID and mutED human minigenes.
We first crossed transgenic mice (mTF+/+,hTF+/–) with mice heterozygous for the mTF gene (mTF+/–) to create offspring heterozygous for both mTF and the hTF minigene (mTF+/–,hTF+/–). Rescue experiments were performed by crossing mTF+/–,hTF+/– mice with mTF+/– mice. In this breeding, mTF–/–,hTF+/– (rescued) and mTF–/–,hTF–/– (null) embryos were produced at equal frequency, which permitted direct comparison of the numbers of mice in each group. Pups were sacrificed at postnatal day 1 (P1) and genotyped.
CHO cell transfection.
CHO cells were cultured in DMEM supplemented with 10% newborn calf serum (Gemini BioProducts Inc., Calabasas, California, USA), 2 mmol/L L-glutamine and 1 × nonessential amino acids. Cells were transfected with 10 μg of each minigene containing plasmid (wild type, mutID, or mutED) or 10 μg of vector using the MBS mammalian transfection kit (Stratagene Cloning Systems). Transfected cells were cultured for a further 48 hours in complete CHO media and then harvested by scraping into PBS. Cell pellets were stored at –20°C before analysis.
Isolation of RNA and RT-PCR.
RNA was isolated from mouse tissue or transfected CHO cells using Trizol reagent (Life Technologies Inc., Gaithersburg, Maryland, USA) and quantified by measuring absorption at 260 nanometers. Expression of hTF mRNA was determined by semiquantitative RT-PCR using primers spanning intron 1 that yielded a 628-bp product (5′ primer 182–207 bp, 3′ primer 783–810 bp; numbering from ref. 37). PCR was performed for 35 cycles, which is in the linear range of the reaction (36). Contaminating genomic DNA yielded a product of 1.7 kb. Primers specific for mouse GAPDH (CLONTECH Laboratories Inc., Palo Alto, California, USA) were used as a control. RT was performed using a Superscript preamplification system (Life Technologies Inc.) with 2 μg of total RNA per cDNA synthesis reaction. The PCR amplification reaction contained 10 mM Tris-HCl (pH 8.3), 1.5 mM MgCl2, 50 mM KCl, 0.4 μM of each primer, 0.2 mM of each dNTP, 2 μl cDNA template, and 2.5 U of Taq polymerase (Boehringer Mannheim Biochemicals, Indianapolis, Indiana, USA). Each sample was subjected to 35 cycles of PCR.
Determination of hTF antigen.
hTF antigen was measured in brain extracts and E9.5 embryos using a mAb capture system ELISA (IMUBIND TF ELISA kit; American Diagnostica Inc., Greenwich, Connecticut, USA), which does not detect mTF (36). Brain extracts were prepared by resuspending pulverized frozen tissue samples (50–150 mg wet weight) in TBS (pH 8.5) containing 1% Triton X-100 followed by incubation for 14 hours at 4°C with agitation. After centrifugation at 12,000 g for 5 minutes, the protein concentration of the supernatant was determined using a μBCA assay (Pierce Chemical Co., Rockford, Illinois, USA). The amount of hTF in 50 or 100 μg of total protein was determined according to the instructions of the manufacturer. Transfected CHO cells were lysed in TBS (pH 8.5) containing 6 mM CHAPS before protein determination and ELISA.
Results
Construction of mutated human TF minigenes.
We previously reported a novel mouse model system in which a wild-type hTF minigene, which contained the hTF promoter and hTF cDNA, directed a low level (∼1% relative to mouse TF) of hTF expression in transgenic mice (36). To examine the role of FVII/VIIa binding to the TF extracellular domain and formation of a proteolytic active TF:VIIa complex in embryogenesis, we introduced three mutations into the wild-type TF minigene to create the TF mutED minigene (Figure 1). These three mutations in the hTF protein reduce human FVII/VIIa binding by four orders of magnitude and reduce clotting in mouse plasma by two orders of magnitude (27). To determine whether the cytoplasmic domain of the hTF protein was required for embryogenesis, we made a TF mutID minigene that expressed hTF lacking the cytoplasmic domain. The DNA sequence in the wild-type minigene coding for Lys244 of hTF was mutated to a stop codon by site-directed mutagenesis. The mutID minigene expresses hTF lacking the COOH-terminal 20 amino acids of the cytoplasmic domain (Figure 1). This deletion removes Ser253 and Ser258, which are phosphorylated in response to various agonists (26), and Cys245, a palmitylated residue that contributes to membrane anchoring.
Figure 1.

Schematic representation of the hTF minigenes. Genomic DNA is indicated by a line and includes intron 1. The bent arrow indicates the start site of transcription at +1. Filled boxes indicate hTF noncoding and coding sequences (taller portion). Construction of the wild-type minigene has been described previously (36). The TF mutED minigene was generated by introducing the mutations Lys20→Ala, Asp44→Ala and Phe140→Ala into the wild-type minigene by site-directed mutagenesis. The TF mutID minigene was generated by mutating Lys244 (boxed) to a stop codon thereby deleting amino acids 245–263. The boundary of the transmembrane domain is indicated. WT, wild-type.
Expression of mutated human TF minigenes in CHO cells.
To confirm that the mutant TF proteins were expressed from the mutID and mutED minigenes, we transfected plasmids into CHO cells and determined the level of hTF mRNA and protein. hTF specific primers were used in a RT-PCR reaction with RNA prepared from cells transfected with the wild-type, TF mutID, or TF mutED minigenes. A PCR fragment of the expected size (628 bp) was obtained from cells transfected with all three minigenes (Figure 2a). Next, we determined the level of hTF protein in transfected CHO cells using a hTF specific ELISA. As shown in Figure 2b, hTF was expressed at 63 ± 2% and 47 ± 1% of the level of the wild-type minigene in the mutID and mutED minigenes, respectively. The differences in the expression level between the wild-type and mutED minigenes may be due, in part, to the affinity of the anti-human TF antibodies for the mutated proteins expressed by the mutED minigene.
Figure 2.

Expression of wild-type, mutID, and mutED minigenes in CHO cells. CHO cells were transfected with 10 μg of plasmid DNA containing each of the three minigenes (WT, mutID, and mutED) or empty vector (Mock). (a) Total RNA was prepared from transfected cells and assayed for hTF mRNA expression by RT-PCR. The position of the 628-bp hTF PCR product is indicated. Primers specific for GAPDH mRNA were used as a control. A typical result from three independent experiments is shown. A higher molecular weight product (1.7 kb) was generated by amplification of contaminating genomic DNA (asterisk). (b) Expression of hTF protein in CHO cells transfected with each of the three minigenes was determined by ELISA. Results from three independent transfections are shown (mean ± SD).
Generation of transgenic mice.
Our previous study (36) with the wild-type hTF minigene indicated that higher levels of hTF expression were observed with low transgene copy number lines compared with high transgene copy number lines. Therefore, transgenic lines exhibiting low and intermediate transgene copy numbers were selected for further study. Screening of 59 offspring from the injection of the TF mutID minigene into fertilized mouse embryos identified nine founder mice (Figure 3a). Two founder lines, TF mutID4 and TF mutID30, were used for further study. Similarly, screening of 81 offspring from the injection of the TF mutED minigene identified 11 founder mice (Figure 3b). Two founder lines, TF mutED6 and TF mutED25, were used for further study. Analysis of the offspring from crosses between founder mice and wild-type C57BL/6 mice revealed germline transmission of the four minigenes chosen for further study (data not shown). No phenotypic abnormalities were associated with the presence of either the TF mutID or TF mutED minigenes.
Figure 3.

Southern blot analysis of DNA from mutID and mutED founders. (a) TF mutID founders. (b) TF mutED founders. DNA (10 μg) was digested with EcoRI, separated by electrophoresis, and Southern blotted. Membranes were hybridized with a 32P-labeled hTF cDNA probe under conditions of high stringency, which eliminates cross hybridization with murine TF DNA. The position of the expected hybridizing fragment (2.3 kb) is indicated. Founders used for additional experiments are indicated by the open squares.
Expression of the TF mutID and TF mutED minigenes in vivo.
Our previous studies demonstrated that the wild-type minigene expressed the highest level of hTF in the brain of transgenic mice (36). We therefore characterized expression of hTF in the brain of mice containing the TF mutID (ID4 and ID30) and TF mutED (ED6 and ED25) minigenes. A 628-bp PCR product was observed with hTF specific primers using total RNA from brains of TF mutID4, TF mutED6, and TF mutED25 mice but not from the TF mutID30 line or a nontransgenic mouse (Figure 4a). TF mutID4 expressed the highest levels of hTF mRNA. Next, we determined the level of hTF antigen in brain extracts from transgenic mice. hTF antigen was detected in brain extracts from TF mutID4 transgenic mice but not in the other founder lines examined (Figure 4b), which indicates that hTF expression by TF mutID30, TF mutED6, and TF mutED25 was below the detectable threshold (< 10 pg/mL) of the hTF-specific ELISA assay.
Figure 4.

In vivo expression of hTF from TF mutID and TF mutED transgenic mouse lines. (a) Expression of hTF mRNA in the brain of transgenic mice. Total RNA was extracted from the brain of transgenic mice expressing either the TF mutID minigene (founder lines ID4 and ID30), the TF mutED minigene (founder lines ED6 and ED25), or a nontransgenic (Non-Tg) mouse. hTF mRNA levels were determined by RT-PCR using hTF-specific primers followed by Southern blotting with an hTF cDNA probe. A 628-bp product was observed with TF mutID4, TF mutED6, and TF mutED25 but not with TF mutID30 or the nontransgenic mouse. (b) Detection of hTF antigen in the brain of transgenic mice. Levels of hTF antigen were measured in brain extracts of transgenic mouse and in a nontransgenic mouse (Non-Tg) using an hTF specific ELISA. The mean of duplicate determinations are shown.
Rescue of mTF null embryos with the TF mutID and TF mutED minigenes.
Targeted disruption of the mTF gene results in approximately 90% embryonic lethality of mTF null embryos between E9.5 and E10.5 (14–16). Importantly, expression of hTF from the wild-type minigene present in three independent transgenic lines (Tg27, Tg31, and Tg47) quantitatively rescues the embryonic lethality of mTF null embryos (ref. 36; G.C.N. Parry and N. Mackman, unpublished observations).
To determine whether hTF lacking the cytoplasmic domain (TF mutID) or hTF with a mutated extracellular domain (TF mutED) could rescue mTF null embryos in a manner similar to wild-type hTF, we generated mice that were heterozygous for both mTF and hTF (mTF+/–,hTF+/–) (where hTF+ designates the presence of either the TF mutID or TF mutED minigene). These mice were crossed with mice heterozygous for mTF (mTF+/–) (Table 1). Crosses were performed using either male or female mice containing the minigene. If the mutated minigene rescued the embryonic lethality of mTF null embryos, 14% (0.125/1–[0.125 × 0.9]) of live born pups (P1) would be expected to survive due to the presence of the hTF minigene. This breeding produced mTF null embryos at 12.5%, although we expect the majority (∼90%) of these mTF null embryos to die at E10.5.
Table 1.
Offspring from mTF+/–,hTF+/– x mTF+/–,hTF–/–

Screening of 132 P1 pups from a breeding with the TF mutID4 line identified 13 pups (9.8%) that were homozygous for the targeted mTF gene and contained the TF mutID minigene (Table 2). Similarly, screening of 108 P1 pups from a breeding with the TF mutID30 line identified 19 (17.6%) rescued pups. These results demonstrate that hTF lacking the cytoplasmic domain expressed by two independent transgenic lines quantitatively rescued the embryonic lethality of mTF null embryos and permitted birth of live born pups. In contrast to results using the TF mutID minigene, screening of 257 P1 pups from a breeding with the TF mutED6 line identified only 1 pup (0.4%) that was homozygous for the mTF gene and contained the TF mutED minigene (Table 2). Screening of 133 P1 pups from a breeding with the TF mutED25 line produced no rescued pups. These data indicated that expression of the TF extracellular mutant from two independent transgenic lines failed to rescue the embryonic lethality of mTF null embryos.
Table 2.
Genotype of P1 pups from rescue experiments

In the mutID breedings, we identified 1 live born pup (0.4%; 1/240) that was homozygous for the targeted mTF gene without the hTF minigene. In the mutED breeding, we identified 8 pups (2.1%; 8/390) that were homozygous for the targeted mTF gene but did not contain the mutED minigene. The observed frequency of live born TF null pups in all these breedings is 1.43% (9/630 or 11.4% of the expected number). These results are consistent with approximately 90% lethality of TF null embryos at E9.5–10.5 and with the frequency of live born TF null pups (7.6% of expected) observed in a previous study (38).
Expression of hTF mRNA and protein in E9.5 embryos containing the TF mutED minigene.
To exclude the possibility that the different results obtained using the TF mutID and TF mutED minigenes were due to a lack of expression of the hTF mutED minigene at E9.5, we examined hTF mRNA and protein expression at E9.5 in embryos containing TF mutED25. Timed breeds were performed between female mice heterozygous for mTF (mTF+/–) and male mice from the transgenic lines TF mutED6, TF mutED25, and TF mutID30 that were heterozygous for both mTF and hTF (mTF+/–,hTF+/–). Thus, any hTF expression must be from the embryo and cannot be from contaminating maternal tissue.
Human TF mRNA expression was detected by RT-PCR analysis of RNA from E9.5 embryos. Figure 5a (upper panel) shows a representative embryo containing the TF mutED25 minigene that expressed hTF mRNA (ED25-2), whereas another embryo from the same litter (ED25-1) did not contain the minigene and expressed only mTF mRNA. Similar results were obtained in breedings of TF mutED6 mice (data not shown). Importantly, we observed hTF antigen expression by an E9.5 embryo containing the TF mutED25 minigene (Figure 5b), which indicated that the failure of the mutED minigene to rescue mTF null embryos is not due to an absence of expression of the mutant protein at E9.5. For comparison, we analyzed hTF mRNA expression in E9.5 embryos containing the mutID30 minigene because this minigene rescued mTF null embryos in the absence of detectable hTF mRNA or protein expression in the brains of transgenic mice. Figure 5a (lower panel) shows a low level of hTF mRNA expression in an embryo (ID30-2) containing the TF mutID30 minigene, whereas another embryo from the same litter (ID30-1) did not contain the minigene and expressed only mTF mRNA. hTF mRNA expression at E9.5 by TF mutID30 is consistent with the successful rescue of the embryonic lethality of mTF null embryos. hTF protein in embryos and the visceral yolk sac could not be detected by immunohistochemistry, presumably due to the low level of expression of hTF protein. We conclude that both hTF mRNA and protein were expressed by the TF mutED minigene at E9.5 of embryogenesis, which is the stage in embryogenesis at which an absence of mTF results in embryonic lethality.
Figure 5.

Expression of hTF in E9.5 embryos containing the TF mutED25 minigene. Timed-breeds were performed by crossing female mice heterozygous for mTF with male mice heterozygous for mTF and hTF. E9.5 embryos were carefully dissected, and total RNA was prepared using Trizol. The expression of hTF (H), mTF (M), and GAPDH (G) was determined by RT-PCR using specific primers. Molecular weight markers are shown (left side). (a) Representative result obtained with two embryos from a breeding with the mutED25 minigene (upper panel). Representative results obtained with two embryos from a breeding with the mutID30 minigene (lower panel). (b) Detection of hTF antigen in E9.5 embryos. Expression of hTF protein in tissue extracts of E9.5 embryos containing the TF mutED25 minigene (ED25) or nontransgenic (non-Tg) embryos was determined using an hTF specific ELISA.
Postpartum survival of mTF null mice rescued with the TF mutID minigene.
Our previous study (36) using the wild-type transgenic line Tg47 demonstrated long-term survival of rescued mice (mTF–/–,hTF+ Tg47). In an independent experiment, we determined the survival at day 21 of mTF null mice rescued with the TF mutID30 minigene. We observed less than 50% of the expected frequency of rescued mice at day 21 (6/129 = 4.7% at day 21 versus the expected frequency of 14%), which suggested that mice rescued with the TF mutID minigene were dying before day 21. To analyze the survival of mTF–/–,hTF+ mutID30 mice in more detail, we generated a survival curve of these mice. We found that many rescued mice (hTF+ mutID30) (∼80%) died shortly after birth, and all rescued mice were dead by 10 weeks. Similar results were observed with rescued mice containing TF mutID4 (data not shown). Neonates exhibited abdominal hemorrhages (Figure 6), whereas the majority of the older rescued mice died of brain hemorrhages (data not shown).
Figure 6.
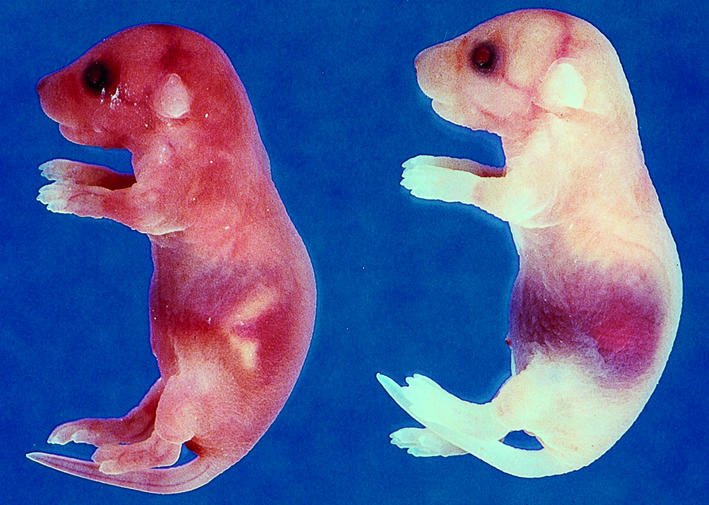
Postpartum hemorrhage of neonates rescued with the TF mutID30 minigene. The majority of rescued neonates died at day 1 due to abdominal hemorrhage (right). A wild-type neonate is shown (left).
The postpartum death of rescued mice containing the minigene expressing the TF cytoplasmic domain mutant may be due to a low level expression of the mutant TF in pups from these two transgenic lines or could suggest a role for the cytoplasmic domain of TF in postpartum hemostasis. For comparison, we determined whether two other transgenic lines (Tg27 and Tg31) that express wild-type hTF at similar levels to Tg47 (36) could rescue mTF null embryos beyond day 1. Similar to the results observed with the TF mutID30 and TF mutID4, mTF null pups rescued with either the wild-type Tg31 or Tg27 minigenes died by 9 weeks (data not shown). These data suggest the spontaneous hemorrhage observed in mice rescued with the TF mutID minigene is due to low levels of hTF expression at a site(s) required for hemostasis and is not due to an absence of cytoplasmic domain.
Discussion
We have used a transgenic approach to analyze the role of TF in mouse embryogenesis. Expression of wild-type hTF from a minigene rescued mTF null embryos from death at approximately E10.5. In this study, we made two mutant minigenes that expressed either hTF without the cytoplasmic domain (mutID) or hTF with three mutations in the extracellular domain (mutED), which reduces FVII/VIIa binding and proteolytic activity of the TF:VIIa complex. Two independent transgenic lines expressing hTF lacking the cytoplasmic domain rescued mTF null embryos from embryonic death, which indicated that the cytoplasmic domain was not required for embryogenesis. In contrast, the TF extracellular domain mutant expressed by two independent transgenic lines containing the mutED minigene failed to rescue mTF null embryos from embryonic death.
The strength of the transgenic approach is the ability to manipulate the cloned minigene in vitro and to introduce multiple mutations into the TF protein. A weakness of this approach is that the transgene is randomly inserted into the genome, and therefore expression of the minigene may be affected by the insertion site. However, three independent founder lines (Tg27, Tg31, and Tg47) expressing the wild-type minigene all rescued the embryonic lethality of mTF null embryos (data not shown). Similarly, two independent founder lines (mutID4 and mutID30) expressing the TF cytoplasmic domain mutant both rescued mTF null embryos. These results suggest that expression of the hTF minigene during embryogenesis is independent of the insertion site.
The failure of the TF extracellular domain mutant to rescue mTF null embryos may be due to a lack of hTF expression at E9.5. We excluded this possibility by demonstrating hTF mRNA and protein expression by the mutED25 minigene at E9.5. In fact, the mutED25 minigene expressed hTF mRNA at higher levels than the mutID30 minigene, but only the mutID30 minigene successfully rescued mTF null embryos. The low levels of hTF protein in rescued embryos and their visceral yolk sacs at E9.5 prohibited analysis of the cell type–specific expression of hTF protein. Therefore, we cannot formally exclude the possibility that hTF protein was not expressed at the appropriate cellular sites to permit rescue of mTF null embryos. Our study analyzed two independent transgenic lines (mutED6 and mutED25) expressing the TF extracellular domain mutant, and both failed to rescue mTF null embryos. Although it is possible that the extracellular domain of TF is binding a ligand other than FVII/VIIa, it seems unlikely because the three mutations of the TF extracellular domain were designed specifically to reduce FVII/VIIa binding. Taken together, our data suggest that binding of FVII/VIIa to the extracellular domain of TF and formation of a proteolytically active TF:VIIa complex are required for mouse embryogenesis.
We observed a high postpartum mortality of mTF null pups rescued with the TF mutID minigene. This result was in contrast with the long-term survival of mTF null pups rescued with the wild-type minigene in Tg47 line and suggested a role for the TF cytoplasmic domain in postpartum hemostasis. However, similar postpartum deaths were observed with mTF null embryos rescued with the wild-type minigene present in two independent founder lines (Tg27 and Tg31). Moreover, Carmeliet and colleagues have used the cre-lox technology to generate mice expressing normal levels of mTF without a cytoplasmic domain (P. Carmeliet, personal communication). These TF cytoplasmic domain–deleted mice are viable and exhibit no spontaneous hemorrhage. Taken together, these results indicate that the postpartum death of mTF mice rescued with the mutID minigene is not due to deletion of the cytoplasmic domain but is due to a low level of hTF expression that is below a threshold required for postpartum hemostasis. Interestingly, the abdominal and subdural hemorrhages observed in rescued neonates containing the mutID minigene were similar to those observed in FVII–/– neonates (20). The hemorrhagic death of mTF null pups rescued with wild-type (Tg27 and Tg31) or mutant (mutID4 and mutID30) minigenes contrasts with the survival of these pups during embryogenesis. We propose that the nonhemostatic role of TF in the maintenance of the yolk sac vasculature during embryogenesis requires very low levels of hTF that are insufficient to maintain hemostasis postpartum.
It is provocative that embryos deficient in TF, FV, prothrombin, and PAR-1 die at approximately E10.5 (14–19, 24, 25). These results suggest a link between TF-dependent activation of the coagulation protease cascade and PAR-1 signaling. The survival of FVII–/– embryos (20) may be due to the transfer of small amounts of maternal FVII to null embryos. We hypothesize that TF may function in embryogenesis to generate thrombin, which then activates PAR-1–dependent intracellular signaling in the visceral yolk sac. This TF-thrombin–PAR-1 pathway appears to mediate maintenance of the integrity of the yolk sac blood vessels. The higher level of lethality observed with the TF null embryos (90%) compared with the level of lethality of PAR-1 null embryos (50%) may be due to either the activation of other pathways by upstream serine proteases, such as FVIIa and FXa, or a compensation for the loss of PAR-1 by other PARs. Our results with the hTF mutID and mutED minigenes support the hypothesis that proteolytic activity of the TF:VIIa complex is required for embryogenesis. Further studies are required to elucidate how thrombin activation of PAR-1, and possibly of other PARs, is required for mouse embryogenesis. To date, no vascular abnormalities have been reported in adult PAR-1–/– mice during normal life, although it cannot be excluded that these mice have subtle defects in their vasculature (24, 25). In addition, PAR-1–/– mice showed normal skin wound healing (39). However, a recent study demonstrated that PAR-1–/– mice exhibit altered responses to arterial injury, with less medial and intimal area and a decreased lumen diameter compared with wild-type mice (40). Thus, we speculate that in adult mice the TF-thrombin–PAR-1 pathway may contribute to the maintenance of vessels and may play an important role in response to vascular injury.
Acknowledgments
We acknowledge helpful discussions in breeding strategies by J. Erlich; critical reading of the manuscript by W. Ruf and M. Riewald; preparation of the manuscript by J. Robertson; and expert technical support by D. Hsia, P. McGovern, A. Hollis, and W. Lee. This work was supported by a grant from the National Institutes of Health (HL16411 to both authors) and by an Established Investigatorship from the American Heart Association (to N. Mackman).
References
- 1.Bach RR. Initiation of coagulation by tissue factor. CRC Crit Rev Biochem. 1988;23:339–368. doi: 10.3109/10409238809082548. [DOI] [PubMed] [Google Scholar]
- 2.Edgington TS, Mackman N, Brand K, Ruf W. The structural biology of expression and function of tissue factor. Thromb Haemost. 1991;66:67–79. [PubMed] [Google Scholar]
- 3.Drake TA, Morrissey JH, Edgington TS. Selective cellular expression of tissue factor in human tissues. Am J Pathol. 1989;134:1087–1097. [PMC free article] [PubMed] [Google Scholar]
- 4.Fleck RA, Rao LVM, Rapaport SI, Varki N. Localization of human tissue factor antigen by immunostaining with monospecific, polyclonal anti-human tissue factor antibody. Thromb Res. 1990;57:765–781. doi: 10.1016/0049-3848(90)90148-6. [DOI] [PubMed] [Google Scholar]
- 5.Leatham EW, Bath PMW, Tooze JA, Camm AJ. Increased monocyte tissue factor expression in coronary disease. Br Heart J. 1995;73:10–13. doi: 10.1136/hrt.73.1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Osterud B, Flaegstad T. Increased tissue thromboplastin activity in monocytes of patients with meningococcal infection: related to an unfavourable prognosis. Thromb Haemost. 1983;49:5–7. [PubMed] [Google Scholar]
- 7.Egan RW, Rickles FR, Cronlund M. Abnormalities of blood coagulation in patients with cancer. Mononuclear cell tissue factor generation. J Lab Clin Med. 1981;98:917–928. [PubMed] [Google Scholar]
- 8.Contrino J, Hair G, Kreutzer DL, Rickles FR. In situ detection of tissue factor in vascular endothelial cells: correlation with the malignant phenotype of human breast disease. Nat Med. 1996;2:209–215. doi: 10.1038/nm0296-209. [DOI] [PubMed] [Google Scholar]
- 9.Zhang Y, et al. Tissue factor controls the balance of angiogenic and antiangiogenic properties of tumor cells in mice. J Clin Invest. 1994;94:1320–1327. doi: 10.1172/JCI117451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mueller BM, Reisfeld RA, Edgington TS, Ruf W. Expression of tissue factor by melanoma cells promotes efficient hematogenous metastasis. Proc Natl Acad Sci USA. 1992;89:11832–11836. doi: 10.1073/pnas.89.24.11832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bromberg ME, Konigsberg WH, Madison JF, Pawashe A, Garen A. Tissue factor promotes melanoma metastasis by a pathway independent of blood coagulation. Proc Natl Acad Sci USA. 1995;92:8205–8209. doi: 10.1073/pnas.92.18.8205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luther T, et al. Tissue factor expression during human and mouse development. Am J Pathol. 1996;149:101–113. [PMC free article] [PubMed] [Google Scholar]
- 13.Soifer SJ, Peters KG, O’Keefe J, Coughlin SR. Disparate temporal expression of the prothrombin and thrombin receptor genes during mouse development. Am J Pathol. 1994;144:60–69. [PMC free article] [PubMed] [Google Scholar]
- 14.Toomey JR, Kratzer KE, Lasky NM, Stanton JJ, Broze GJ., Jr Targeted disruption of the murine tissue factor gene results in embryonic lethality. Blood. 1996;88:1583–1587. [PubMed] [Google Scholar]
- 15.Bugge TH, et al. Fatal embryonic bleeding events in mice lacking tissue factor, the cell-associated initiator of blood coagulation. Proc Natl Acad Sci USA. 1996;93:6258–6263. doi: 10.1073/pnas.93.13.6258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carmeliet P, et al. Role of tissue factor in embryonic vessel development. Nature. 1996;385:73–75. doi: 10.1038/383073a0. [DOI] [PubMed] [Google Scholar]
- 17.Cui J, O’Shea KS, Purkayastha A, Saunders TL, Ginsburg D. Fatal haemorrhage and incomplete block to embryogenesis in mice lacking coagulation factor V. Nature. 1996;384:66–68. doi: 10.1038/384066a0. [DOI] [PubMed] [Google Scholar]
- 18.Sun WY, et al. Prothrombin deficiency results in embryonic and neonatal lethality in mice. Proc Natl Acad Sci USA. 1998;95:7597–7602. doi: 10.1073/pnas.95.13.7597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xue J, et al. Incomplete embryonic lethality and fatal neonatal hemorrhage caused by prothrombin deficiency in mice. Proc Natl Acad Sci USA. 1998;95:7603–7607. doi: 10.1073/pnas.95.13.7603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Albrecht S, et al. Mice lacking factor VII develop normally but suffer fatal perinatal bleeding. Nature. 1997;390:290–294. doi: 10.1038/36862. [DOI] [PubMed] [Google Scholar]
- 21.Suh TT, et al. Resolution of spontaneous bleeding events but failure of pregnancy in fibrinogen-deficient mice. Genes Dev. 1995;9:2020–2033. doi: 10.1101/gad.9.16.2020. [DOI] [PubMed] [Google Scholar]
- 22.Shivdasani RA, et al. Transcription factor NF-E2 is required for platelet formation independent of the actions of thrombopoietin/MGDF in megakaryocyte development. Cell. 1995;81:695–704. doi: 10.1016/0092-8674(95)90531-6. [DOI] [PubMed] [Google Scholar]
- 23.Shivdasani RA, Fielder P, Keller G-A, Orkin SH, de Sauvage FJ. Regulation of the serum concentration of thrombopoietin in thrombocytopenic NF-E2 knockout mice. Blood. 1997;90:1821–1827. [PubMed] [Google Scholar]
- 24.Connolly AJ, Ishihara H, Kahn ML, Farese RVJ, Coughlin SR. Role of the thrombin receptor in development and evidence for a second receptor. Nature. 1996;381:516–519. doi: 10.1038/381516a0. [DOI] [PubMed] [Google Scholar]
- 25.Darrow AL, et al. Biological consequences of thrombin receptor deficiency in mice. Thromb Haemost. 1996;76:860–866. [PubMed] [Google Scholar]
- 26.Ruf W, Mueller BM. Tissue factor signaling. Thromb Haemost. 1999;82:175–182. [PubMed] [Google Scholar]
- 27.Mueller BM, Ruf W. Requirement for binding of catalytically active factor VIIa in tissue factor-dependent experimental metastasis. J Clin Invest. 1998;101:1372–1378. doi: 10.1172/JCI930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fischer EG, Ruf W, Mueller BM. Tissue factor-initiated thrombin generation activates the signaling thrombin receptor on malignant melanoma cells. Cancer Res. 1995;55:1629–1632. [PubMed] [Google Scholar]
- 29.Ollivier V, Bentolila S, Chabbat J, Hakim J, De Prost D. Tissue-factor-dependent vascular endothelial growth factor production by human fibroblasts in response to activated factor VII. Blood. 1998;91:2698–2703. [PubMed] [Google Scholar]
- 30.Taniguchi T, Kakkar AK, Tuddenham EGD, Williamson RCN, Lemoine NR. Enhanced expression of urokinase receptor induced through the tissue factor-factor VIIa pathway in human pancreatic cancer. Cancer Res. 1998;58:4461–4467. [PubMed] [Google Scholar]
- 31.Camerer E, et al. Coagulation factors VIIa and Xa induce cell signaling leading to up-regulation of the egr-1 gene. J Biol Chem. 1999;274:32225–32233. doi: 10.1074/jbc.274.45.32225. [DOI] [PubMed] [Google Scholar]
- 32.Zioncheck TF, Roy S, Vehar GA. The cytoplasmic domain of tissue factor is phosphorylated by a protein kinase C–dependent mechanism. J Biol Chem. 1992;267:3561–3564. [PubMed] [Google Scholar]
- 33.Mody RS, Carson SD. Tissue factor cytoplasmic domain peptide is multiply phosphorylated in vitro. Biochemistry. 1997;36:7869–7875. doi: 10.1021/bi9701235. [DOI] [PubMed] [Google Scholar]
- 34.Abe K, et al. Regulation of vascular endothelial growth factor production and angiogenesis by the cytoplasmic tail of tissue factor. Proc Natl Acad Sci USA. 1999;96:8663–8668. doi: 10.1073/pnas.96.15.8663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ott I, Fischer EG, Miyagi Y, Mueller BM, Ruf W. A role for tissue factor in cell adhesion and migration mediated by interaction with actin binding protein 280. J Cell Biol. 1998;140:1241–1253. doi: 10.1083/jcb.140.5.1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Parry GCN, Mackman N. Low levels of tissue factor are compatible with development and hemostasis in mice. J Clin Invest. 1998;101:560–569. doi: 10.1172/JCI814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morrissey JH, Gregory SA, Mackman N, Edgington TS. Tissue factor regulation and gene organization. Oxf Surv Eukaryot Genes. 1989;6:67–84. [PubMed] [Google Scholar]
- 38.Toomey JR, Kratzer KE, Lasky NM, Broze GJ., Jr Effect of tissue factor deficiency on mouse and tumor development. Proc Natl Acad Sci USA. 1997;94:6922–6926. doi: 10.1073/pnas.94.13.6922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Connolly AJ, Suh DY, Hunt TK, Coughlin SR. Mice lacking the thrombin receptor, PAR1, have normal skin wound healing. Am J Pathol. 1997;151:1199–1204. [PMC free article] [PubMed] [Google Scholar]
- 40.Cheung WM, D’Andrea MR, Andrade-Gordon P, Damiano BP. Altered vascular injury responses in mice deficient in protease-activated receptor-1. Arterioscler Thromb Vasc Biol. 1999;19:3014–3024. doi: 10.1161/01.atv.19.12.3014. [DOI] [PubMed] [Google Scholar]