

Abstract
An effective high-speed countercurrent chromatography (HSCCC) method was established for further separation and purification of four minor flavonols in addition to five major flavonols which were reported by our previous study from extracts of Flos Gossypii. HSCCC was performed with three two-phase solvent systems composed of n-hexane-ethyl acetate-methanol-water (7.5:15:6:7, v/v), (2.5:15:2:7, v/v) and (0:1:0:1, v/v). The separation was repeated 3 times, and 3.8 mg of 8-methoxyl-kaempferol-7-O-β-D-rhamnoside (HPLC purity 98.27%), 6.7 mg of astragalin (HPLC purity 94.18%), 3.3 mg of 4′-methoxyl-quercetin-7-O-β-D-glucoside (HPLC purity 94.30%) and 8.2 mg of hyperoside (HPLC purity 93.48%) were separated from 150 mg of the crude sample. The chemical structures of the flavonols were confirmed by MS, 1H NMR and 13C NMR. Meanwhile, the results indicated that the target compound with smaller K value (<0.5) can be separated by increasing column length of HSCCC. And four separation rules of flavonols according to the present study and references were summarized, which can be used as a useful guide for separation of flavonols by HSCCC.
Keywords: HSCCC, Flos Gossypii, flavonol, separation rules of flavonols
INTRODUCTION
As a Uighur’s traditional medicine, Flos Gossypii flowers have been used for tranquilization, detumescence, treatment of pruritus, alleviating burn pain and replenishing heart and brain for a thousand years in Xinjiang, China[1]. Up to now, however, few phytochemical studies on this plant have been described in the literature except by our group[2, 3].
Traditional separation methods, such as silica gel column chromatography and preparative thin-layer chromatography (PTLC) are time consuming and some minor samples are often irreversibly adsorbed onto the solid support. High-speed countercurrent chromatography (HSCCC) is a liquid-liquid separation method that does not require a solid sorbent and therefore it is possible to totally recover the introduced samples. Consequently, HSCCC has been successfully applied to the isolation of various natural products [4, 5], especially for flavonols [6-9]. Its unique properties promise a future of preparation and exploitation of natural compounds. HSCCC has been considered as a powerful tool for the separation and purification of bioactive compounds from traditional Chinese herbs and other natural products.
In our previous study, we have successfully isolated six major flavonols from Flos Gossypii by HSCCC [2, 3]. But we have not separated many minor compounds detected by HPLC in the crude sample In order to isolate the minor constituents, K values of these minor compounds were measured thoroughly by HPLC. Consequently, four minor flavonols, in addition to five flavonols which had been reported in the past [2, 3], were isolated and purified from Flos Gossypii by HSCCC.
Although HSCCC has been widely used in preparative separation of flavonols for the past two decades, its separation rules have never been reported before. In our present study, the separation rules of flavonols were made according to the results of the present study together with references. These rules will serve as a useful guidance for the isolation and separation of flavonols from various natural resources by HSCCC.
EXPERIMENTAL
Apparatus
The preparative HSCCC instrument employed in this study is a model HHS–400A high-speed countercurrent chromatograph (Shanghai Tonghong Machine Co., Ltd., Shanghai, China). The apparatus were equipped with four polytetrafluoroethylene (PTFE) preparative coils (inner diameter of 1.6 mm, each 125 mL in capacity with a total capacity of 500 mL) which are symmetrically mounted on the rotary frame to attain stable balancing of the centrifuge system. Four preparative coils can be operated in parallel or series or as individual coil. Different column capacities are available according to the requirement of peak resolution of the target compounds. The revolution radius was 6.5 cm, and the β-values varied from 0.25 at the internal terminal to 0.9 at the external terminal (β = r/R, where r is the distance from the coil to the holder shaft). An optimum speed of 800 rpm was used thourghout this study.
The solvent was pumped into the column with a Model NS-1007 constant-flow pump (Beijing Institute of New Technology Application, Beijing, China). Continuous monitoring of the effluent was achieved with a Model 8823A-UV monitor (Beijing Institute of New Technology Application) at 254 nm. A manual sample injection valve with a 20-mL loop for the preparative HSCCC (Tianjin High New Science Technology, Tianjin, China) was used to introduce the sample into the column.. A portable recorder (Yokogawa Model 3057, Sichuan Instrument Factory, Chongqing, China) was used to draw the chromatogram.
The high-performance liquid chromatography (DIONEX, USA) equipment used was a DIONEX system including a P680 pump, an ASI-100 Automated sample injector, a TCC-100 Thermostatted column compartment, a UVD170U detector. The analysis was carried out with an inertsil ODS-SP column (5 μm, 4.6×250 mm GL Sciences Inc, Japan). Evaluation and quantification were made on a Chromeleon WorkStation.
Reagents
All organic solvents used for HSCCC were of analytical grade and purchased from Tianjin chemical Factory (Tianjin, China). Methanol used for HPLC was of HPLC-grade and purchased from Fisher Scientific Company (Fair Lawn, NJ, USA). Flos gossypii was purchased from a local store in Urumqi of China.
Preparation of Crude Sample [2]
Flos gossypii purchased from a local store in China was powdered and extracted with aqueous ethanol in reflux for 3 h (three times). All the extracts were combined and evaporated under reduced pressure at 60°C, and the residue was dissolved in water, which was in turn loaded into a glass column packed with AB-8 macroporous resin. The column was first eluted with water until the eluent becomes colorless, followed by stepwise elution with 50% and 70% aqueous ethanol to elute out the target compound. These fractions were combined, evaporated to dryness. The residues were dissolved in ethyl acetate. After filtration the ethyl acetate extract was evaporated to dryness, yielding the crude sample for HSCCC separation.
Measurement of Partition Coefficient (K) [6]
The two-phase solvent systems were selected according to the partition coefficient (K) of the target components. Different ratios of n-hexane-ethyl acetate-methanol -water were prepared and equilibrated in a separation funnel at room temperature. The K values were determined by HPLC analysis as follows: a suitable amount of samples (1 mg) was added to 4.0 mL mixture of equal volume of each phase of the two-phase solvent system followed by thorough mixing of the contents. After equilibration was established, aliquots of the upper and the lower phases were separately analyzed by HPLC. The peak area of the upper phase was recorded as AU and that of the lower phase as AL. The K value was calculated according to the following equation: K=AU/AL. (Table 1)
Table 1.
Partition coefficient (K) of eleven flavonoids in different radio of n-hexane-ethyl acetate-methanol-water
| Compound | Solvent system radio | K value |
|---|---|---|
| I | 7.5:15:6:7, v/v | 0.29 |
| II | 0.43 | |
| X | 2.78 | |
| III | 2.5:15:2:7, v/v | 0.78 |
| IV | 0.93 | |
| V | 1.23 | |
| VI | 0:1:0:1, v/v | --- |
| VII | 0.85 | |
| VIII | 1.03 | |
| IX | 1.55 | |
HSCCC Separation
The preparative HSCCC was performed with a model HHS-400A HSCCC instrument as follows: the multiplayer coiled column was first entirely filled with the upper phase as stationary phase. After rotation at 800 rpm, the sample solution (50 mg of the crude sample in 20 mL of a mixture of upper and lower phases) was injected through the sample port. The lower phase was pumped into the head end of the HSCCC coil column, and the effluent from the outlet of the column was monitored with a UV detector at 254 nm. Peak fractions were manually collected according to the chromatogram.
HPLC Analysis and Identification of Crude Sample and the Peak Fractions from HSCCC [11]
The crude sample and the peak fractions from HSCCC were analyzed by HPLC. The analyses were performed with an inertsil ODS-SP column(4.6 mm I.D. × 250 mm, 5 μm)at column temperature of 35 °C. The mobile phase was eluted with a linear gradient of acetonitrile (A), methanol (B) and 0.2% formic acid (C) that follows: A-B-C (10:10:80, v/v) to A-B-C (15:15:70, v/v) in 15 min, then to A-B-C (0:55:45, v/v) in 35 min, then to A-B-C (0:80:20, v/v) in 6 min, and finally to A-B-C (0:80:20, v/v) in 4 min. The flow-rate was 1.0 mL min−1 and the effluent was monitored at 254 nm by a UV detector.
Identification of the HSCCC peak fractions was carried out by 1H nuclear magnetic resonance (1H NMR) and 13C nuclear magnetic resonance (13C NMR).
RESULTS AND DISCUSSION
HPLC Analysis of the Crude Sample
The crude extract of Flos gossypii was first analyzed by HPLC. The result indicated that it contained several flavonols, including kaempferol (retention time 49.33 min), kaempferol-3-O-β-D-(6″-O-p-coumaroyl)-glucoside (retention time 46.89 min), 8-methoxyl-kaempferol-7-O-β-D-rhamnoside (retention time 43.47 min), quercetin (retention time 38.53 min), 4′-methoxyl-quercetin-7-O-β-D-glucoside (retention time 33.38 min), quercetin-3′-O-β-D-glucoside (retention time 29.58 min), astragalin (retention time 25.28 min), quercetin-3-O-β-D-glucoside (retention time 20.21 min), hyperoside (retention time 19.73 min), and quercetin-7-O-β-D-glucoside (retention time 17.64 min), as shown in Fig. 1A.
Fig. 1.
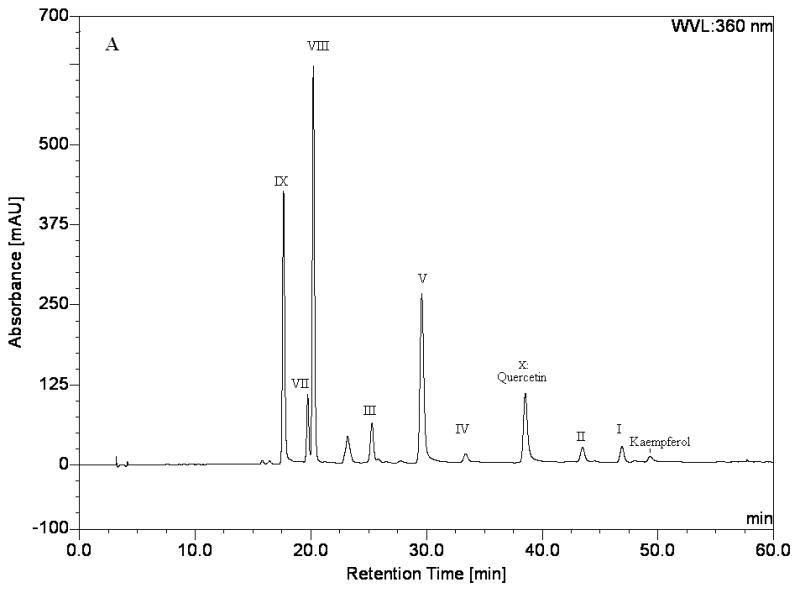
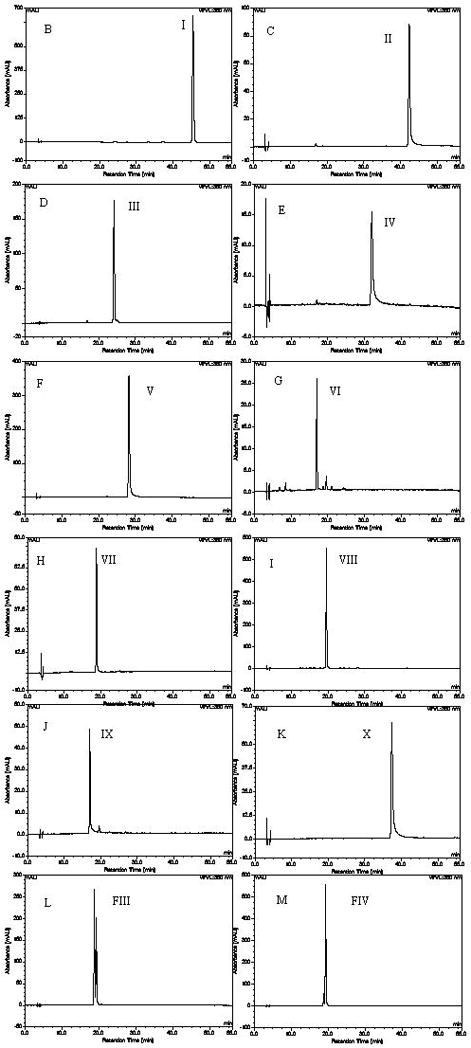
HPLC analysis of the crude sample and peak fractions from HSCCC. Separation column: an inertsil ODS-SP column (4.6mmI.D. × 250mm, 5μm); column temperature: 35 °C; the mobile phase: a linear gradient of acetonitrile (A), methanol (B) and 0.2% formic acid (C) that follows: A-B-C (10:10:80, v/v) to A-B-C (15:15:70, v/v) in 15 min, then to A-B-C (0:55:45, v/v) in 35 min, then to A-B-C (0:80:20, v/v) in 6 min, and finally to A-B-C (0:80:20, v/v) in 4 min. The flow-rate: 1.0 mL min−1; detection wavelength: 360 nm. A) crude sample, B) peak I in Fig. 2A, C) peak II in Fig. 2A, D) peak III in Fig. 2B, E) peak IV in Fig. 2B, F) peak V in Fig. 2B, G) peak VI in Fig. 2C, H) peak VII in Fig. 2C, I) peak VIII in Fig. 2C, J) peak IX in Fig. 2C, K) peak X in Fig. 4L) fraction III in Fig. 2C, M) fraction IV in Fig. 2C.
Selection of Two-Phase Solvent System and Other Conditions of HSCCC
In HSCCC, successful separation highly depends upon the selection of a suitable two-phase solvent system, which requires the following considerations: retention of the stationary phase should be satisfactory, which is judged with short settling time of the solvent system (<25 sec)[12], and the partition coefficient of the target compound is between 0.4-2.5[13]. The solute with a smaller K value will be eluted closer to the solvent front with low peak resolution while the solute with a larger K value tends to give better resolution but with broader and more dilute peaks[14]. Because natural products are complex, it will be better to apply the slightly longer elution time to attain better resolution with a higher K value ≤3.5.
Several monographs, review articles and book chapters described various two-phase solvent systems successfully used for CCC. Refs. [15-17] provide lists of the two-phase solvent systems for HSCCC. Several two-phase solvent systems of n-hexane-ethyl acetate-methanol-water were tested based on these references and the properties of flavonols. As shown in Table 1, three solvent systems composed of n-hexane-ethyl acetate-methanol-water (7.5:15:6:7, v/v), (2.5:15:2:7, v/v) and (0:1:0:1, v/v) were selected to separate the target compounds. Two compounds had suitable K values in the first solvent system; three compounds had suitable K values in the second solvent system; and another three compounds had suitable K values in the third solvent system.
Fig. 2A shows the chromatogram obtained from 50 mg of the crude sample by preparative HSCCC using a two-phase solvent system composed of n-hexane-ethyl acetate-methanol-water (7.5:15:6:7, v/v). The peaks I & II and Fraction I (FI) were isolated at a flow rate of .5 mL·min−1. The purities of the peaks were detected by HPLC (Fig. 1B, C). Fig. 2B shows the chromatogram obtained from Fraction I using n-hexane-ethyl acetate-methanol-water (2.5:15:2:7, v/v). The peaks III, IV, V and Fraction II (FII) were isolated at a flow rate of 1.5 mL·min−1. The purities of the peaks were evaluated by HPLC (Fig. 1D–F). Fig. 2C shows the chromatogram obtained from Fraction II using n-hexane-ethyl acetate-methanol-water (0:1:0:1, v/v). The peaks VI, VII, VIII, IX and Fraction III (FIII), IV (FIV) were isolated. FIII and IV were the mixture of quercetin-3-O-β-D-glucoside and hyperoside (Figs. L and M). But a small amount of quercetin-3-O-β-D-glucoside and hyperoside can be obtained from the cut peak. The flow rate was 1.5 mL·min−1. The retention of stationary phase was 63.38%, 56.55% and 42.76%, respectively (Fig. 1E–J). In addition to flavonols previously reported, four minor components, peaks II, III, IV and VII, were newly separated from Flos gossypii.
Fig. 2.
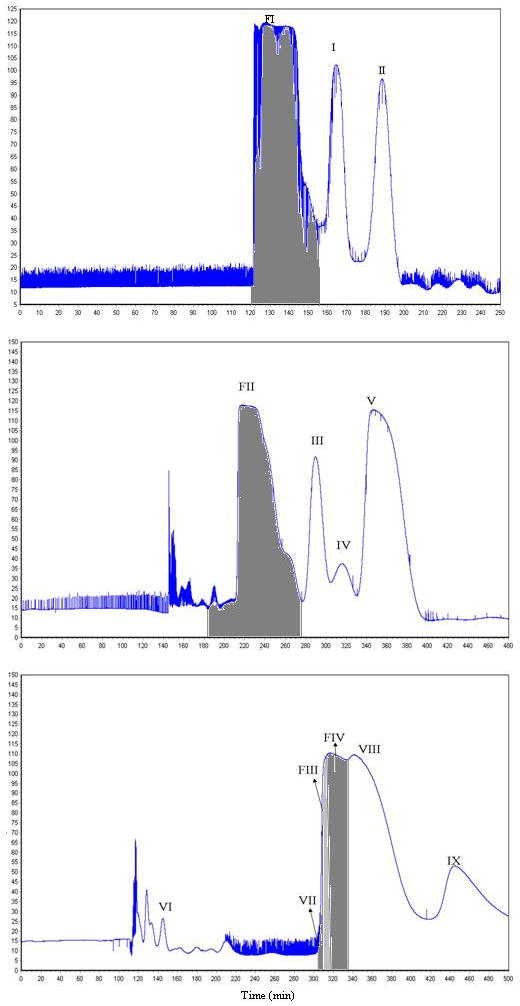
HSCCC chromatograms of crude sample from Flos Gossypii. revolution speed: 800 rpm; detection wavelength: 254 nm; flow rate: 1.5 mL min−1; solvent systems: (A) n-hexane-ethyl acetate-methanol-water (7.5:15:6:7, v/v), (B) n-hexane-ethyl acetate-methanol-water (2.5:15:2:7, v/v); (C) n-hexane-ethyl acetate-methanol-water (0:1:0:1, v/v); samples: (A) crude sample I, (B) fraction I, (C) fraction II.
Structural Identification [18, 19]
The structural identifications of HSCCC peaks were carried out by MS, 1H NMR and 13C NMR. The HSCCC separation was repeated 3 times, and the fractions from the same peak were combined based on the HPLC analysis. Peak I (2.9 mg, HPLC purity 95.77%) was identified as Kaempferol-3-O-β-D-(6″-O-p-coumaroyl)-glucoside which has been reported by us [3]. Peak II (3.8 mg, HPLC purity 98.27%) was identified as 8-methoxyl-kaempferol-7-O-β-D-rhamnoside compared with Ref. [20]. Peak III (6.7 mg, HPLC purity 94.18%) was identified as astragalin according to Ref. [21]. Peak IV (3.3 mg, HPLC purity 94.30%) was identified as 4′-methoxyl-quercetin-7-O-β-D-glucoside compared with Ref. [22]. Peak V (16.2 mg, HPLC purity 99.12%) was identified as quercetin-3′-O-β-D-glucoside which has been reported in our previous studies [2, 10]. Peak VI (HPLC purity 85.25%) was not identified because of its low purity and content. Peak VII (8.2 mg, HPLC purity 98.47%) was identified as hyperoside according to Ref. [10, 21]. Peak VIII (40.1 mg, HPLC purity 93.48%) was identified as quercetin-3-O-β-D-glucoside which has been reported in our previous studies [2, 10]. Peak IX (34.3 mg, HPLC purity 93.75%) was identified as quercetin-7-O-β-D-glucoside which has been reported in our previous studies [2, 10].
Study on the Resolution under the Different Column Capacity of HSCCC
As mentioned earlier, the preparative HSCCC instrument employed in this study is a model HHS–400A high-speed countercurrent chromatograph. The apparatus was equipped with four polytetrafluoroethylene (PTFE) preparative coils (1.6 mm ID tubing, each 125 mL in capacity with a total capacity of 500 mL). Four preparative coils can be operated in parallel or series or as an individual coil. Thus, various column capacities are available according to the need for the separations.
The partition coefficient (K) is the ratio of solute distributed between the mutually equilibrated two solvent phases. Ref. [15] indicated that the suitable K values of target compounds for HSCCC are 0.5≤K≤1.0. And another Ref. [13] indicated the proper range of K value is between 0.4–2.5. However, complex natural products can be isolated with larger K values of over 3.0 by HSCCC, although the procedure was somewhat time-consuming [23, 24]. If their separation factor (α) is large enough, the column capacity can be decreased to separate the target compounds, namely the length of the column can be reduced to shorten the separation time and minimize solvent consumption. But if the K value is lower than 0.4, it is difficult to resolve the target compounds in the crude sample [13, 15]. In any case, if their separation factor (α) is not large enough, they are difficult to be resolved. Usually we can improve the resolution of closely related compounds by increasing the length of the column. In this study, the K values of Kaempferol-3-O-β-D-(6″-O-p-coumaroyl)-glucoside and 8-methoxyl-kaempferol-7-O-β-D-rhamnoside in the two-phase solvent system composed of n-hexane-ethyl acetate-methanol-water (7.5:15:6:7, v/v) (Table 1) were 0.28 and 0.43 with their separation factor (α) at 1.5 which is not large enough for the separation with the standard column presently employed..
A series of studies was performed to examine the resolution (Rs) of peaks I and II with the different column length. Fig. 4 shows the comparative analysis of HSCCC chromatograms obtained at different column lengths. The resolution of target compounds is increased with the applied column volume as expected. The different column capacities were selected to separate these two targets from 125 mL to 500 mL. The results clearly show that the four coils operated in series yielded the best peak resolution. For the isolation of peak I, the full volume (500 mL), four coils operated in series, should be selected, while for the peak II, 3/4 volume (375 mL), three coils operated in series, is sufficient. But if we want to get the peak X, the 1/4 volume (125 mL), individual coil, is the best choice to save time and solvent.
Fig. 4.
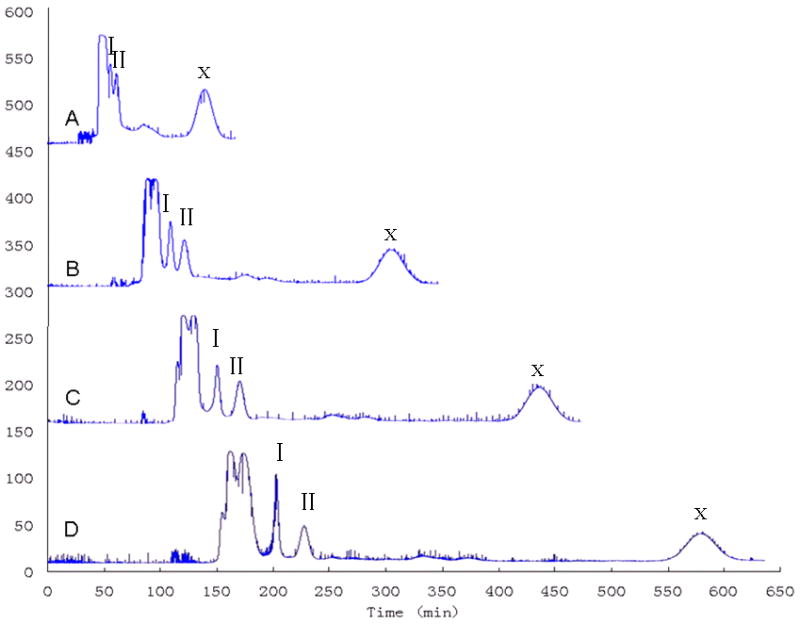
Comparative analysis of HSCCC chromatogram of 50 mg of the crude sample at different column volumes: (A) 125 mL capacity (B) 250 mL capacity (C) 375 mL capacity (D) 500 mL capacity. Solvent systems: n-hexane–ethyl acetate–methanol–water (7.5:15:6:7, v/v). Note: The apparatus was equipped with four PTFE preparative coils (1.6 mm ID tubing, each 125 mL capacity with a total capacity 500 mL), symmetrically mounted on the rotary frame. Four preparative coils can be operated in parallel or series or as individual coil.
Four Separation Rules of Flavonols in the Two-Phase Solvent System of n-Hexane-Ethyl Acetate-Methanol-Water
Flavonols and their conjugates form a very large group of natural products. They are found in many plant tissues, where they are present inside the cells or on the surfaces of different plant organs. The separation of flavonols and their conjugates is one of the most important areas in the field of instrumental analytical methods, helping solve problems in biological and medical sciences. Although HSCCC has been widely used in preparative separation of flavonols for the past two decades, its separation rules have never been reported. In the present study, ten flavonols were separated from Flos gossypii by HSCCC with the two-phase solvent system of n-hexane-ethyl acetate-methanol-water at different volume ratios. Fortunately, they had the same mother structure. According to the separation patterns of these compounds and some references about flavonols, four separation rules are prpposed. In our study where the stationary phase was the upper organic phase of n-hexane-ethyl acetate-methanol-water, the elution order of flavonols is hyperoside, quercetin-3-O-β-D-glucoside, quercetin-7-O-β-D-glucoside, astragalin, 4′-methoxyl-quercetin-7-O-β-D-glucoside, quercetin-3′-O-β-D-glucoside, kaempferol-3-O-β-D-(6″-O-p-coumaroyl)-glucoside, 8-methoxyl-kaempferol-7-O-β-D-rhamnoside, quercetin and kaempferol in turn.
Further analysis of the results above indicated that kaempferol is retained in the column longer than quercetin, and astragalin is retained longer than quercetin-3-O-β-D-glucoside. Comparing the structure of kaempferol and quercetin, quercetin has a 3′-hydroxyl, while kaempferol doesn’t. So does the structure of astragalin and quercetin-3-O-β-D-glucoside (see Fig. 3.). Based on the above findings, Rule 1 states “Flavonol with more hydroxyls is eluted out earlier”. Ref. [8] describes the separation and purification of flavonol glycosides from leave extracts of Ampelopsis grossedentata by HSCCC with n-hexane-ethyl acetate-methanol-water, where 5,7-dihydroxy-3′,4′-trihydroxyflavone-3-O-6″-rhamnose was eluted out ahead of 5,7-dihydroxy-3′,4′-dihydroxyflavone-3-O-6″-rhamnose, according with Rule 1. The results also showed that quercetin-3-O-β-D-glucoside, quercetin-7-O-β-D-glucoside, astragalin, 4′-methoxyl-quercetin-7-O-β-D-glucoside, quercetin-3′-O-β-D-glucoside, 8-methoxyl-kaempferol-7-O-β-D-rhamnoside were eluted earlier than kaempferol and quercetin (see Fig. 2. and Fig. 3). Rule 2 is “Flavonol glycoside with more glycosidic link is eluted out earlier”. The result of ref. [25] can prove this rule again. And from the results that quercetin-7-O-β-D-glucoside was eluted earlier than 4′-methoxyl-quercetin-7-O-β-D-glucoside (see Fig. 2B), Rule 3 states “When the hydroxyl of flavonol turns to methoxyl, the retention time is increased”. Quercetin-3-O-β-D-glucoside was eluted before isorhamnetin 3-O-β-D-glucoside in the Ref. [9], which agrees well with Rule 3. Quercetin-3-O-β-D-glucoside, quercetin-7-O-β-D-glucoside and quercetin-3′-O-β-D-glucoside have the same mother structure and the same glycoside substituent, the only difference is the link position. Rule 4 is “Flavonol with 3-glycoside is eluted out earlier than flavonol with 7-glycoside or 3′-glycoside”.
Fig. 3.
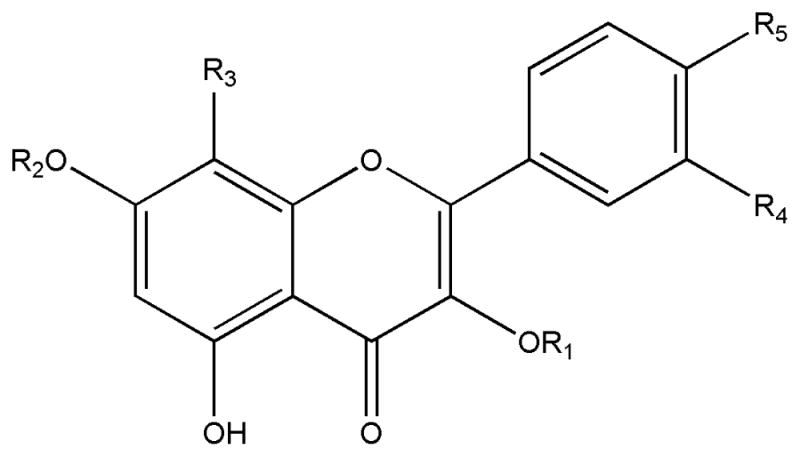
Chemical structures of ten flavonols from Flos Gossypii isolated by HSCCC
| No. | compound | R1 | R2 | R3 | R4 | R5 |
| I | Kaempferol-3-O-β-D-(6″-O-p-coumaroyl)-glucoside | (6″-p-coumaroyl) –Glu | H | H | OH | OH |
| II | 8-methoxyl-kaempferol-7-O-β-D-rhamnoside | H | Rha | OMe | H | H |
| III | Astragalin | Glu | H | H | H | H |
| IV | 4′-methoxyl-quercetin-7-O-β-D-glucoside | H | Glu | H | OH | OMe |
| V | Quercetin-3′-O-β-D-glucoside | H | H | H | OGlu | H |
| VI | -- | -- | -- | -- | -- | -- |
| VII | Hyperoside | Gal | H | H | OH | H |
| VIII | Quercetin-3-O-β-D-glucoside | Glu | H | H | OH | H |
| IX | Quercetin-7-O-β-D-glucoside | H | Glu | H | OH | H |
| X | Quercetin | H | H | H | OH | H |
Acknowledgments
This work was financially supported by the Chinese Academy of Sciences Innovative Research International Partnership Project and a grant from National Key Technology R&D Program (code: 2007BAI30B00) and National Natural Science Foundation of China (code: 30873455).
References
- 1.China Pharmacopoeia Committee. Materia medica criterion, the division of Uigur materia medica. Xinjiang Science and Sanitation Press; Urumqi, China: 1999. p. 98. [Google Scholar]
- 2.Yang Y, Wu T, Yang W, Aisa HA, Zhang T, Ito Y. Preparative Isolation and Purification of Four Flavonoids from Flos Gossypii by High-Speed Counter-current Chromatography. J Liq Chromatogr Related Technol. 2008;31:1523–1531. [Google Scholar]
- 3.Wu T, Lin J, Yang Y, Abdulla R, Chen J, Aisa HA. Preparative isolation of three flavonoids from Flos Gossypii by high-speed counter-current chromatography. Sep Purif Technol. 2009;66:295–298. [Google Scholar]
- 4.Gu D, Yang Y, Zhong J, Aisa HA, Zhang T. High-Speed Counter-Current Chromatography Combined with Column Chromatography for Isolation of Methyllycaconitine from Delphinium pseudocyanthum. Chromatographia. 2007;66:949–951. [Google Scholar]
- 5.Yang Y, Gu D, Wu H, Aisa HA, Zhang T, Ito Y. Application of Preparative High-Speed Countercurrent Chromatography for Separation of Elatine from Delphinium shawurense. J Liq Chromatogr Related Technol. 2008;31:3012–3019. doi: 10.1080/10826070802424956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang Y, Huang Y, Gu D, Yili A, Sabir G, Aisa HA. Separation and Purification of Three Flavonoids from Helichrysum arenarium (L.) Moench by HSCCC. Chromatographia. 2009;69:963–967. [Google Scholar]
- 7.Sannomiya M, Rodrigues CM, Coelho RG, Dos Santos LC, Hiruma-Lima CA, Brito ARMS, Vilegas W. Application of preparative high-speed counter-current chromatography for the separation of flavonoids from the leaves of Byrsonima crassa Niedenzu (IK) J Chromatogr A. 2004;1035:47–51. doi: 10.1016/j.chroma.2004.02.050. [DOI] [PubMed] [Google Scholar]
- 8.Du Q, Chen P, Jerz G, Winterhalter P. Preparative separation of flavonoid glycosides in leaves extract of Ampelopsis grossedentata using high-speed counter-current chromatography. J Chromatogr A. 2004;1040:147–149. doi: 10.1016/j.chroma.2004.03.062. [DOI] [PubMed] [Google Scholar]
- 9.Gutzeit D, Wray V, Winterhalter P, Jerz G. Preparative Isolation and Purification of Flavonoids and Protocatechuic Acid from Sea Buckthorn Juice Concentrate (Hippophaë rhamnoides L. ssp. rhamnoides) by High-Speed Counter-Current Chromatography. Chromatographia. 2007;65:1–7. [Google Scholar]
- 10.Wu T, Abdulla R, Yang Y, Aisa HA. Flavonoids from Gossypium hirsutum flowers. Chem Nat Comp. 2008;44:361–362. [Google Scholar]
- 11.Wu T, Abdulla R, Zhao Y, Yang Y, Chen J, Aisa HA. Simultaneous Quantification of Seven Flavonoids in Flos Gossypii by LC. Chromatographia. 2008;68:467–470. [Google Scholar]
- 12.Oka H, Goto Y, Ito Y, Hashimoto H, Harada K, Suzuki M, Iwaya M, Fuji K, Ito Y. Purification of Food Color Red No. 106 (acid red) using pH-zone-refining counter-current chromatography. J Chromatogr. 2002;A 946:157–162. doi: 10.1016/s0021-9673(01)01548-5. [DOI] [PubMed] [Google Scholar]
- 13.Froesen JB, Pauli GG. G.U.E.S.S.—A generally useful estimate of solvent systems for CCC. J Liq Chromatogr Related Technol. 2005;28:2777–2806. [Google Scholar]
- 14.Oka H, Harada K, Ito Y. Separation of antibiotics by counter-current chromatography. J Chromatogr A. 2008;812:35–52. doi: 10.1016/s0021-9673(97)01277-6. [DOI] [PubMed] [Google Scholar]
- 15.Ito Y. Golden rules and pitfalls in selecting optimum conditions for high-speed counter-current chromatography. J Chromatogr A. 2005;1065:145–168. doi: 10.1016/j.chroma.2004.12.044. [DOI] [PubMed] [Google Scholar]
- 16.Ito Y, Conway WD. High-speed Countercurrent Chromatography. Wiley-Interscience; New York: 1996. pp. 36–39. [Google Scholar]
- 17.Ito Y, Heftmann E. Journal of Chromatography Library. Chapter 2. Elsevier; Amsterdam: 1992. Chromatography V, Part A; pp. A69–A107. [Google Scholar]
- 18.Ternai B, Markham KR. Carbon-13 NMR studies of flavonoids—I: Flavones and flavonols. Tetrahedron. 1976;32:565–569. [Google Scholar]
- 19.Markham KR, Ternai B, Stanley R, Geiger H, Mabry TJ. Carbon-13 NMR studies of flavonoids—III: Naturally occurring flavonoid glycosides and their acylated derivatives. Tetrahedron. 1978;34:1389–1397. [Google Scholar]
- 20.Elliger CA. Sexangularetin 3-glucoside-7-rhamnoside from gossypium hirsutum. phytochemistry. 1984;23:1199–1201. [Google Scholar]
- 21.Xiao Z, Wu H, Wu T, Shi H, Ba H, Aisa HA. Kaempferol and quercetin flavonoids from Rosa rugosa. Chem Nat Comp. 2006;42:736–737. [Google Scholar]
- 22.Tschesche R, Delhvi S, Sepulveda S. Tamarixentin glycosides from the flowers of Verbascum phlomoides. Phytochemistry. 1979;18:1248–1249. [Google Scholar]
- 23.Du Q, Zheng J, Xu Y. Composition of anthocyanins in mulberry and their antioxidant activity. J Food Compos Anal. 2008;21:390–395. [Google Scholar]
- 24.Ma X, Tian W, Wu L, Cao X, Ito Y. Isolation of quercetin-3-O-l-rhamnoside from Acer truncatum Bunge by high-speed counter-current chromatography. J Chromatogr A. 2005;1070:211–214. doi: 10.1016/j.chroma.2005.02.052. [DOI] [PubMed] [Google Scholar]
- 25.Zhou X, Peng J, Fan G, Wu Y. Isolation and purification of flavonoid glycosides from Trollius ledebouri using high-speed counter-current chromatography by stepwise increasing the flow-rate of the mobile phase. J Chromatogr A. 2005;1092:216–221. doi: 10.1016/j.chroma.2005.07.064. [DOI] [PubMed] [Google Scholar]