

Abstract
Motivation: Datasets from high-throughput sequencing technologies have yielded a vast amount of data about organisms in environmental samples. Yet, it is still a challenge to assess the exact organism content in these samples because the task of taxonomic classification is too computationally complex to annotate all reads in a dataset. An easy-to-use webserver is needed to process these reads. While many methods exist, only a few are publicly available on webservers, and out of those, most do not annotate all reads.
Results: We introduce a webserver that implements the naïve Bayes classifier (NBC) to classify all metagenomic reads to their best taxonomic match. Results indicate that NBC can assign next-generation sequencing reads to their taxonomic classification and can find significant populations of genera that other classifiers may miss.
Availability: Publicly available at: http://nbc.ece.drexel.edu.
Contact: gailr@ece.drexel.edu
1 INTRODUCTION
After acquiring a sample and using next-generation technology to perform shotgun sequencing, the next step in metagenomic analysis it to assess the taxonomic content of the sample. This methodology, also known as phylogenetic analysis, gives a simple look at ‘Who is in this sample?’ The first tool ever used (which is still widely used) for taxonomic assessment is Basic Local Alignment Search Tool (BLAST; Altschul et al., 1990). In recent years, several specialized webservers have been made available to the public to ease the process of taxonomically classifying reads, namely Phylopythia (McHardy et al., 2007), CAMERA (Seshadri et al., 2007), WebCARMA (Gerlach et al., 2009), MG-RAST (Meyer et al., 2008) and Galaxy (Pond et al., 2009). Unlike BLAST, Phylopythia and WebCARMA return more specific taxonomic information and assign reads to higher level taxonomic levels using a consensus of BLAST top-hit taxonomies [aka ‘last common ancestor’ algorithms (Huson et al., 2007)]. In this article, we focus our comparison to remote stand-alone webservers and not to methods that only have locally installable software. Ultimately, all the metagenomic analysis webservers aim to ease analysis of complex environmental samples for users that do not have resources to maintain their own databases and systems.
Phylopythia was the first taxonomic classification webserver to be implemented. Phylopythia is based on a support vector machine (SVM) classification method and produces very good accuracy for long (≥ 1 Kbp) reads (McHardy et al., 2007). WebCARMA is a homology-based approach that matches environmental gene tags to protein families and reports good results for long and ultrashort 35-bp reads using (i) BLASTX to find candidate environmental gene tags (EGTs) and (ii) using Pfam (protein family) hidden Markov models (HMMs) to match the EGTs against protein families during an EGT candidate selection process. MG-RAST (Metagenome Rapid Annotation using Subsystem Technology) (Meyer et al., 2008), CAMERA (Community Cyberinfrastructure for Advanced Microbial Ecology Research and Analysis) (Seshadri et al., 2007) and the Galaxy Project (Pond et al., 2009) are high-throughput metagenomic pipelines that aim to be an all-in-one one-stop analysis for metagenomic samples. For taxonomic classification of shotgun sequencing, MG-RAST offers a homology-based approach, SEED (Overbeek et al., 2005). CAMERA and Galaxy provide high-throughput implementations and custom databases for BLASTN. BLASTN yields best hit sequence matches and is known to have reasonable accuracy (Rosen et al., 2009).
Previously, Rosen et al. have explored a machine learning method, naïve Bayes classifier (NBC), as a possible way to classify fragments that can annotate more sequences than BLAST (Rosen et al., 2008). We now implement the algorithm on a webserver for public use and benchmark it against other web sites.
2 METHODS AND MATERIALS
We selected a previously benchmarked dataset (Gerlach et al., 2009): the Biogas reactor dataset (Schlüter et al., 2008), composed of 353 213 reads of average 230 bp length. We selected a real dataset as opposed to a synthetic one because we did not want to tailor the dataset to any specific database, since the database will vary on each web site. This comparison fairly assesses each webserver's performance on a ‘real’ dataset containing known and novel organisms.
We conducted our tests against NBC and five other webservers in July and August of 2010. WebCARMA and MG-RAST require no parameters. Phylopythia requires the type of model to match against. MG-RAST requires an E-value cutoff under the SEED viewer (which we selected the highest). We selected default BLAST parameters for the NT database for Galaxy. For NBC, we used an Nmer size of 15 and the default 1032 organism genome-list. For CAMERA, we only retained the best top-hit organism for each read and used the ‘All Prokaryotes’ BLASTN database (and used the default parameters for the rest).
We implement the NBC approach in Rosen et al. (2008) that assigns each read a log-likelihood score. We introduce two functions of NBC: (i) the novice functionality and (ii) the expert functionality. We expect that most users will fit into the ‘novice’ category, which will enable them to upload their FASTA file of reads and obtain a file of summarized results matching each read to its most likely organism, given the training database. The parameters that (expert and novice) users can choose from are as follows:
Upload File: the FASTA formatted file of metagenomic reads. The webserver also accepts .zip, .gz and .tgz of several FASTA files.
Genome list: the algorithm speed depends linearly on the number of genomes that one scores against. So, if an expert user has prior knowledge about the expected microbes in the environment, he/she can select only those microbes that should be scored against. This will both speed up the computation time and reduce false positives of the algorithm.
Nmer length: the user can select different Nmer feature sizes, but it is recommended that the novice user use N = 15 since it works well for both long and short reads (Rosen et al., 2008).
Email: The user's email address is required so that they can be notified as to where to retrieve the results when the job is completed.
Output: For a beginner, we suggest to (i) upload a FASTA file with the metagenomic reads and (ii) enter an email address. The output is a link to a directory that contains your original upload file (renamed as userAnalysisFile.txt), the genomes that were scored against (masterGenomeList.txt) and a summary of the matches for each read (summarized_results.txt). The expert user may be particularly interested in the *.csv.gz files where he/she can analyze the ‘score distribution’ of each read more in depth.
3 DISCUSSION
In Figure 1, we show the percentage of reads (out of the whole dataset) that ranked in the top eight genera for each algorithm. We see that all methods are in unanimous agreement for Clostridium and Bacillus, while most methods (except Galaxy) agree for prominence of Methanoculleus. CAMERA supports NBC's findings of Pseudomonas and Burkholderia, known to be found in sewage treatment plants (Vinneras et al., 2006). [The biogas reactor contained ∼2% chicken manure so it can have the traits of sludge waste (Schlüter et al., 2008)]. In Hery et al. (2010), Pseudomonas and Sorangium have been found in sludge wastes. Streptosporangium and Streptomyces are commonly found in vegetable gardens (Nolan et al., 2010), which is quite reasonable since this is an agricultural bioreactor. Therefore, NBC potentially has found significant populations of genera that other classifiers have missed. Thermosinus is not in NBC's completed microbial training database and therefore, it did not find any matches.
Fig. 1.
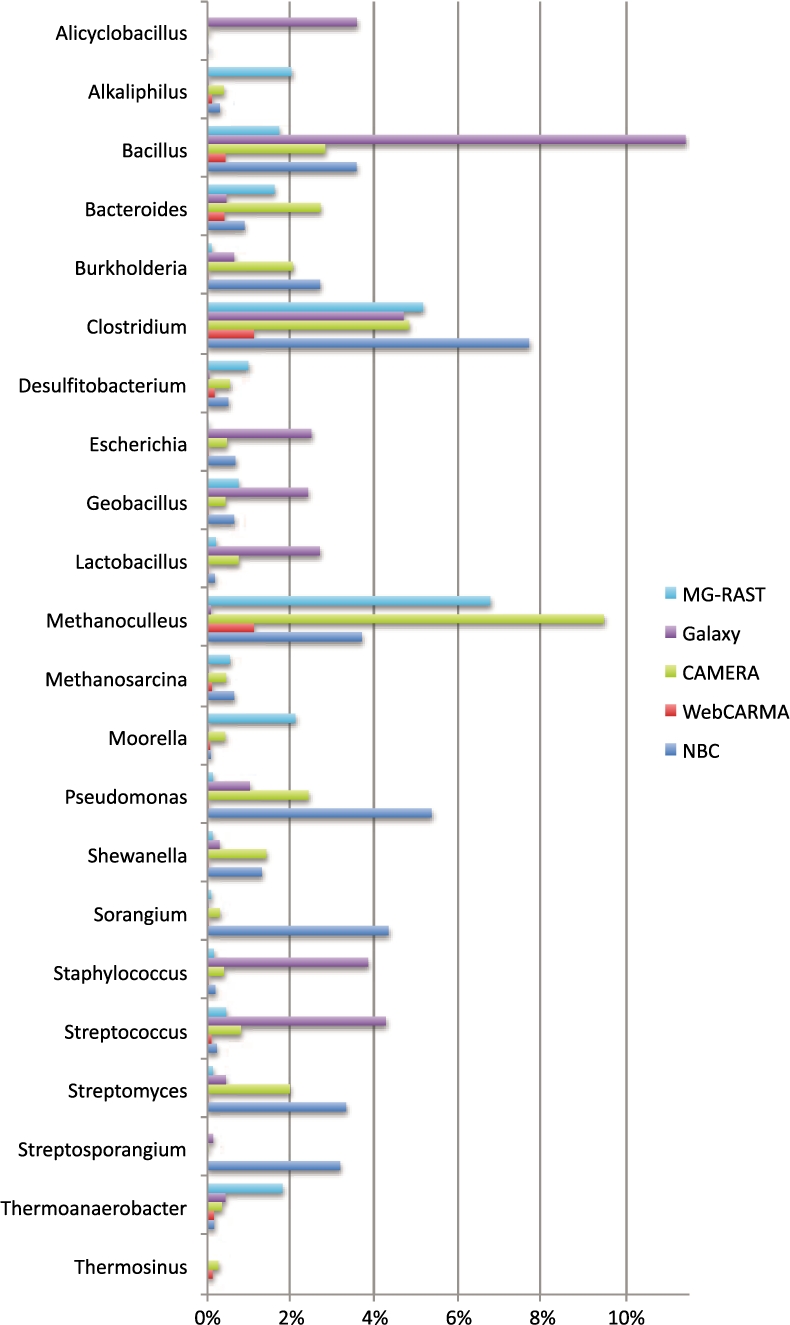
Percentage of reads that are assigned to a particular genera out of all 454 reads from the Biogas reactor community. CAMERA and NBC tend to agree for over 70% of the genera shown while MG-RAST agrees with CAMERA and NBC near 50%. WebCARMA bins fewers reads, and Galaxy has high variability. For the first 5602 reads (1.5 Mb web site limit), Phylopythia only classifies eight reads to the phylum level and is not included in the graph due to its inability to make assignments at the genus level.
NBC took 21 h to run and classified all 100% of the reads compared with 12 h/23% for WebCARMA, 5 h/99% for CAMERA, 2–3 h/140% for Galaxy 1, and a few weeks 2/56.2% for MG-RAST. NBC runs on a 4-core Intel machine and speed would linearly increase with distributed computing in the future.
4 CONCLUSION
The naïve Bayes classification tool is implemented on a web site for public use. We demonstrate that the tool can handle a complete pyrosequencing dataset, and it gives the full taxonomy for each read, so that users can easily analyze the taxonomic composition of their datasets. NBC classifies every read unlike other tools and is easy to use, runs an entire dataset in a reasonable amount of time and yields competitive results.
ACKNOWLEDGEMENT
We thank Christopher Cramer for the scoring code and binary packages.
Funding: Supported in part by the National Science Foundation CAREER award #0845827 and Department of Energy award DE-SC0004335.
Conflict of Interest: none declared.
Footnotes
1In Galaxy, the number of BLAST hits is greater than the original # of reads.
2There was a lengthy wait queue for MG-RAST. It is difficult to assess true run times due to each site's different hardware and usage.
REFERENCES
- Altschul SF, et al. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Gerlach W, et al. Webcarma: a web application for the functional and taxonomic classification of unassembled metagenomic reads. BMC Bioinformatics. 2009;10 doi: 10.1186/1471-2105-10-430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hery M, et al. Monitoring of bacterial communities during low temperature thermal treatment of activated sludge combining dna phylochip and respirometry techniques. Water Res. 2010 doi: 10.1016/j.watres.2010.07.003. [Epub ahead of print, doi: 10.1016/j.watres.2010.07.003.] [DOI] [PubMed] [Google Scholar]
- Huson DE, et al. Megan analysis of metagenomic data. Genome Res. 2007;17:377–386. doi: 10.1101/gr.5969107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHardy AC, et al. Accurate phylogenetic classification of variable-length dna fragments. Nat. Methods. 2007;4:63–72. doi: 10.1038/nmeth976. [DOI] [PubMed] [Google Scholar]
- Meyer F, et al. The metagenomics rast server - a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics. 2008;9:386. doi: 10.1186/1471-2105-9-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan M, et al. Complete genome sequence of streptosorangium roseum type strain (ni 9100t) Stand. Genomic Sci. 2010;2:1. doi: 10.4056/sigs.631049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overbeek R, et al. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucelic Acids Res. 2005;33:5691–5702. doi: 10.1093/nar/gki866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pond SK, et al. Windshield splatter analysis with the galaxy metagenomic pipeline. Genome Res. 2009;19:2144–2153. doi: 10.1101/gr.094508.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen GL, et al. Signal processing for metagenomics: extracting information from the soup. Curr. Genomics. 2009;10:493–510. doi: 10.2174/138920209789208255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen GL, et al. Metagenome fragment classification using n-mer frequency profiles. Adv. Bioinformatics. 2008;2008 doi: 10.1155/2008/205969. Article ID 205969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlüter A, et al. The metagenome of a biogas-producing microbial community of a production-scale biogas plant fermenter analysed by the 454-pyrosequencing technology. J. Biotechnol. 2008;136:77–90. doi: 10.1016/j.jbiotec.2008.05.008. [DOI] [PubMed] [Google Scholar]
- Seshadri R, et al. Camera: A community resource for metagenomics. PLoS Biol. 2007;5 doi: 10.1371/journal.pbio.0050075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinneras B, et al. Identification of the microbiological community in biogas systems and evaluation of microbial risks from gas usage. Sci. Total Environ. 2006;367:606–615. doi: 10.1016/j.scitotenv.2006.02.008. [DOI] [PubMed] [Google Scholar]