

Abstract
With an ever increasing number of people taking numerous medications, the need to safely administer drugs and limit unintended side effects has never been greater. Antidote control remains the most direct means to counteract acute side effects of drugs, but, unfortunately, it has been challenging and cost prohibitive to generate antidotes for most therapeutic agents. Here we describe the development of a set of antidote molecules that are capable of counteracting the effects of an entire class of therapeutic agents based upon aptamers. These universal antidotes exploit the fact that, when systemically administered, aptamers are the only free extracellular oligonucleotides found in circulation. We show that protein-and polymer-based molecules that capture oligonucleotides can reverse the activity of several aptamers in vitro and counteract aptamer activity in vivo. The availability of universal antidotes to control the activity of any aptamer suggests that aptamers may be a particularly safe class of therapeutics.
Oligonucleotide therapeutics (that is, small interfering RNAs (siRNAs) and aptamers) have gained much attention as potential treatments for a variety of diseases1-6. Recent scientific advancements have led to an exponential increase in the number of such molecules going through clinical trials. Aptamers are single-stranded nucleic acid molecules that bind and inhibit protein targets7. We were the first to show the potential therapeutic utility of aptamers by inhibiting HIV-replication with a TAR aptamer8 and others continue to develop this clinically9. Building on this principle and using combinatorial libraries of nucleic acids and the systematic evolution of ligands by exponential enrichment (SELEX) method10,11, many aptamers have been developed that have potential therapeutic value7.
A number of aptamers targeting coagulation factors have been described that inhibit clot formation and represent potential anticoagulant therapeutics7,12,13. Regrettably, anticoagulants engender complications, such as considerable bleeding, that increase patient morbidity and mortality14. Currently, only one anticoagulant and antidote pair, heparin and protamine, is routinely used in clinics15,16. Antidote control provides the safest means to regulate drug action and minimize side effects. Unfortunately, it has been challenging to develop antidotes for antibody and small-molecule drugs, owing to the inability to distinguish therapeutic antibodies or small-molecule drugs from their endogenous counterparts in a patient’s body.
Previously, we described how customized antidote oligonucleotides could be created to specifically reverse the activity of particular aptamers targeting coagulation factor IXa (FIXa) and von Willebrand factor (vWF) by exploiting Watson-Crick base pairing to generate duplex RNAs1,16-18. This approach yielded rationally designed drug-antidote pairs but has several limitations15,16,19. First, it is cost prohibitive to develop such customized and expensive oligonucleotide antidotes for every therapeutic aptamer. In addition, we have observed that it is challenging to develop such antidotes to highly structured aptamers. Finally, because the resulting aptamer-antidote complex is a double stranded RNA, the complex may activate the innate immune response, leading to inflammation20.
Here we describe an approach that overcomes these limitations in antidote design and yields universal antidote molecules that can control the activity of many aptamers, regardless of their sequence. We recognized that oligonucleotide-based drugs are different from antibody and small-molecule drugs in that no endogenous counterparts for oligonucleotide drugs are normally free in circulation. Thus, we hypothesized that molecules that can sequester oligonucleotides in a sequence-independent manner should be able to function as universal antidotes for extracellular oligonucleotide-based drugs in animals and humans.
RESULTS
Because two aptamers selected to different targets (thrombin and vascular endothelial growth factor) have been shown to interact with the heparin-binding domain on their target proteins5,21, we first investigated whether protamine, the antidote for heparin, could reverse the activity of two aptamers that target FIXa (aptamer 9.3t) and FXa (aptamer 11F7T). The activated partial thromboplastin time (APTT) assay is used for monitoring anticoagulation therapy, and we have previously shown that this assay can also be used to follow the anticoagulant effects of both FIXa and FXa aptamers16,19 (J. Layzer and B.A.S., unpublished data). These aptamers have markedly different primary sequences and secondary structures (as predicted by mFold, Fig. 1a)22 and target different proteins in the coagulation cascade. We found that aptamer 9.3t is a potent anticoagulant (Fig. 1b). However, addition of protamine neutralized the anticoagulant effects of this aptamer within 5 min (Fig. 1b). Similarly, aptamer 11F7T is a potent anticoagulant, and protamine also rapidly neutralized the activity of this aptamer (Fig. 1c). In both cases, protamine (2.5 μg) was able to totally reverse the aptamers’ activity in an APTT clotting assay (Fig. 1d) at a 50% lower concentration than is routinely used to reverse heparin’s anticoagulant activity. Moreover, protamine was able to rapidly reverse the anticoagulant activity of the two aptamers simultaneously (Fig. 1e), and such reversal was maintained for at least 1 h.
Figure 1.

The structures of aptamers used in this study and protamine-mediated reversal of anticoagulant aptamer function. (a) The primary sequence and predicted secondary structures of FIXa (9.3t), FXa (11F7T), VWF (9.3), FII (R9D-14), VWF (9.14), FVII (7S-1), FX (7K-5) and FIX (9D-6) aptamers. (b) APTT clotting time of normal human plasma in the presence of aptamer 9.3t (150 nM), with and without the addition of protamine (2.5 μg per 160 μl). (c) APTT clotting time of normal human plasma in the presence of aptamer 11F7T (250 nM), with and without the addition of protamine (2.5 μg per 160 μl). (d) APTT clotting time of normal human plasma in the presence of aptamer 9.3t (150 nM) and increasing concentrations of protamine. (e) APTT clotting time of normal human plasma in the presence of aptamers 9.3t and 11F7T, with and without the addition of protamine. The data are plotted as the means ± s.e.m. for three independent measurements.
Although protamine is routinely used to reverse the activity of heparin after cardiopulmonary bypass surgery, protamine administration is associated with several side effects, including increased pulmonary artery pressure, decreased systolic and diastolic blood pressure, impaired myocardial oxygen consumption, and reduced cardiac output, heart rate and systemic vascular resistance15,23-26. Therefore, we sought to identify other agents that could rapidly reverse the activity of aptamers. We first screened a number of nucleic acid binding polymers for their ability to act as antidotes for aptamer 9.3t (Supplementary Table 1). Several of the polymers were able to completely reverse the activity of the aptamer within 5 min (Fig. 2a). To better understand why certain polymers were more effective than others, we measured the interactions of the different polymers with aptamer 9.3t by isothermal titration calorimetry (ITC). We used a two-site model to interpret the ITC data for the interactions between the cationic polymers and aptamer 9.3t. Binding constants and some thermodynamic parameters are summarized in Supplementary Table 2. As expected, all of the interactions were entropy driven, except for with polyamidoamine dendrimer, 1,4-diaminobutane core, G3 (PAMAM), and most were also enthalpy driven. Although no conclusions can be drawn about the mechanisms of the interactions, the trends of binding strength, in the two-site model, were consistent with the results from APTT screening. Protamine, β-cyclodextrin–containing polycation (CDP), β-cyclodextrin–containing polycation, imadazole-containing variant (CDP-Im), polyphosphoramidate polymer (PPA-DPA) and PAMAM showed considerable and similar affinities for aptamer 9.3t, whereas the binding constants of polybrene and spermine were orders of magnitude lower (Supplementary Table 2). Thus, there is a direct correlation between a polymer’s affinity for the aptamer and its potency as an antidote.
Figure 2.
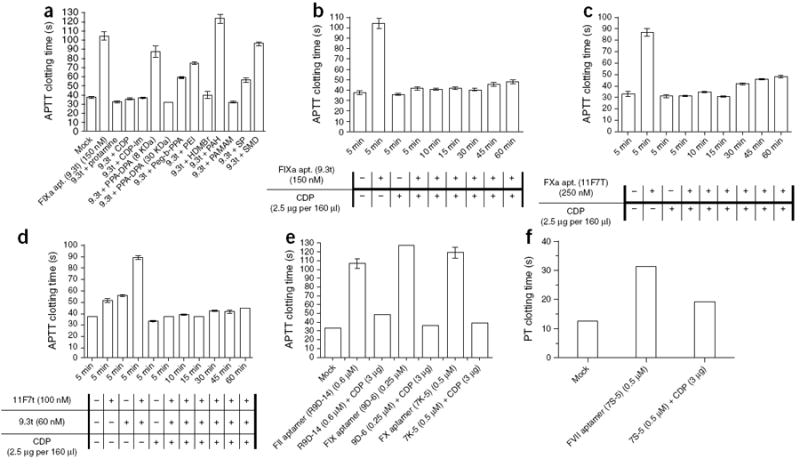
Polymer-mediated reversal of anticoagulant aptamer function. (a) APTT clotting time of normal human plasma in the presence of aptamer 9.3t (150 nM), with and without the addition of protamine or 11 different polymers (2.5 μg per 160 μl). PEI, polyethyleneimine; PAH, poly(allylamine hydrochloride); HDMBr, hexadimethrine bromide; SP, spermine; SMD, spermidine. (b–e) APTT clotting time of normal human plasma in the presence of aptamer 9.3t (150 nM) (b), aptamer 11F7T (c), aptamers 9.3t and 11F7 (d), aptamers R9D-14, 9D-6 and 7K-5 (e), with and without the addition of CDP. (f) Prothrombin time (PT) clotting time of normal human plasma in the presense of aptamer 7S-1 with and without the addition of CDP. The data are plotted as the means ± s.e.m. for three independent measurements.
Because CDP has high binding affinity for the aptamer and is known to have low toxicity, we tested this polymer in time-course experiments27. Similarly to protamine, CDP could rapidly and durably reverse the activity of these two distinct anticoagulant aptamers (9.3t and 11F7T) in vitro (Fig. 2b–d). Subsequently, we examined CDP’s ability to reverse the activity of four additional aptamers that target FII, FIX, FX and FVII. CDP could rapidly reverse the activity of each of these aptamers (Fig. 2e,f).
Next, we tested CDP and PPA-DPA for their ability to neutralize the antiplatelet effects of VWF aptamer 9.3 and vWF aptamer 9.14 (Fig. 1a) in a platelet function assay (PFA-100)17. vWF aptamers 9.3 and 9.14 have no sequence or structure similarity to the previously tested aptamers, and both could inhibit platelet function in whole blood (Fig. 3a,b). Addition of either CDP or PPA-DPA resulted in rapid reversal of vWF aptamer 9.3 antiplatelet activity, with CDP achieving complete reversal at an order of magnitude lower amount than PPA-DPA (Fig. 3a,b). Moreover, CDP was able to rapidly reverse the activity of vWF aptamer 9.14 at this same concentration (Fig. 3c). These experiments further demonstrate that CDP and PPA-DPA can act as sequence-independent antidotes for aptamers. Moreover, these results point to the broad applicability of this approach, as the antidotes work in both plasma and whole blood against eight different aptamers.
Figure 3.
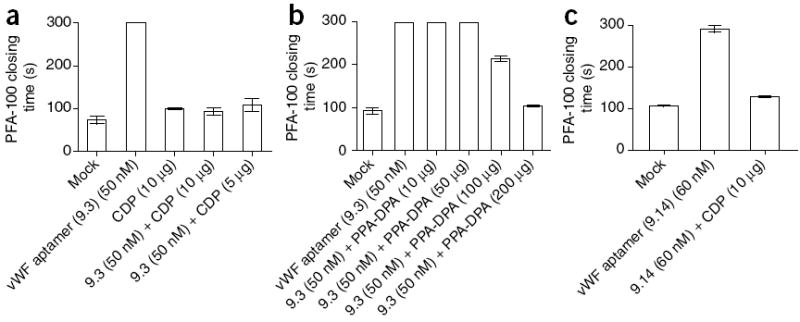
Polymer-mediated reversal of antiplatelet aptamer function. (a,b) PFA-100 closing time of normal whole blood in the presence of vWF aptamer 9.3 with and without the addition of CDP (a) or PPA-DPA (30 kDa) (b). (c) PFA closing time of normal whole blood in the presence of vWF aptamer 9.14 with and without the addition of CDP. The data are plotted as the means ± s.e.m. for three independent measurements.
Next, we sought to determine whether such universal antidotes are able to reverse aptamer activity in vivo. Results from in vitro experiments (gel electrophoresis and dynamic light scattering) using the same concentrations as anticipated for use in mice showed that CDP is able to bind the aptamer and form a composite entity (Supplementary Fig. 1), and we observed that CDP-Im formed a complex with siRNA when sequentially injected into mice (Supplementary Fig. 2). Therefore, we evaluated the activity of the universal antidotes in a swine anticoagulation model. Pigs (n = 5) were anticoagulated with the FIXa aptamer (Ch-9.3t) (0.5 mg per kg body weight) that had been modified with a cholesterol at its 5′ end to improve its circulating half-life19 (Fig. 4a). We observed an immediate increase in the activated clotting time (ACT) (from 105 ± 5 s to 150 ± 5 s) for the treated pigs. When no antidote was administered, the level of anticoagulation only gradually decreased over the 90-min time frame of the experiment (Fig. 4a). However, administration of protamine (10 mg per kg body weight) resulted in a total reversal of the anticoagulant effect within 5 min (n = 5) (Fig. 4b). In addition, this reversal was sustained for the remainder of the experiment, 60 min (Fig. 4b). Similarly, CDP (n = 5) (2.5 mg per kg body weight) was also able to rapidly and durably reverse the activity of this aptamer in vivo (Fig. 4c). Furthermore, we did not observe any toxicities after administration of these antidotes during the experiment (Supplementary Fig. 3). All vital signs stayed within error of their baseline levels, with the exception that protamine induced a mild hypotension and CDP a mild hypertension (<15% change; Supplementary Fig. 3d,e). These results indicate that both protamine and CDP can act as antidotes for aptamers in vivo.
Figure 4.
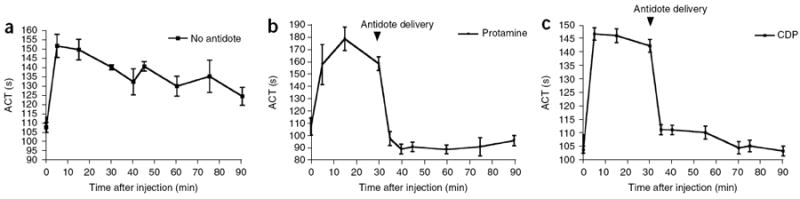
In vivo aptamer and antidote activity. (a–c) ACT clotting times of blood from swine treated with the cholesterol-modified FIXa aptamer 9.3t (Ch-9.3t) in pigs (n = 5) alone (a) or with the addition of protamine (b) or CDP (c). The data are plotted as the means ± s.e.m. for duplicate measurements from each pig.
DISCUSSION
Between 1998 and 2005, the number of serious adverse drugs events reported to the US Food and Drug Administration increased 2.6-fold, and fatal adverse events increased 2.7-fold to 15,107 events in 2005 (refs. 28,29). Therefore, there is a pressing medical need to develop safer and more controllable therapeutic strategies. Unfortunately, it has been both technically challenging and very expensive to develop antidote molecules to counteract the side effects of most medicines. We have shown that characteristics unique to oligonucleotides can be used to design universal antidotes that can sequester aptamers and reverse their activity, regardless of the aptamer’s primary sequence and folded structure. Our initial studies showed that protamine, a commonly used and inexpensive heparin reversal agent with well-known side effects, can be used as an antidote for multiple aptamers30,31. Furthermore, the observation that protamine can neutralize aptamer activity indicates that protamine should be used with caution in patients being treated with oligonucleotide-based drugs, as protamine may unintentionally reverse their activity.
To find universal antidotes with more favorable characteristics such as low toxicity, we tested a number of polymeric gene carriers for their ability to reverse the activity of aptamers. The field of nonviral gene therapy has stimulated the synthesis of many polymers for the delivery of plasmid DNA and siRNA. The rigidity, hydrophobicity and hydrophilicity, charge density, biodegradability and molecular weight of the polymer chain are all parameters that can be adjusted to achieve an optimal complexation with oligonucleotides32,33. Moreover, it has previously been shown that a small cationic porphyrin can act as an antidote to a G-quartet–containing thrombin aptamer34. Therefore, we screened a number of DNA or siRNA delivery polymers and found that several of them can reverse the activity of multiple aptamers in vitro, and that CDP can also rapidly reverse the activity of an anticoagulant aptamer in pigs.
Previously, we developed a strategy to reverse the activity of aptamers using Watson-Crick base-pairing rules to create a customized antidote oligonucleotide for each aptamer16,19. This customization is very costly, because for each aptamer a new antidote oligonucleotide has to be developed, tested and manufactured. Another concern with this approach is that when antidote oligonucleotides bind to aptamers, a double-stranded RNA is formed that may stimulate the innate immune system20,35. Although both the RNA aptamer and the antidote oligonucleotide are often comprised of modified oligonucleotides (that is, 2-F and 2-OMe RNA), there is still a danger that these molecules may activate Toll-like receptor 3, as short 2’Ome siRNA duplexes have been reported to induce such effects20. Finally, because most indications will not require an antidote for the reversal of drug action 100% of the time, the added cost of developing a customized antidote is difficult to justify for most drugs that usually are safe but are associated with relatively rare but serious side effects. Thus, we believe that the universal antidote approach will be more broadly applicable than the customized antidote oligonucleotide approach we have described. As with any new therapeutic agent, the safety of the universal antidote molecules will have to be tested in the clinic. Recent studies evaluating the toxicity of CDP-Im in nonhuman primates have shown that such compounds have a favorable safety profile36. Thus, we are optimistic that the universal antidote strategy can be rapidly translated into the clinic and believe that safer therapeutic agents may be forthcoming.
Methods
Methods and any associated references are available in the online version of the paper at http://www.nature.com/naturemedicine/.
Supplementary Material
Acknowledgments
We thank S.M. Nimjee and J. Layzer for helpful discussions and Calando Pharmaceuticals for providing the CDP and CDP-Im polymers. This work was supported by a grant from the US National Institutes of Health (HL065222 to B.A.S.), a predoctoral fellowship from the American Heart Association (0615443U to S.O.) and a grant from the US National Cancer Institute (CA 119347 to M.E.D).
Footnotes
AUTHOR CONTRIBUTIONS S.O. designed and performed research, analyzed data and wrote the manuscript; R.T.S.L designed and performed research and analyzed data; K.M.B. performed research; C.M.B. performed research and analyzed data; G.Q. performed research; J.D.H. designed and performed research, provided useful reagents, analyzed data and provided useful discussions; J.Y.-C.L. performed research; B.C.M. performed research; M.E.D. provided useful reagents and discussions; K.W.L. provided useful reagents and discussions; B.A.S. suggested the universal antidote idea, designed and coordinated research, analyzed data and wrote the manuscript.
COMPETING INTERESTS STATEMENT The authors declare competing financial interests: details accompany the full-text HTML version of the paper at http://www.nature.com/naturemedicine/.
Note: Supplementary information is available on the Nature Medicine website.
References
- 1.Dyke CK, et al. First-in-human experience of an antidote-controlled anticoagulant using RNA aptamer technology: a phase 1a pharmacodynamic evaluation of a drug-antidote pair for the controlled regulation of factor IXa activity. Circulation. 2006;114:2490–2497. doi: 10.1161/CIRCULATIONAHA.106.668434. [DOI] [PubMed] [Google Scholar]
- 2.EyetechStudyGroup. Preclinical and phase 1A clinical evaluation of an anti-VEGF pegylated aptamer (EYE001) for the treatment of exudative age-related macular degeneration. Retina. 2002;22:143–152. doi: 10.1097/00006982-200204000-00002. [DOI] [PubMed] [Google Scholar]
- 3.EyetechStudyGroup. Anti-vascular endothelial growth factor therapy for subfoveal choroidal neovascularization secondary to age-related macular degeneration: phase II study results. Ophthalmology. 2003;110:979–986. doi: 10.1016/S0161-6420(03)00085-X. [DOI] [PubMed] [Google Scholar]
- 4.Gilbert JC, et al. First-in-human evaluation of anti von Willebrand factor therapeutic aptamer ARC1779 in healthy volunteers. Circulation. 2007;116:2678–2686. doi: 10.1161/CIRCULATIONAHA.107.724864. [DOI] [PubMed] [Google Scholar]
- 5.Lee JH, et al. A therapeutic aptamer inhibits angiogenesis by specifically targeting the heparin binding domain of VEGF165. Proc Natl Acad Sci USA. 2005;102:18902–18907. doi: 10.1073/pnas.0509069102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McNamara JO, et al. Multivalent 4–1BB binding aptamers costimulate CD8+ T cells and inhibit tumor growth in mice. J Clin Invest. 2008;118:376–386. doi: 10.1172/JCI33365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nimjee SM, Rusconi CP, Sullenger BA. Aptamers: an emerging class of therapeutics. Annu Rev Med. 2005;56:555–583. doi: 10.1146/annurev.med.56.062904.144915. [DOI] [PubMed] [Google Scholar]
- 8.Sullenger BA, Gallardo HF, Ungers GE, Gilboa E. Overexpression of TAR sequences renders cells resistant to human immunodeficiency virus replication. Cell. 1990;63:601–608. doi: 10.1016/0092-8674(90)90455-n. [DOI] [PubMed] [Google Scholar]
- 9.Anderson J, et al. Safety and efficacy of a lentiviral vector containing three anti-HIV genes—CCR5 ribozyme, tat-rev siRNA, and TAR decoy—in SCID-hu mouse–derived T cells. Mol Ther. 2007;15:1182–1188. doi: 10.1038/sj.mt.6300157. [DOI] [PubMed] [Google Scholar]
- 10.Ellington AD, Szostak JW. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990;346:818–822. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- 11.Tuerk C, Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990;249:505–510. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- 12.Becker RC, Rusconi C, Sullenger B. Nucleic acid aptamers in therapeutic anticoagulation. Technology, development and clinical application. Thromb Haemost. 2005;93:1014–1020. doi: 10.1160/TH04-12-0790. [DOI] [PubMed] [Google Scholar]
- 13.Nimjee SM, Rusconi CP, Harrington RA, Sullenger BA. The potential of aptamers as anticoagulants. Trends Cardiovasc Med. 2005;15:41–45. doi: 10.1016/j.tcm.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 14.Fanikos J, et al. Adverse drug events in hospitalized cardiac patients. Am J Cardiol. 2007;100:1465–1469. doi: 10.1016/j.amjcard.2007.06.041. [DOI] [PubMed] [Google Scholar]
- 15.Nimjee SM, et al. A novel antidote-controlled anticoagulant reduces thrombin generation and inflammation and improves cardiac function in cardiopulmonary bypass surgery. Mol Ther. 2006;14:408–415. doi: 10.1016/j.ymthe.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 16.Rusconi CP, et al. RNA aptamers as reversible antagonists of coagulation factor IXa. Nature. 2002;419:90–94. doi: 10.1038/nature00963. [DOI] [PubMed] [Google Scholar]
- 17.Oney S, et al. Antidote-controlled platelet inhibition targeting von Willebrand factor with aptamers. Oligonucleotides. 2007;17:265–274. doi: 10.1089/oli.2007.0089. [DOI] [PubMed] [Google Scholar]
- 18.Rusconi CP, Yeh A, Lyerly HK, Lawson JH, Sullenger BA. Blocking the initiation of coagulation by RNA aptamers to factor VIIa. Thromb Haemost. 2000;84:841–848. [PubMed] [Google Scholar]
- 19.Rusconi CP, et al. Antidote-mediated control of an anticoagulant aptamer in vivo. Nat Biotechnol. 2004;22:1423–1428. doi: 10.1038/nbt1023. [DOI] [PubMed] [Google Scholar]
- 20.Kleinman ME, et al. Sequence- and target-independent angiogenesis suppression by siRNA via TLR3. Nature. 2008;452:591–597. doi: 10.1038/nature06765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.White R, et al. Generation of species cross-reactive aptamers using “toggle” SELEX. Mol Ther. 2001;4:567–573. doi: 10.1006/mthe.2001.0495. [DOI] [PubMed] [Google Scholar]
- 22.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hird RB, et al. Direct effects of protamine sulfate on myocyte contractile processes. Cellular and molecular mechanisms. Circulation. 1995;92:11433–11446. doi: 10.1161/01.cir.92.9.433. [DOI] [PubMed] [Google Scholar]
- 24.Porsche R, Brenner ZR. Allergy to protamine sulfate. Heart Lung. 1999;28:418–428. doi: 10.1016/s0147-9563(99)70031-2. [DOI] [PubMed] [Google Scholar]
- 25.Shigeta O, et al. Low-dose protamine based on heparin-protamine titration method reduces platelet dysfunction after cardiopulmonary bypass. J Thorac Cardiovasc Surg. 1999;118:354–360. doi: 10.1016/S0022-5223(99)70227-8. [DOI] [PubMed] [Google Scholar]
- 26.Welsby IJ, et al. Hemodynamic changes after protamine administration: association with mortality after coronary artery bypass surgery. Anesthesiology. 2005;102:308–314. doi: 10.1097/00000542-200502000-00011. [DOI] [PubMed] [Google Scholar]
- 27.Gonzalez H, Hwang SJ, Davis ME. New class of polymers for the delivery of macromolecular therapeutics. Bioconjug Chem. 1999;10:1068–1074. doi: 10.1021/bc990072j. [DOI] [PubMed] [Google Scholar]
- 28.Moore TJ, Cohen MR, Furberg CD. Serious adverse drug events reported to the Food and Drug Administration, 1998–2005. Arch Intern Med. 2007;167:1752–1759. doi: 10.1001/archinte.167.16.1752. [DOI] [PubMed] [Google Scholar]
- 29.Lazarou J, Pomeranz BH, Corey PN. Incidence of adverse drug reactions in hospitalized patients: a meta-analysis of prospective studies. J Am Med Assoc. 1998;279:1200–1205. doi: 10.1001/jama.279.15.1200. [DOI] [PubMed] [Google Scholar]
- 30.Carr JA, Silverman N. The heparin-protamine interaction. A review. J Cardiovasc Surg (Torino) 1999;40:659–666. [PubMed] [Google Scholar]
- 31.Stanker LH, Wyrobek A, McKeown C, Balhorn R. Identification of the binding site of two monoclonal antibodies to human protamine. Mol Immunol. 1993;30:1633–1638. doi: 10.1016/0161-5890(93)90436-f. [DOI] [PubMed] [Google Scholar]
- 32.Davis ME, et al. Self-assembling nucleic acid delivery vehicles via linear, water-soluble, cyclodextrin-containing polymers. Curr Med Chem. 2004;11:179–197. doi: 10.2174/0929867043456179. [DOI] [PubMed] [Google Scholar]
- 33.Mao HQ, Leong KW. Design of polyphosphoester-DNA nanoparticles for nonviral gene delivery. Adv Genet. 2005;53:275–306. [PubMed] [Google Scholar]
- 34.Joachimi A, Mayer G, Hartig JS. A new anticoagulant-antidote pair: control of thrombin activity by aptamers and porphyrins. J Am Chem Soc. 2007;129:3036–3037. doi: 10.1021/ja0677822. [DOI] [PubMed] [Google Scholar]
- 35.Schröder M, Bowie AG. TLR3 in antiviral immunity: key player or bystander? Trends Immunol. 2005;26:462–468. doi: 10.1016/j.it.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 36.Heidel JD, et al. Administration in non-human primates of escalating intravenous doses of targeted nanoparticles containing ribonucleotide reductase subunit M2 siRNA. Proc Natl Acad Sci USA. 2007;104:5715–5721. doi: 10.1073/pnas.0701458104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.