

Abstract
Synaptic plasticity of different inputs converging onto CA3 pyramidal neurons is central to theories of hippocampal function. The mossy fibre (MF) input to these neurons is thought to exhibit plasticity that is in nearly all aspects fundamentally different from plasticity in other brain regions: in particular, when induced by high frequency presynaptic stimulation, plasticity at these synapses is independent of NMDA receptor (NMDAR) activation and presynaptically expressed. Here, we show that different stimulation protocols that depend on the close timing of MF activity and postsynaptic spikes induce bidirectional plasticity in CA3 neurons in 3-week-old rats. Long-term potentiation (LTP) is observed when an excitatory postsynaptic potential (EPSP), evoked by MF stimulation, precedes a single postsynaptic action potential (AP) or a brief AP burst by 10 ms. Instead, timing-dependent long-term depression (LTD) requires the pairing of a single AP to an EPSP with a delay of 30 ms. The pairing of APs to synaptic activity is required for plasticity induction, since the application of unpaired APs or EPSPs did not alter synaptic strength. Furthermore, our results demonstrate that both timing-dependent LTP and LTD critically depend on the activation of NMDARs. Specifically blocking postsynaptic NMDARs prevents plasticity, demonstrating that NMDARs important to spike-timing-dependent plasticity in CA3 neurons are required at postsynaptic sites. In summary, this study shows that the close timing of APs to MF excitatory synaptic input can alter synaptic efficacy in CA3 neurons in a bidirectional manner.
Introduction
The hippocampal formation is critically involved in memory storage and retrieval (Amaral et al. 1990; Squire, 2004). Key components of this hippocampal network are the pyramidal neurons of the CA3 region, which are ideally placed to integrate and complete information patterns that are created in neocortex and relayed in the hippocampal input structures (Lorincz & Buzsaki, 2000; Leutgeb et al. 2007). In agreement with their role in pattern completion, the CA3 neurons represent a highly associative (recurrent) network on which a plethora of pathways converge, for example temporoammonic (TA) projections, mossy fibre (MF) projections, and associational-commissural (A/C) fibres. All these pathways display plasticity upon high frequency stimulation (HFS) (Harris & Cotman, 1986; Williams & Johnston, 1988; Zalutsky & Nicoll, 1990; Tsukamoto et al. 2003). In contrast, the predominant spiking activity observed in vivo in CA3 pyramidal neurons and in their afferent neurons is rather sparse with single spikes and brief spike bursts at low rates (McNaughton et al. 1983; Jung & McNaughton, 1993; Hahn et al. 2007). Hence, in vivo, synaptic input from any of these pathways can be expected to coincide at times with CA3 neuron firing.
Timing of spikes to synaptic input has a powerful influence on synaptic strength (Magee & Johnston, 1997; Markram et al. 1997; Dan & Poo, 2004), and there is increasing evidence that such spike-timing-dependent plasticity (STDP) may also exist in the hippocampal CA3 region. Specifically, for recurrent excitatory connections between CA3 neurons and for GABAergic transmission at immature MF–CA3 synapses, STDP was recently shown (Debanne et al. 1998; Sivakumaran et al. 2009). Surprisingly, it is not known, if the excitatory MF–CA3 projection as one of the key entry sites of cortical information to the hippocampus displays such STDP. Evidence actually exists that this MF–CA3 projection holds vital requirements for STDP. First, NMDA receptor channels, which act as crucial coincidence detectors for STDP, mediate substantial currents at the MF–CA3 synapse (Jonas et al. 1993; Weisskopf & Nicoll, 1995). These NMDARs have long been thought to be unimportant to MF plasticity (Harris & Cotman, 1986; Williams & Johnston, 1988; Zalutsky & Nicoll, 1990), but recently a new involvement of NMDARs in the induction of plasticity of MF-NMDA currents by HFS was demonstrated (Kwon & Castillo, 2008; Rebola et al. 2008). Second, NMDARs are likely to mediate a substantial Ca2+ influx into postsynaptic spines of CA3 neurons during the coincidence of synaptic activity and backpropagation of action potentials (Reid et al. 2001), which could represent an important triggering signal for STDP induction (Magee & Johnston, 1997; Nevian & Sakmann, 2006; Wittenberg & Wang, 2006). MF–CA3 synapses are thought to be unique amongst excitatory synapses due to several features: giant presynaptic terminals, multiple transmitter release sites, prominent paired-pulse facilitation (Henze et al. 2000; Bischofberger et al. 2006) and presynaptic expression mechanisms of plasticity (Staubli et al. 1990; Zalutsky & Nicoll, 1990; Kobayashi et al. 1996; Tzounopoulos et al. 1998). Also, the induction site of HFS-induced plasticity was suggested to be presynaptic (Nicoll & Schmitz, 2005), although some studies indicated that postsynaptic depolarization and Ca2+ rises contribute to MF LTP (Jaffe & Johnston, 1990; Urban & Barrionuevo, 1996; Yeckel et al. 1999 and see Sokolov et al. (2003) for postsynaptic hyperpolarization) and MF LTD (Domenici et al. 1998; Lei et al. 2003).
The aim of the present study was to investigate whether precisely timed postsynaptic action potentials in CA3 neurons and presynaptic activation of the MF pathway are capable of altering excitatory transmission at these unique synapses. Furthermore, we also asked whether NMDAR channels might play a role in MF–CA3 STDP. We present the first evidence that depending on the precise spike-timing to MF input, either LTP or LTD is induced in CA3 neurons, and that postsynaptic NMDARs are critically involved in the induction of this bidirectional synaptic plasticity.
Methods
Hippocampal slice preparation and electrophysiology
All experimental procedures were performed according to the animal welfare guidelines of the Max-Planck-Society. Transverse or sagittal hippocampal slices (300 μm) were obtained from P19–23 Wistar rats. Animals were deeply anaesthetized with isoflurane, the brain was dissected out, and acute slices were prepared either in a sucrose based ice-cold solution or in a potassium gluconate based ice-cold solution (pH 7.2). Here, a custom-built slicer was used, whose design minimized vertical vibrations of the cutting blade to less than 3 μm (peak-to-peak; according to Geiger et al. 2002). The sucrose based solution contained (in mm): 120 sucrose, 64 NaCl, 25 NaHCO3, 10 glucose, 7 MgCl2, 2.5 KCl, 1.25 NaH2PO4, 0.5 CaCl2. The potassium gluconate based solution contained (in mm): 140 potassium gluconate, 10 Hepes, 15 sodium gluconate, 0.2 EGTA, 4 NaCl. Slices were incubated for recovery at 35°C for at least 30 min in artificial cerebrospinal fluid (ACSF) containing (in mm): 125 NaCl, 25 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 1 MgCl2, 2 CaCl2, 25 glucose, saturated with 95% O2–5% CO2, and were then kept at room temperature until use.
Slices were bathed at near physiological temperature (30–32°C) in ACSF added with (in mm): 0.01 picrotoxin (or 0.01 bicuculline methiodide) as GABAA receptor (GABAAR) antagonist and 0.01 glycine as NMDAR coagonist. For whole-cell current-clamp recordings in CA3 pyramidal neurons, patch pipettes were pulled from borosilicate glass capillaries and had resistances of 5–8 MΩ when filled with (in mm): 130 potassium gluconate, 10 Hepes, 10 phosphocreatine, 10 sodium gluconate, 4 MgATP, 0.3 GTP, 4 NaCl (pH 7.2). Liquid junction potential (LJP) was −15.2 mV and not corrected for. Cells were held at resting membrane potential and were excluded from the analysis if the input resistance (monitored by −25 pA current steps for 200 ms) changed >30% over the entire experiment. Postsynaptic APs were elicited with somatic current injection through the recording electrode (1–1.5 nA, typically 5 ms, rarely up to 10 ms). For STDP, either single APs or AP bursts with a frequency of 50 Hz were paired with EPSPs (60 repetitions; 0.1 Hz).
Electrical stimulation
EPSPs were evoked at 0.1 Hz by extracellular stimulation with a bipolar tungsten (WPI, Saratosa, FL, USA) or platinum/iridium electrode (Science Products, Hofheim, Germany). To estimate how specific MF inputs to CA3 cells were activated under our experimental conditions, EPSPs were evoked in CA3 pyramidal cells by placing the stimulation electrode either in the stratum (s.) radiatum (>100 μm from the edge of the pyramidal cell layer in transverse slices), in the s. lucidum (30–40 μm from the edge of the pyramidal cell layer in transverse slices) or in the hilus (close to granule cell layer in sagittal slices). EPSPs were analysed in terms of paired-pulse ratio (PPR, 50 ms inter-stimulus interval; the mean amplitude of the second EPSP was measured relative to the voltage value preceding the second stimulus and was divided by the mean amplitude of the first EPSP) (Kim & Alger, 2001), rise time (20–80%) and sensitivity to activation of group II metabotropic glutamate receptors (mGluRs) with either (2S,2′R,3′R)-2-(2′,3′-dicarboxycyclopropyl)glycine (DCG- IV; 4 μm) or (2S,1′S,2′S)-2-(carboxycyclopropyl) glycine (l-CCG-I; 20 μm) (Kamiya et al. 1996; Castillo et al. 1997).
For STDP experiments with s. lucidum stimulation, stimulation strength was adapted to evoke single-peak EPSPs and recordings were only accepted when the EPSPs displayed a single rising phase with a non-variable latency (<3 ms) indicating their monosynaptic origin (for review, see Henze et al. 2000; Nicoll & Schmitz, 2005). Recordings were accepted for the MF experimental series (n = 69) only if the evoked EPSP displayed a pronounced PPR (>1.4, 1.9 ± 0.1) and a brief rise time (<4 ms, 2.9 ± 0.1 ms) at the beginning of each experiment as well as a substantial inhibition by group II mGluR agonists (2–4 μm DCG-IV or 20 μm l-CCG-I) at the end of the experiment (>70%, peak analysis, 80 ± 2%; slope analysis, 83.4 ± 1.4%) (Kamiya et al. 1996; Lawrence et al. 2004). Recordings were classified as s. lucidum-stimulated non-MF inputs when the evoked EPSP showed no (less than 12%) reduction upon group II mGluR agonist application. Whole-cell current clamp recordings in the presence of group II mGluR agonists were often unstable under our ionic conditions, which is potentially due to the agonist's activating effect on NMDARs and the resulting depolarization (Ishida et al. 1993). Therefore, Ca2+ and Mg2+ concentrations were raised to 4 and 2 mm, respectively, before washing in the group II mGluR agonist, which improved the recording stability. In a few experiments (n = 4) in the presence of the NMDA antagonist d-AP5, mossy fibre origin of responses was demonstrated by induction of NMDAR-independent LTP by applying tetanic stimulation at the end of the STDP experiment (3 times 100 Hz, 1 s).
Data acquisition and analysis
Data were filtered at 2 kHz and digitized at 10 kHz with either an EPC-9 controlled by Pulse or an EPC-10 controlled by Patchmaster (Heka Elektronik, Lambrecht, Germany). Deactivation kinetics of postsynaptic potentials were fitted monoexponentially. Spike timing was defined as the difference between the onset of the EPSP and the AP peak. In case of the pairing of an EPSP with a burst of three APs, spike timing was defined as the time between EPSP onset and peak of AP closest in time to the EPSP. Change in synaptic efficacy was calculated as the ratio of the average EPSP peak amplitude or EPSP slope at 20–30 min after induction protocol over the average EPSP peak amplitude or EPSP slope during 10 min baseline. Data are presented as means ± s.e.m. Student's t test and ANOVA with Fisher's post hoc analysis were used for statistical analysis as appropriate (*P < 0.05; **P < 0.01).
Chemicals
DCG-IV, l-CCG-I, d-APV and (5R,10S)-(+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cylcohepten- 5,10-imine maleate (MK-801) were products of Tocris (Ellisville, MO, USA). All other compounds were purchased from Sigma-Aldrich (St Louis, MO, USA). l-CCG-I was dissolved in 0.1 m NaOH at a stock concentration of 100 mm. MK-801 was dissolved in the intracellular solution.
Results
Isolation of mossy fibre inputs to CA3 pyramidal neurons
CA3 pyramidal neurons were investigated in acute hippocampal slices of 3-week-old rats. Routinely, recordings were performed in the presence of a GABAAR antagonist in order to isolate the excitatory projection from the MF pathway. To ensure that responses in CA3 neurons are of MF origin, many previous plasticity studies were conducted in ACSF with high concentrations of Ca2+ and Mg2+ (for reviews, see Henze et al. 2000; Nicoll & Schmitz, 2005) to largely prevent polysynaptic inputs (Del Castillo & Katz, 1954). Furthermore, studies using HFS to induce plasticity were often conducted in the presence of the NMDAR antagonist d-APV. Since NMDAR activation is reduced by high Mg2+ concentrations, but at the same time thought to be essential for STDP across many brain regions (Magee & Johnston, 1997; Bi & Poo, 1998; Debanne et al. 1998; Pawlak & Kerr, 2008), plasticity experiments in the present study were done in ACSF with a more physiological Ca2+/Mg2+ concentration. This required a first series of experiments to quantify how specific MF inputs to CA3 cells are activated under our experimental conditions (Fig. 1). EPSPs were evoked by extracellular stimulation in s. lucidum or in the hilus, which should activate the soma-close MF synapses, or in s. radiatum, which should activate non-MF synapses via associational-commissural (Claiborne et al. 1986) and/or entorhinal inputs (Berzhanskaya et al. 1998; Witter & Amaral, 2004). Stimulation in s. lucidum or hilus when compared to stimulation in s. radiatum evoked EPSPs with higher PPRs (2.2 ± 0.1, n = 15 or 2.5 ± 0.4, n = 7 versus 1.2 ± 0.1, n = 13; P < 0.05) and faster rise times (2.8 ± 0.2 ms, n = 15 or 2.3 ± 0.2 ms, n = 7 versus 4.6 ± 0.5 ms, n = 13; P < 0.01). EPSP amplitude was for s. lucidum 3.92 ± 0.36 mV (n = 15), for hilus 2.21 ± 0.37 mV (n = 7) and for s. radiatum 7.00 ± 1.31 mV (n = 13). In addition, the peak amplitudes of EPSPs evoked by s. lucidum or hilus stimulation showed a high sensitivity to application of group II mGluR agonists, which is consistent with the specific expression of mGluRII receptors at MF terminals (DCG-IV, 70.1 ± 4.1%, n = 8 or 69.2 ± 6.5%, n = 7 versus 10.0 ± 18.0%, n = 7 for s. radiatum; P < 0.05 and l-CCG-I, 61.8 ± 8.4%, n = 7 for s. lucidum versus−13.6 ± 9.2%, n = 6 for s. radiatum; P < 0.01). These differences are in agreement with previous studies (Jonas et al. 1993; Kapur et al. 1998; Yeckel et al. 1999) and allowed us to distinguish EPSPs that were mainly of MF origin from EPSPs that were mainly of non-MF origin.
Figure 1. Isolation of mossy fibre inputs to CA3 pyramidal neurons.
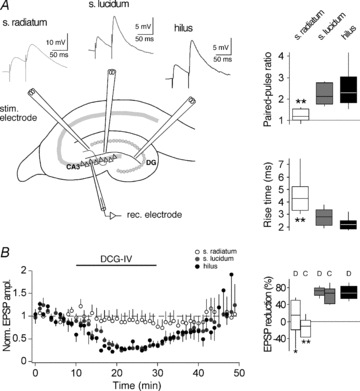
A, left, schematic drawing of a hippocampal section where electrodes were placed to stimulate distinct afferents of a CA3 pyramidal cell. Top, examples of EPSPs evoked in a paired-pulse protocol (50 ms interstimulus interval) by stimulation in the s. radiatum (activating presumably non-MFs, light grey trace), in the s. lucidum (activating presumably MFs, dark grey trace) and in the hilus (activating presumably MFs, black trace). Notably, rise times of s. radiatum EPSPs were slower. Right, box-and-whisker plots of EPSP parameters measured with s. radiatum, s. lucidum and hilus stimulation. In each box, the mid-line shows the median, the top and the bottom show the upper and lower percentiles (75th and 25th) and the whiskers show 90th and 10th percentiles. B, left, plot showing the effect of 4 μm DCG-IV on EPSPs evoked by stimulation in s. radiatum (white circles), s. lucidum (grey circles) and hilus (black circles). Each data point represents the average of 6 EPSPs during 1 min. Right, box-and-whisker plot showing EPSP reduction upon application of either 4 μm DCG-IV (D) or 20 μm l-CCG-I (C). Asterisks indicate that parameters recorded with radiatum stimulation were significantly different from those recorded with either lucidum stimulation or hilus stimulation (unpaired t test; *P < 0.05; **P < 0.01). The lack of significant EPSP amplitude changes when evoked by s. radiatum stimulation indicates that both group II mGluR agonists at the concentrations used were unlikely to have had unspecific effects.
For the following STDP experiments, MF origin of responses evoked by s. lucidum stimulation was verified for every single recording (n = 69) by determining the PPR (1.9 ± 0.1) and the rise time (2.9 ± 0.1 ms) at the beginning and group II mGluR agonist sensitivity (peak analysis, 80 ± 2%; slope analysis, 83.4 ± 1.4%) of EPSPs at the end of each experiment.
Then, we investigated the composition of responses evoked by MF stimulation by recording postsynaptic potentials (PSPs) with and without the GABAAR inhibitor bicuculline methiodide (10 μm) in the ACSF. Blocking GABAA receptors increased the peak amplitude of MF–CA3 PSPs by 40% (3.32 ± 1.38 mV vs. 4.65 ± 1.55 mV, n = 6; P < 0.05) without affecting the decay time (15.85 ± 2.22 ms vs. 15.70 ± 1.78 ms, n = 6; P = 0.93). To determine the contribution of NMDARs to EPSPs recorded under our conditions, we additionally perfused the NMDAR blocker d-AP5 (50 μM). d-AP5 did not affect the peak amplitude (4.65 ± 1.55 mV vs. 4.69 ± 1.57 mV, n = 6; P > 0.86) but decreased the decay time (15.70 ± 1.78 ms vs. 11.44 ± 1.22 ms, n = 6; P < 0.0005). Hence, plasticity of NMDAR-mediated currents should primarily affect the decay of MF EPSPs, whereas plasticity of AMPAR-mediated currents should rather affect the peak amplitude of MF EPSPs.
Close timing of AP bursts to synaptic activation induces timing-dependent LTP but not timing-dependent LTD in CA3 neurons
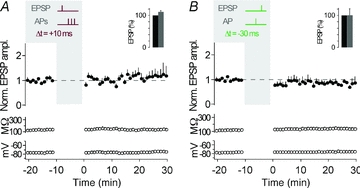
Because CA3 neurons fire single APs and AP bursts in vivo (McNaughton et al. 1983; Hahn et al. 2007), we first asked how the timing of an AP burst, as opposed to the timing of a single AP, would affect synaptic strength in these neurons. Timing-dependent LTP (t-LTP) was induced when presynaptic stimulation in s. lucidum was followed by a burst of three postsynaptic action potentials (APs at 50 Hz) with short positive time delays (Δt = +10 to +40 ms). Such pairing was repeated 60 times at 0.1 Hz, which is close to the firing rate of granule cells in freely moving rats (Jung & McNaughton, 1993; Henze et al. 2002). After induction of t-LTP, the amplitude ratio of EPSPs was 1.65 ± 0.09 (determined 20–30 min after the STDP protocol; measured as the ratio of mean EPSP amplitude after/before; n = 10; P < 0.01). Also, the slope of the EPSPs was increased after the STDP protocol (slope ratio, 1.69 ± 0.16, n = 10; P < 0.01). Changes in EPSPs were not due to changes in membrane properties such as input resistance and resting membrane potential (Fig. 2A and B, see also Methods for criteria).
Figure 2. Pairing of synaptic activation with three action potentials.
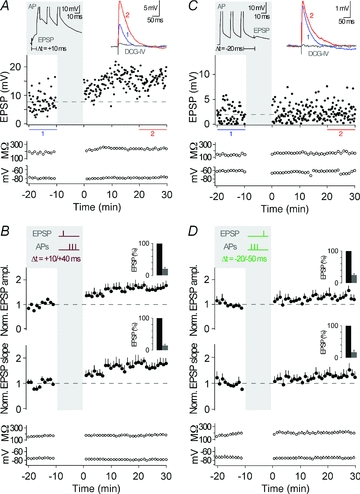
A, representative recording from a CA3 neuron showing t-LTP. The plasticity induction protocol consisted of an EPSP followed by an AP burst with a time delay of 10 ms, repeated 60 times at 0.1 Hz (see grey inset, Δt = +10 ms; APs are clipped). EPSP amplitude, input resistance and membrane potential are plotted against time. The coloured EPSP traces (top right inset) are averages of all EPSPs recorded during the time periods indicated by the correspondingly coloured bars or recorded in the presence of group II mGluR agonists (in grey). B, average of all recordings, where a STDP protocol consisting of an EPSP followed by three APs was applied (as shown by the schematic drawing in the grey inset; Δt = +10/+40 ms). t-LTP was induced (n = 10, P < 0.01). The bar insets illustrate the reduction of EPSP amplitudes or EPSP slopes by the group II mGluR agonist; EPSPs were set to 100% before washing in the group II mGluR agonist. C, representative recording from a CA3 neuron showing no change in synaptic strength when an AP burst was followed by an EPSP with a time delay of 20 ms (Δt = −20 ms). D, average of all recordings, where a STDP protocol consisting of an AP burst before an EPSP (Δt = −20/−50 ms) was applied. No changes in synaptic efficacy (n = 5, P > 0.05) were observed. Dashed lines (here, and in the following figures) indicate average of EPSP amplitudes or EPSP slope during baseline.
Activation of EPSPs alone did not change the synaptic efficacy (peak, 1.05 ± 0.10, n = 6; P > 0.05). To address whether AP bursts alone represent a sufficiently strong stimulus to induce plasticity, we performed another control experiment in which the AP burst alone was applied (60 times at 0.1 Hz). This led to no changes in the average EPSP ratio (peak, 1.08 ± 0.14, slope, 1.05 ± 0.11, n = 6; P > 0.05), and thus AP bursts need to be timed to presynaptic activation to induce t-LTP. The occurrence of t-LTP raises the question of whether the reversal of the two events, APs and evoked EPSPs, reverses the direction of plasticity (Dan & Poo, 2004). Such reversal, specifically when the AP burst was followed by an EPSP with a time delay of 20–50 ms (Δt = −20 to −50 ms), induced no significant plasticity (peak, 1.30 ± 0.15, slope, 1.32 ± 0.17, n = 5; P > 0.05) (Fig. 2C and D). Next, we tested if t-LTD could be induced with a longer time delay between AP bursts and the EPSP (Debanne et al. 1994; Sourdet & Debanne, 1999). However, t-LTD was not induced with a time delay of Δt = −100 ms, nor was it induced when the time delay was extended to Δt = −200 ms (Δt = −100 ms, peak, 1.09 ± 0.19, slope, 1.03 ± 0.21, n = 4; P > 0.05; Δt = −200 ms, peak, 1.04 ± 0.21, slope, 1.02 ± 0.15, n = 5; P > 0.05; Supplemental Fig. 1). Thus, burst pairing protocols with time delays between −20 ms and −200 ms did not result in t-LTD.
Close timing of single APs to synaptic activation induces both t-LTP and t-LTD in CA3 neurons
At many synapses, the timing of AP bursts relative to synaptic activation, as opposed to timing of single APs, is essential for t-LTP (Magee & Johnston, 1997; Thomas et al. 1998; Pike et al. 1999; but see Feldman, 2000; Pawlak & Kerr, 2008). In CA3 neurons, pairing of presynaptic MF stimulation and a single postsynaptic AP with a time delay of 10 ms (Δt = +10 ms) was sufficient to induce t-LTP (peak, 1.55 ± 0.22, slope, 1.43 ± 0.16, n = 7; P < 0.05) (Fig. 3A and B). The amount of LTP induced was not different between single AP t-LTP and AP burst t-LTP (P > 0.05). And, although burst pairing protocols did not result in consistent t-LTD (see above), timing of single APs 30 ms before the evoked EPSPs (Δt = −30 ms) depressed synaptic strength (peak, 0.70 ± 0.09, slope, 0.71 ± 0.11, n = 7; P < 0.05) (Fig. 3C and D). Also, close timing relative to presynaptic activity was required for single APs to induce synaptic plasticity, as 60 single postsynaptic APs at 0.1 Hz without presynaptic stimulation caused no plasticity (peak, 1.17 ± 0.11, slope, 1.05 ± 0.16, n = 7; P > 0.05).
Figure 3. Pairing of synaptic activation with a single action potential.
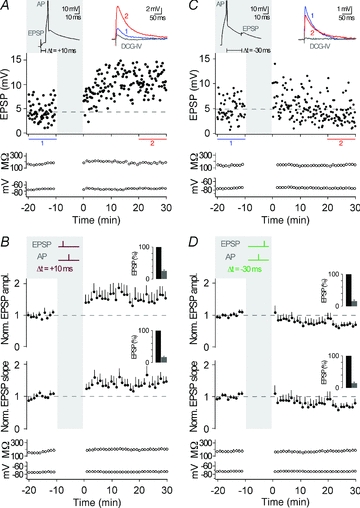
A, representative recording from a CA3 neuron showing t-LTP when single APs were evoked 10 ms after an EPSP, repeated 60 times at 0.1 Hz (see grey inset, Δt = +10 ms; AP is clipped). EPSP amplitude, input resistance and membrane potential are plotted against time. The coloured EPSP traces (top right inset) are averages of all EPSPs recorded during the time periods indicated by the correspondingly coloured bars or recorded in the presence of group II mGluR agonists (in grey). B, average of all recordings, where a STDP protocol consisting of an EPSP followed by a single AP was applied (as shown by the schematic drawing in the grey inset; Δt = +10 ms). t-LTP was induced (n = 7, P < 0.05). The bar insets illustrate the reduction of EPSP amplitudes or EPSP slopes by the group II mGluR agonist; EPSPs were set to 100% before washing in the group II mGluR agonist. C, representative recording from a CA3 neuron showing t-LTD when a single AP was followed by an EPSP with a time delay of 30 ms (Δt = −30 ms). D, average of all recordings, where a STDP protocol consisting of a single AP before an EPSP (Δt = −30 ms) was applied. t-LTD was induced (n = 7, P < 0.05).
In all cases in which we observed t-LTP or t-LTD, the amount of plasticity did not correlate with the initial size of the EPSPs (Pearson's correlation coefficient for t-LTP: r = −0.23, P > 0.05, n = 17; for t-LTD: r = −0.29, P > 0.05, n = 7). This indicates that the initial number of activated MF inputs to CA3 neurons is unlikely to affect their capability to undergo STDP (for comparison, see Sjostrom et al. 2001). Furthermore for t-LTP (n = 17), peak amplitude as well as slope of EPSPs increased by 62 ± 11% (P < 0.01) and 58 ± 12% (P < 0.05), respectively, and EPSP decay became slower by 24 ± 7% (P < 0.05). For t-LTD (n = 7), peak amplitude as well as slope of the EPSPs decreased by 30 ± 9% (P < 0.05) and 29 ± 11% (P < 0.05), respectively, whereas the decay kinetics of the EPSPs did not significantly change (13 ± 11% increase, P = 0.56). Overall, changes in EPSP slope and peak amplitude were correlated (Pearson's correlation coefficient: t-LTP, r = 0.89, P < 0.01 and t-LTD, r = 0.95, P < 0.01). Collectively, our data demonstrate the occurrence of t-LTP and t-LTD of excitatory transmission in CA3 neurons when MF stimulation is appropriately timed to APs. The changes of EPSP amplitude and slope indicate an increase in AMPAR-mediated transmission after pre-post pairings (Δt = +10 ms) and a decrease in AMPAR-mediated transmission after post–pre pairings (Δt = −30 ms, single APs). The slower decay kinetics after pre–post pairings (Δt = +10 ms, see above) might suggest a concurrent potentiation of NMDAR-mediated transmission, which would be in agreement with previous findings (Kwon & Castillo, 2008; Rebola et al. 2008).
NMDARs act as coincidence detectors in STDP in CA3 neurons
Depending on the pathway stimulated and the applied stimulation protocol, the locus of plasticity expression in CA3 neurons may be pre- or postsynaptic (Nicoll & Schmitz, 2005). To investigate the locus of expression of the STDP described here, we examined the PPR of EPSPs, which often changes when plasticity is expressed presynaptically (Zalutsky & Nicoll, 1990). PPR was determined before and after (30 min) the STDP induction protocol. The PPR, which is inversely correlated with release probability (Zucker & Regehr, 2002), was calculated for experiments showing either t-LTP with one or three APs during pairing (Fig. 4A) or t-LTD with one AP during pairing (Fig. 4B). For both, t-LTP and t-LTD, the PPR did not undergo significant changes after STDP (t-LTP, 3 APs: PPR before, 2.27 ± 0.35; after, 2.16 ± 0.15; n = 8, P > 0.05; t-LTP, 1 AP: PPR before, 1.93 ± 0.27; after, 1.92 ± 0.19; n = 6, P > 0.05 and t-LTD: PPR before, 1.64 ± 0.06; after, 1.53 ± 0.14; n = 7, > 0.05). Then, we compared the coefficient of variation (c.v.) of EPSPs recorded before and after STDP induction. After burst-induced t-LTP, c.v. was reduced (before, 0.52 ± 0.06; after 0.34 ± 0.03; n = 10, P < 0.01), whereas after single AP-induced t-LTP, c.v. was not significantly reduced (before, 0.44 ± 0.06; after, 0.38 ± 0.07; n = 7, P = 0.35). However, the c.v. changes were not correlated to the amount of burst-induced t-LTP (3 APs: r = −0.47, P = 0.17; for comparison 1 AP: r = −0.52, P = 0.22). For t-LTD, c.v. remained unchanged (before, 0.32 ± 0.03; after, 0.32 ± 0.06; n = 7, P > 0.05). In summary, the combined analysis of PPR and c.v. suggests that a purely presynaptic locus of plasticity expression, as is often observed after HFS-induced MF plasticity, is unlikely for t-LTD and t-LTP.
Figure 4. Unchanged PPR after STDP and involvement of postsynaptic NMDARs.
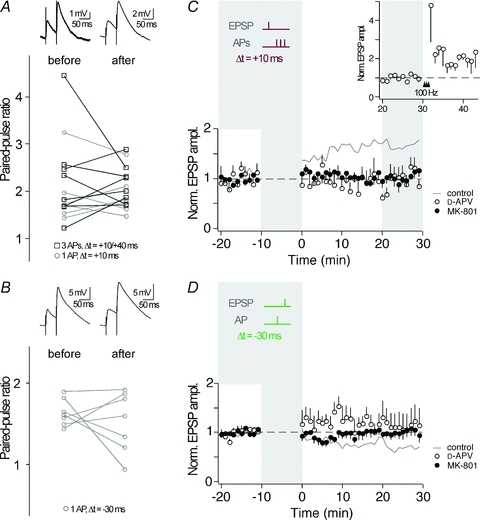
A, the paired-pulse ratio (50 ms interstimulus interval) as determined before and 30 min after induction of t-LTP (open squares, 3 AP pairing; open circles, single AP pairing), was not homogeneously changed. Top panel shows example traces of EPSPs evoked in a paired-pulse protocol (average of >5 traces). B, after induction of t-LTD with single AP pairing, the paired-pulse ratio was not homogeneously changed. Top panel shows example traces of EPSPs evoked in a paired-pulse protocol (average of >5 traces). C, a STDP protocol inducing t-LTP in control conditions (grey line, n = 10; same data as in Fig. 2B) failed in inducing synaptic plasticity when NMDARs were blocked by d-APV (50 μm, open circles, n = 5) or by intracellular MK-801 (1 mm, full circles, n = 5). The inset shows 4 out of the 5 experiments in d-APV, in which tetanic stimulations (100 Hz) 30 min following the STDP protocol increased synaptic efficacy. D, a STDP protocol inducing t-LTD in control conditions (grey line, n = 7; same data as in Fig. 3D) failed in inducing synaptic plasticity when NMDARs were blocked by d-APV (50 μm, open circles, n = 4) or by intracellular MK-801 (1 mm, full circles, n = 6).
Due to their dual dependence on ligand-binding and depolarization for activation, NMDARs are ideal sensors for correlated pre- and postsynaptic activity during AP–EPSP pairing. To examine whether NMDAR activity plays a role in STDP at MF synapses, we applied the EPSP–AP pairing protocol that had induced t-LTP before (Δt = +10 ms, AP bursts), in the presence of the NMDAR blocker d-APV (50 μm). Under these conditions, no t-LTP was observed (peak, 0.97 ± 0.11, slope, 1.05 ± 0.23, n = 5; P > 0.05) (Fig. 4C). To investigate if ‘conventional’ NMDAR-independent MF-LTP (Nicoll & Schmitz, 2005) is observed under our conditions, we subsequently applied high frequency stimulation to the MF input in a subset of experiments (Fig. 4C, inset). This high frequency stimulation in the presence of d-APV potentiated EPSPs (peak, 1.98 ± 0.31, n = 4; P = 0.05), indicating that under our conditions ‘conventional’ NMDAR-independent MF-LTP is expressed. A STDP protocol inducing t-LTD in control conditions (Δt = −30 ms, single APs) failed to induce t-LTD in the presence of d-APV (peak, 1.12 ± 0.26, slope 1.04 ± 0.19, n = 4; P > 0.05) (Fig. 4D). This suggests that NMDARs are likely to act as coincidence detectors for the induction of STDP of excitatory transmission during correlated activity of MFs and CA3 neurons.
Postsynaptic NMDARs are well-known to induce synaptic plasticity throughout the CNS (Bliss & Collingridge, 1993), whereas presynaptic NMDARs only recently were found to be involved in synaptic plasticity, particularly in t-LTD (Sjostrom et al. 2003; Bender et al. 2006; Nevian & Sakmann, 2006; Corlew et al. 2007; Rodriguez-Moreno & Paulsen, 2008). Application of d-APV blocks both pre- and postsynaptic NMDARs and therefore could not identify the location of the NMDARs involved in the STDP described here. To investigate whether the NMDARs necessary for timing-dependent plasticity in CA3 neurons are located pre- or postsynaptically, we blocked the postsynaptic NMDAR channels by applying the irreversible channel blocker MK-801 (1 mm) via the patch pipette (Berretta & Jones, 1996). In the presence of MK-801, t-LTP as well as t-LTD was prevented (Δt = +10 ms, 3-AP burst: peak, 1.05 ± 0.10, slope, 1.02 ± 0.11, n = 5; P > 0.05; Δt = −30 ms, single APs: peak, 1.00 ± 0.13, slope, 0.98 ± 0.16, n = 6; P > 0.05). Together, these experiments show the involvement of postsynaptic NMDARs in STDP in CA3 neurons. For comparison, Fig. 5 contains every single recording under every experimental condition mentioned heretofore.
Figure 5. Summary of STDP experiments in CA3 neurons upon MF stimulation.
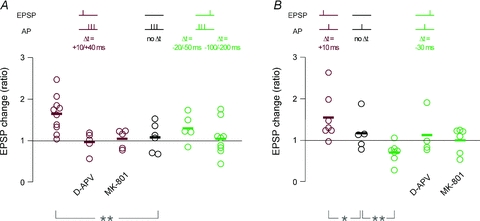
A, all single recordings with AP burst pairing; the change in synaptic efficacy was calculated as ratio of the average EPSP amplitude 20–30 min after induction over the average EPSP amplitude during the 10 min baseline. Colour code indicates pairing with positive time delays (dark red), negative time delays (green) and no pairing (black), as depicted in the schemes above the plots. t-LTP was induced by pairing an EPSP with AP bursts at positive time delays (Δt = +10/+40 ms). This form of t-LTP was not induced when NMDARs were blocked with either extracellular d-APV (50 μm) or intracellular MK-801 (1 mm). Burst pairing at negative time delays (Δt = −20/−50 ms and −100/−200 ms) did not produce significant changes in synaptic efficacy. B, same as in A, for STDP protocols with single AP pairing. Pairing an EPSP with a single AP at positive time delays (Δt = +10 ms) induced t-LTP, whereas pairing at negative time delays (Δt = −30 ms) induced t-LTD. This form of t-LTD was not induced when NMDARs were blocked with either extracellular d-APV (50 μm) or intracellular MK-801 (1 mm). Asterisks indicate statistical significance with ANOVA compared to the experimental series with no pairing (*P < 0.05, **P < 0.01).
Spike-timing protocols that induce t-LTP and t-LTD at MF inputs fail at non-MF inputs
A continuous problem when stimulating MF inputs to CA3 neurons is the exclusion of ‘contamination’ by non-MF inputs, like A/C and/or entorhinal inputs (for review, see Henze et al. 2000). A study in slice cultures by Debanne et al. (1998) has shown that A/C inputs to CA3 neurons display timing-dependent bidirectional plasticity. Therefore, we asked, whether the STDP protocols applied in this study can potentially induce substantial plasticity of ‘contaminating’ non-MF inputs to CA3 neurons. However, if the timing-dependent plasticity described here, was due to the selective potentiation or depression of an EPSP component insensitive to group II mGluR agonists (like for example A/C inputs), one would expect a correlation between mGluRII agonist sensitivity and the amount of plasticity. Such a correlation did not exist (t-LPT, r = −0.37, P = 0.15, n = 16; t-LTD, r = −0.09, P = 0.83, n = 7).
Furthermore, we directly tested whether STDP is expressed at non-MF inputs under our experimental conditions. For this, we stimulated in s. radiatum, which should activate non-MF inputs like A/C and/or entorhinal inputs (Berzhanskaya et al. 1998; Witter & Amaral, 2004), and applied the AP–EPSP pairing protocols that had induced plasticity with s. lucidum stimulation (see above). With s. radiatum stimulation, no plasticity was observed with the pairing protocol that had induced t-LTP of MF inputs (Δt = +10 ms, AP bursts, 1.14 ± 0.18, n = 6; P > 0.05) (Fig. 6A). Likewise, no plasticity was observed upon s. radiatum stimulation with the protocol that had induced t-LTD of MF inputs (Δt = −30 ms, single APs, 0.88 ± 0.09, n = 5; P > 0.05) (Fig. 6B). However, data obtained with s. radiatum stimulation might not allow an optimal estimate of potential non-MF plasticity with s. lucidum stimulation, since the investigated synapses might be of different identity and located at different distances from the soma. Therefore, we investigated a number of recordings obtained with s. lucidum stimulation, which were excluded from Figs 2 and 3, because the EPSPs displayed no reduction when group II mGluR agonists were applied at the end of the experiment (see Methods). Non-MF inputs isolated in this way showed no t-LTP upon the burst pairing protocol with Δt = +10 ms (1.06 ± 0.27, n = 3; P > 0.05), nor did they display t-LTD upon the single-AP pairing protocol with Δt = −30 ms (1.32 ± 0.04, n = 3; P < 0.05). Instead, this protocol (Δt = −30 ms) induced a small potentiation, which might suggest non-standard STDP rules for this special group of non-MF inputs. However, the conclusions that can be drawn from this group are limited due to the small group size. And since experiments with s. lucidum stimulation and no subsequent EPSP reduction upon mGluRII agonist application were rare chance encounters, it was not feasible to further increase the group size.
Figure 6. STDP protocols at non-MF inputs.
A and B, STDP protocols that induced plasticity at MF inputs to CA3 neurons failed in inducing plasticity at s. radiatum stimulated non-MF inputs. Averaged values for normalized EPSP amplitude, input resistance and membrane potential are plotted against time for recordings with three APs paired with an EPSP at positive timing (A, Δt = +10 ms, n = 6) or with single AP pairing at negative timing (B, Δt = −30 ms, n = 5), as shown by the respective schematic drawing in the grey insets. The bar insets illustrate changes of EPSP amplitudes by the group II mGluR agonist; EPSPs were set to 100% before washing in the group II mGluR agonist.
In summary, these experiments demonstrate that indeed primarily the MF component of EPSPs underwent t-LTP or t-LTD when the respective spike-timing protocols were applied to CA3 neurons with s. lucidum stimulation (Figs 2, 3), although our results and analysis cannot fully exclude a concurrent albeit small potentiation/depression of non-MF inputs during s. lucidum stimulation (see Discussion).
Discussion
Requirements for timing-dependent plasticity in CA3 neurons
The present study at juvenile MF–CA3 synapses demonstrates that the precise timing of the two events, postsynaptic APs and presynaptic stimulation, can induce synaptic plasticity of excitatory MF input to CA3 pyramidal neurons. The direction of synaptic modification depended on the temporal order of MF stimulation and somatic spikes of CA3 pyramidal neurons. More specifically, t-LTD was induced when single APs preceded synaptic activation by 30 ms, and t-LTP was induced when synaptic activation preceded either single APs or brief AP bursts by 10 ms. Thus, plasticity that is induced by timing of spikes to synaptic activity at these anatomically and physiologically unusual synapses largely follows learning rules also observed at other excitatory synapses in the brain (Magee & Johnston, 1997; Markram et al. 1997; Feldman, 2000; Pawlak & Kerr, 2008). A finding that has not been reported from other excitatory synapses is the requirement for single APs preceding synaptic activation, as opposed to AP bursts, to induce t-LTD. The underlying mechanisms for this are unclear. Most likely, the spatiotemporal profile of postsynaptic Ca2+ accumulations induced by such burst pairing is not favourable for LTD expression (Lisman, 2001).
In vivo, both single APs and AP bursts occur at a low frequency in granule cells as well as in CA3 neurons (McNaughton et al. 1983; Jung & McNaughton, 1993; Hahn et al. 2007). Given that granule cells do not display the sustained high AP-firing rate that is expected to happen during a high frequency stimulus, the question was raised whether MF LTP can be expected to occur in vivo (Henze et al. 2000). We propose that the near-coincidence of single spikes and spike bursts might be a relevant mechanism for adapting synaptic strength in vivo at the MF–CA3 synapse.
NMDARs play a critical role in STDP at MF–CA3 synapses
Action potentials backpropagating into the dendritic tree may provide the postsynaptic depolarization necessary to relieve the voltage-dependent Mg2+ block of NMDARs. Thus, NMDARs are ideal sensors for correlated pre- and postsynaptic activity during AP–EPSP pairing. Although NMDARs are the long-known coincidence detectors for plasticity induction in nearly all brain regions (Bliss & Collingridge, 1993; Seeburg et al. 1995; Pawlak et al. 2005), they were reported to be unimportant for MF–CA3 synaptic plasticity when induced by HFS (Harris & Cotman, 1986; Williams & Johnston, 1988; Zalutsky & Nicoll, 1990). The present study gives first indication that NMDARs might have a critical role for timing-dependent LTP as well as LTD of MF inputs to CA3 neurons. Hereby, the activation of NMDARs has gained new relevance for MF plasticity, which is in agreement with two recent studies, in which NMDAR activation was required for selective potentiation of MF-NMDAR-mediated transmission (Kwon & Castillo, 2008; Rebola et al. 2008). Our study also indicates the location of the NMDARs relevant to STDP in CA3 neurons as being postsynaptic, as demonstrated by intracellular MK-801 application. The activation of postsynaptic NMDARs during spike timing may mediate the postsynaptic calcium rise (Reid et al. 2001) that was reported as being important for MF plasticity (Urban & Barrionuevo, 1996; Yeckel et al. 1999). Several cortical studies have found that postsynaptic NMDARs are required for t-LTP, but presynaptic NMDARs are required for t-LTD (Sjostrom et al. 2003; Bender et al. 2006; Nevian & Sakmann, 2006; Corlew et al. 2007; Rodriguez-Moreno & Paulsen, 2008).
Among numerous controversies regarding synaptic plasticity of MF synapses, the two areas of agreement were that plasticity is induced independently of NMDARs and presynaptically expressed (for review, see Nicoll & Schmitz, 2005). Two recent studies, though, described postsynaptic expression for LTP of NMDA EPSCs at MF–CA3 synapses (Kwon & Castillo, 2008; Rebola et al. 2008), which is consistent with the expression mechanisms that PPR and c.v. analyses suggest for the STDP observed in this study. The observed NMDAR dependence of STDP in this study discloses new Hebbian aspects of MF plasticity (Hebb, 1949). NMDARs at these synapses appear to be well-suited to detect sparse granule cell activity coinciding with CA3 spiking (McNaughton et al. 1983; Jung & McNaughton, 1993; Hahn et al. 2007). In contrast, higher granule cell activity, as mimicked by HFS, might release Zn2+ from MF terminals (Li et al. 2001), which is known to reduce NMDAR activation (Vogt et al. 2000). In combination with the recording conditions often used for HFS-induced plasticity (see below), this Zn2+ release might further suppress NMDAR activation during tetanic stimulation protocols.
Is timing-dependent plasticity observed exclusively at MF inputs and not at non-MF inputs?
An ongoing debate in mossy fibre research concerns the method of separating distinct inputs to CA3 neurons. An ideal scenario to observe pure MF–CA3 plasticity is to directly record from synaptically connected pairs of neurons, which has not yet been achieved due to the low connectivity in acute slice preparations. One common approach to isolate MF inputs is to perform experiments in high concentrations of divalent cations or in the presence of NMDAR antagonists, an approach that will not support forms of NMDAR-dependent plasticity. Since this study investigated STDP, which is usually NMDAR dependent, ionic and pharmacological conditions were employed that favour NMDAR activation. Although we used widely accepted physiological and pharmacological criteria to identify MF inputs, we cannot fully exclude ‘contamination’ of non-MF origin. Timing-dependent bidirectional plasticity at A/C synapses has indeed been shown in paired recordings from CA3 pyramidal neurons in organotypic slice cultures (Debanne et al. 1998). However, marginally activated non-MF inputs are unlikely to have contributed in a major way to the observed MF plasticity for the following reasons. (1) A correlation between the amount of EPSP sensitivity to group II mGluR agonists and the amount of plasticity observed did not exist. Such a correlation should be expected when for example the observed amount of t-LTP was entirely of non-MF origin. (2) Specifically, we report a mGluR-II agonist sensitivity for our dataset of 80 ± 2% (peak analysis) and 83.4 ± 1.4% (slope analysis). However, with STDP protocols similar to ours, across many experimental configurations and brain regions, usually an average synaptic change of approximately −20% to −30% for t-LTD, and +25% to +50% for t-LTP is observed (Feldman, 2000; Froemke et al. 2005; Bender et al. 2006; Nevian & Sakmann, 2006). If under our conditions exclusively the fraction of presumably ‘contaminating’ non-MF inputs had shown plasticity in this range, one would have observed at best a synaptic change of −6% for t-LTD and +10% for t-LTP. Instead, the amount of plasticity observed in our study was +62% (peak analysis) and +58% (slope analysis) for t-LTP, and −30% (peak analysis) and −29% (slope analysis) for t-LTD. (3) When specifically activating non-MF inputs by s. radiatum stimulation, no plasticity was observed upon the STDP protocols that had induced plasticity at MF inputs with s. lucidum stimulation. In addition, in some rare cases, non-MF inputs were activated by s. lucidum stimulation, as demonstrated by absent EPSP reduction by group II mGluR agonists. Also here, neither t-LTP was induced by a pre–post protocol, nor was t-LTD induced by a post–pre protocol. In summary, the t-LTP and t-LTD described in this study are indeed primarily attributed to MF inputs and not to non-MF inputs.
By failure to observe consistent STDP at non-MF synapses activated by s. radiatum stimulation, our study seemingly contradicts at first glance an important study, in which STDP was observed at associational CA3 inputs (Debanne et al. 1998). However, several differences between studies might explain the different observations, for example our compound non-MF EPSP may contain A/C and entorhinal inputs, whereas Debanne and colleagues demonstrated STDP at associational recurrent CA3 connections in paired recordings. Also, the amount and composition of dendritic voltage-gated channels and thus the dendritic backpropagation of action potentials is likely to be different in acute slices (our study) as opposed to slice cultures (Debanne et al. 1998). In acute slices, backpropagating APs potentially do not provide postsynaptic depolarization sufficient for STDP induction at distally located CA3 synapses as indicated by results obtained at neocortical synapses (Froemke et al. 2005). Similarly, in another study using acute slices, A/C LTP could not be induced by repetitive burst stimulation of the A/C input alone, but required association with MF inputs (Kobayashi & Poo, 2004), which is likely to have provided sufficient postsynaptic depolarization for plasticity induction. Here, it is noteworthy that in our study, non-MF inputs displayed a small albeit not significant trend towards potentiation with a pre–post protocol and a similar trend towards depression with a post–pre protocol. Thus, it is feasible that under our conditions, a stronger postsynaptic AP burst when paired with one (or multiple) EPSPs might induce plasticity of non-MF input when stimulated in s. radiatum. This was not our focus, but is without doubt an interesting subject for future studies.
Physiological implications of MF-STDP
In vivo evidence demonstrates that increased granule cell activity can drive CA3 neurons to spiking threshold (Henze et al. 2002). It may be that during low granule cell activity, for example during explorative behaviour, concurrent granule cell inputs or associative inputs are required to drive a cell to spiking threshold, thereby allowing for plasticity at this suggested ‘teacher’ synapse (Rolls et al. 1989; Jung & McNaughton, 1993; Henze et al. 2002).
How do our observations fit with the view that granule cell and recurrent/associative CA3 activity play a role in the separation and completion of cortical input patterns to the hippocampus (for review, see Nakazawa et al. 2004; McNaughton et al. 2006; Leutgeb et al. 2007)? When the activity of a single granule cell or a granule cell assembly is causally linked to spiking in a CA3 neuron, their connection might undergo potentiation of synaptic strength, thereby gaining importance for pattern separation (O’Reilly & McClelland, 1994). Furthermore, when a granule cell shows low activity, which is still somehow important in certain contexts, recurrent/ associative (or perforant path) inputs could help in bringing the CA3 neuron to spiking threshold, thereby strengthening this specific granule cell–CA3 connection. This process could be important for pattern completion, e.g. during the remapping of place fields (O’Keefe & Dostrovsky, 1971; Bostock et al. 1991; Kentros et al. 1998; for review, see McNaughton et al. 2006).
Acknowledgments
This work was supported by a Max-Planck fellowship to S.A. and by a Deutsche Forschungsgemeinschaft Grant KO 1064/5 to G.K. We thank M. Treviño for help with control experiments and P. H. Seeburg for valuable ideas, comments on the manuscript and generous support.
Glossary
Abbreviations
- AP
action potential
- HFS
high frequency stimulation
- MF
mossy fibre
- PPR
paired-pulse ratio
- STDP
spike-timing-dependent plasticity
- t-LTD/t-LTP
timing-dependent LTD/LTP
Author contributions
S.A. and V.P. contributed equally to the present study. V.P., S.A. and G.K. designed the research, V.P. conceived and started this work, S.A. played a major role in completing this work, and all authors jointly wrote the paper. All authors approved the final version for publication.
Authors' present addresses
V. Pawlak: Network Imaging Group, Max-Planck-Institute for Biological Cybernetics, 72076 Tübingen, Germany.
S. Astori: Department of Cell Biology and Morphology, University of Lausanne, 1005 Lausanne, Switzerland.
References
- Amaral DG, Ishizuka N, Claiborne B. Neurons, numbers and the hippocampal network. Prog Brain Res. 1990;83:1–11. doi: 10.1016/s0079-6123(08)61237-6. [DOI] [PubMed] [Google Scholar]
- Bender VA, Bender KJ, Brasier DJ, Feldman DE. Two coincidence detectors for spike timing-dependent plasticity in somatosensory cortex. J Neurosci. 2006;26:4166–4177. doi: 10.1523/JNEUROSCI.0176-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berzhanskaya J, Urban NN, Barrionuevo G. Electrophysiological and pharmacological characterization of the direct perforant path input to hippocampal area CA3. J Neurophysiol. 1998;79:2111–2118. doi: 10.1152/jn.1998.79.4.2111. [DOI] [PubMed] [Google Scholar]
- Berretta N, Jones RS. Tonic facilitation of glutamate release by presynaptic N-methyl-D-aspartate autoreceptors in the entorhinal cortex. Neuroscience. 1996;75:339–344. doi: 10.1016/0306-4522(96)00301-6. [DOI] [PubMed] [Google Scholar]
- Bi GQ, Poo MM. Synaptic modifications in cultured hippocampal neurons: dependence on spike timing, synaptic strength, and postsynaptic cell type. J Neurosci. 1998;18:10464–10472. doi: 10.1523/JNEUROSCI.18-24-10464.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischofberger J, Engel D, Frotscher M, Jonas P. Timing and efficacy of transmitter release at mossy fiber synapses in the hippocampal network. Pflugers Arch. 2006;453:361–372. doi: 10.1007/s00424-006-0093-2. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Bostock E, Muller RU, Kubie JL. Experience-dependent modifications of hippocampal place cell firing. Hippocampus. 1991;1:193–205. doi: 10.1002/hipo.450010207. [DOI] [PubMed] [Google Scholar]
- Castillo PE, Malenka RC, Nicoll RA. Kainate receptors mediate a slow postsynaptic current in hippocampal CA3 neurons. Nature. 1997;388:182–186. doi: 10.1038/40645. [DOI] [PubMed] [Google Scholar]
- Claiborne BJ, Amaral DG, Cowan WM. A light and electron microscopic analysis of the mossy fibers of the rat dentate gyrus. J Comp Neurol. 1986;246:435–458. doi: 10.1002/cne.902460403. [DOI] [PubMed] [Google Scholar]
- Corlew R, Wang Y, Ghermazien H, Erisir A, Philpot BD. Developmental switch in the contribution of presynaptic and postsynaptic NMDA receptors to long-term depression. J Neurosci. 2007;27:9835–9845. doi: 10.1523/JNEUROSCI.5494-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dan Y, Poo MM. Spike timing-dependent plasticity of neural circuits. Neuron. 2004;44:23–30. doi: 10.1016/j.neuron.2004.09.007. [DOI] [PubMed] [Google Scholar]
- Debanne D, Gahwiler BH, Thompson SM. Long-term synaptic plasticity between pairs of individual CA3 pyramidal cells in rat hippocampal slice cultures. J Physiol. 1998;507:237–247. doi: 10.1111/j.1469-7793.1998.237bu.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debanne D, Gahwiler BH, Thompson SM. Asynchronous pre- and postsynaptic activity induces associative long-term depression in area CA1 of the rat hippocampus in vitro. Proc Natl Acad Sci U S A. 1994;91:1148–1152. doi: 10.1073/pnas.91.3.1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Castillo J, Katz B. The effect of magnesium on the activity of motor nerve endings. J Physiol. 1954;124:553–559. doi: 10.1113/jphysiol.1954.sp005128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domenici MR, Berretta N, Cherubini E. Two distinct forms of long-term depression coexist at the mossy fiber-CA3 synapse in the hippocampus during development. Proc Natl Acad Sci U S A. 1998;95:8310–8315. doi: 10.1073/pnas.95.14.8310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman DE. Timing-based LTP and LTD at vertical inputs to layer II/III pyramidal cells in rat barrel cortex. Neuron. 2000;27:45–56. doi: 10.1016/s0896-6273(00)00008-8. [DOI] [PubMed] [Google Scholar]
- Froemke RC, Poo MM, Dan Y. Spike-timing-dependent synaptic plasticity depends on dendritic location. Nature. 2005;434:221–225. doi: 10.1038/nature03366. [DOI] [PubMed] [Google Scholar]
- Geiger JR, Bischofberger J, Vida I, Frobe U, Pfitzinger S, Weber HJ, Haverkampf K, Jonas P. Patch-clamp recording in brain slices with improved slicer technology. Pflugers Arch. 2002;443:491–501. doi: 10.1007/s00424-001-0735-3. [DOI] [PubMed] [Google Scholar]
- Hahn TT, Sakmann B, Mehta MR. Differential responses of hippocampal subfields to cortical up-down states. Proc Natl Acad Sci U S A. 2007;104:5169–5174. doi: 10.1073/pnas.0700222104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris EW, Cotman CW. Long-term potentiation of guinea pig mossy fiber responses is not blocked by N-methyl D-aspartate antagonists. Neurosci Lett. 1986;70:132–137. doi: 10.1016/0304-3940(86)90451-9. [DOI] [PubMed] [Google Scholar]
- Hebb DO. The Organization of Behavior. New York: Wiley; 1949. [Google Scholar]
- Henze DA, Urban NN, Barrionuevo G. The multifarious hippocampal mossy fiber pathway: a review. Neuroscience. 2000;98:407–427. doi: 10.1016/s0306-4522(00)00146-9. [DOI] [PubMed] [Google Scholar]
- Henze DA, Wittner L, Buzsaki G. Single granule cells reliably discharge targets in the hippocampal CA3 network in vivo. Nat Neurosci. 2002;5:790–795. doi: 10.1038/nn887. [DOI] [PubMed] [Google Scholar]
- Ishida M, Saitoh T, Shimamoto K, Ohfune Y, Shinozaki H. A novel metabotropic glutamate receptor agonist: marked depression of monosynaptic excitation in the newborn rat isolated spinal cord. Br J Pharmacol. 1993;109:1169–1177. doi: 10.1111/j.1476-5381.1993.tb13745.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe D, Johnston D. Induction of long-term potentiation at hippocampal mossy-fiber synapses follows a Hebbian rule. J Neurophysiol. 1990;64:948–960. doi: 10.1152/jn.1990.64.3.948. [DOI] [PubMed] [Google Scholar]
- Jonas P, Major G, Sakmann B. Quantal components of unitary EPSCs at the mossy fibre synapse on CA3 pyramidal cells of rat hippocampus. J Physiol. 1993;472:615–663. doi: 10.1113/jphysiol.1993.sp019965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung MW, McNaughton BL. Spatial selectivity of unit activity in the hippocampal granular layer. Hippocampus. 1993;3:165–182. doi: 10.1002/hipo.450030209. [DOI] [PubMed] [Google Scholar]
- Kamiya H, Shinozaki H, Yamamoto C. Activation of metabotropic glutamate receptor type 2/3 suppresses transmission at rat hippocampal mossy fibre synapses. J Physiol. 1996;493:447–455. doi: 10.1113/jphysiol.1996.sp021395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur A, Yeckel MF, Gray R, Johnston D. L-Type calcium channels are required for one form of hippocampal mossy fiber LTP. J Neurophysiol. 1998;79:2181–2190. doi: 10.1152/jn.1998.79.4.2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kentros C, Hargreaves E, Hawkins RD, Kandel ER, Shapiro M, Muller RV. Abolition of long-term stability of new hippocampal place cell maps by NMDA receptor blockade. Science. 1998;280:2121–2126. doi: 10.1126/science.280.5372.2121. [DOI] [PubMed] [Google Scholar]
- Kim J, Alger BE. Random response fluctuations lead to spurious paired-pulse facilitation. J Neurosci. 2001;21:9608–9618. doi: 10.1523/JNEUROSCI.21-24-09608.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Manabe T, Takahashi T. Presynaptic long-term depression at the hippocampal mossy fiber-CA3 synapse. Science. 1996;273:648–650. doi: 10.1126/science.273.5275.648. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Poo MM. Spike train timing-dependent associative modification of hippocampal CA3 recurrent synapses by mossy fibers. Neuron. 2004;41:445–454. doi: 10.1016/s0896-6273(03)00873-0. [DOI] [PubMed] [Google Scholar]
- Kwon HB, Castillo PE. Long-term potentiation selectively expressed by NMDA receptors at hippocampal mossy fiber synapses. Neuron. 2008;57:108–120. doi: 10.1016/j.neuron.2007.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence JJ, Grinspan ZM, McBain CJ. Quantal transmission at mossy fibre targets in the CA3 region of the rat hippocampus. J Physiol. 2004;554:175–193. doi: 10.1113/jphysiol.2003.049551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei S, Pelkey KA, Topolnik L, Congar P, Lacaille JC, McBain CJ. Depolarization-induced long-term depression at hippocampal mossy fiber-CA3 pyramidal neuron synapses. J Neurosci. 2003;23:9786–9795. doi: 10.1523/JNEUROSCI.23-30-09786.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leutgeb JK, Leutgeb S, Moser MB, Moser EI. Pattern separation in the dentate gyrus and CA3 of the hippocampus. Science. 2007;315:961–966. doi: 10.1126/science.1135801. [DOI] [PubMed] [Google Scholar]
- Li Y, Hough CJ, Frederickson CJ, Sarvey JM. Induction of mossy fiber→CA3 long-term potentiation requires translocation of synaptically released Zn2+ J Neurosci. 2001;21:8015–8025. doi: 10.1523/JNEUROSCI.21-20-08015.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman JE. Three Ca2+ levels affect plasticity differently: the LTP zone, the LTD zone and no man's land. J Physiol. 2001;532:285. doi: 10.1111/j.1469-7793.2001.0285f.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorincz A, Buzsaki G. Two-phase computational model training long-term memories in the entorhinal-hippocampal region. Ann N Y Acad Sci. 2000;911:83–111. doi: 10.1111/j.1749-6632.2000.tb06721.x. [DOI] [PubMed] [Google Scholar]
- Magee JC, Johnston D. A synaptically controlled, associative signal for Hebbian plasticity in hippocampal neurons. Science. 1997;275:209–213. doi: 10.1126/science.275.5297.209. [DOI] [PubMed] [Google Scholar]
- Markram H, Lubke J, Frotscher M, Sakmann B. Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science. 1997;275:213–215. doi: 10.1126/science.275.5297.213. [DOI] [PubMed] [Google Scholar]
- McNaughton BL, Barnes CA, O’Keefe J. The contributions of position, direction, and velocity to single unit activity in the hippocampus of freely-moving rats. Exp Brain Res. 1983;52:41–49. doi: 10.1007/BF00237147. [DOI] [PubMed] [Google Scholar]
- McNaughton BL, Battaglia FP, Jensen O, Moser EI, Moser MB. Path integration and the neural basis of the ‘cognitive map’. Nat Rev Neurosci. 2006;7:663–678. doi: 10.1038/nrn1932. [DOI] [PubMed] [Google Scholar]
- Nakazawa K, McHugh TJ, Wilson MA, Tonegawa S. NMDA receptors, place cells and hippocampal spatial memory. Nat Rev Neurosci. 2004;5:361–372. doi: 10.1038/nrn1385. [DOI] [PubMed] [Google Scholar]
- Nevian T, Sakmann B. Spine Ca2+ signaling in spike-timing-dependent plasticity. J Neurosci. 2006;26:11001–11013. doi: 10.1523/JNEUROSCI.1749-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll RA, Schmitz D. Synaptic plasticity at hippocampal mossy fibre synapses. Nat Rev Neurosci. 2005;6:863–876. doi: 10.1038/nrn1786. [DOI] [PubMed] [Google Scholar]
- O’Keefe J, Dostrovsky J. The hippocampus as a spatial map. Preliminary evidence from unit activity in the freely-moving rat. Brain Res. 1971;34:171–175. doi: 10.1016/0006-8993(71)90358-1. [DOI] [PubMed] [Google Scholar]
- O’Reilly RC, McClelland JL. Hippocampal conjunctive encoding, storage, and recall: avoiding a trade-off. Hippocampus. 1994;4:661–682. doi: 10.1002/hipo.450040605. [DOI] [PubMed] [Google Scholar]
- Pawlak V, Jensen V, Schupp BJ, Kvello A, Hvalby O, Seeburg PH, Köhr G. Frequency-dependent impairment of hippocampal LTP from NMDA receptors with reduced calcium permeability. Eur J Neurosci. 2005;22:476–484. doi: 10.1111/j.1460-9568.2005.04226.x. [DOI] [PubMed] [Google Scholar]
- Pawlak V, Kerr JN. Dopamine receptor activation is required for corticostriatal spike-timing-dependent plasticity. J Neurosci. 2008;28:2435–2446. doi: 10.1523/JNEUROSCI.4402-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike FG, Meredith RM, Olding AW, Paulsen O. Rapid report: postsynaptic bursting is essential for ‘Hebbian’ induction of associative long-term potentiation at excitatory synapses in rat hippocampus. J Physiol. 1999;518:571–576. doi: 10.1111/j.1469-7793.1999.0571p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebola N, Lujan R, Cunha RA, Mulle C. Adenosine A2A receptors are essential for long-term potentiation of NMDA-EPSCs at hippocampal mossy fiber synapses. Neuron. 2008;57:121–134. doi: 10.1016/j.neuron.2007.11.023. [DOI] [PubMed] [Google Scholar]
- Reid CA, Fabian-Fine R, Fine A. Postsynaptic calcium transients evoked by activation of individual hippocampal mossy fiber synapses. J Neurosci. 2001;21:2206–2214. doi: 10.1523/JNEUROSCI.21-07-02206.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Moreno A, Paulsen O. Spike timing-dependent long-term depression requires presynaptic NMDA receptors. Nat Neurosci. 2008;11:744–745. doi: 10.1038/nn.2125. [DOI] [PubMed] [Google Scholar]
- Rolls ET, Miyashita Y, Cahusac PM, Kesner RP, Niki H, Feigenbaum JD, Bach L. Hippocampal neurons in the monkey with activity related to the place in which a stimulus is shown. J Neurosci. 1989;9:1835–1845. doi: 10.1523/JNEUROSCI.09-06-01835.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeburg PH, Burnashev N, Köhr G, Kuner T, Sprengel R, Monyer H. The NMDA receptor channel: molecular design of a coincidence detector. Recent Prog Horm Res. 1995;50:19–34. doi: 10.1016/b978-0-12-571150-0.50006-8. [DOI] [PubMed] [Google Scholar]
- Sivakumaran S, Mohajerani MH, Cherubini E. At immature mossy-fiber-CA3 synapses, correlated presynaptic and postsynaptic activity persistently enhances GABA release and network excitability via BDNF and cAMP-dependent PKA. J Neurosci. 2009;29:2637–2647. doi: 10.1523/JNEUROSCI.5019-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjostrom PJ, Turrigiano GG, Nelson SB. Rate, timing, and cooperativity jointly determine cortical synaptic plasticity. Neuron. 2001;32:1149–1164. doi: 10.1016/s0896-6273(01)00542-6. [DOI] [PubMed] [Google Scholar]
- Sjostrom PJ, Turrigiano GG, Nelson SB. Neocortical LTD via coincident activation of presynaptic NMDA and cannabinoid receptors. Neuron. 2003;39:641–654. doi: 10.1016/s0896-6273(03)00476-8. [DOI] [PubMed] [Google Scholar]
- Sokolov MV, Rossokhin AV, M Kasyanov A, Gasparini S, Berretta N, Cherubini E, Voronin LL. Associative mossy fibre LTP induced by pairing presynaptic stimulation with postsynaptic hyperpolarization of CA3 neurons in rat hippocampal slice. Eur J Neurosci. 2003;17:1425–1437. doi: 10.1046/j.1460-9568.2003.02563.x. [DOI] [PubMed] [Google Scholar]
- Sourdet V, Debanne D. The role of dendritic filtering in associative long-term synaptic plasticity. Learn Mem. 1999;6:422–447. doi: 10.1101/lm.6.5.422. [DOI] [PubMed] [Google Scholar]
- Squire LR. Memory systems of the brain: a brief history and current perspective. Learn Mem. 2004;82:171–177. doi: 10.1016/j.nlm.2004.06.005. [DOI] [PubMed] [Google Scholar]
- Staubli U, Larson J, Lynch G. Mossy fiber potentiation and long-term potentiation involve different expression mechanisms. Synapse. 1990;5:333–335. doi: 10.1002/syn.890050410. [DOI] [PubMed] [Google Scholar]
- Thomas MJ, Watabe AM, Moody TD, Makhinson M, O’Dell TJ. Postsynaptic complex spike bursting enables the induction of LTP by theta frequency synaptic stimulation. J Neurosci. 1998;18:7118–7126. doi: 10.1523/JNEUROSCI.18-18-07118.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukamoto M, Yasui T, Yamada MK, Nishiyama N, Matsuki N, Ikegaya Y. Mossy fibre synaptic NMDA receptors trigger non-Hebbian long-term potentiation at entorhino-CA3 synapses in the rat. J Physiol. 2003;546:665–675. doi: 10.1113/jphysiol.2002.033803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzounopoulos T, Janz R, Sudhof TC, Nicoll RA, Malenka RC. A role for cAMP in long-term depression at hippocampal mossy fiber synapses. Neuron. 1998;21:837–845. doi: 10.1016/s0896-6273(00)80599-1. [DOI] [PubMed] [Google Scholar]
- Urban NN, Barrionuevo G. Induction of hebbian and non-hebbian mossy fiber long-term potentiation by distinct patterns of high-frequency stimulation. J Neurosci. 1996;16:4293–4299. doi: 10.1523/JNEUROSCI.16-13-04293.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt K, Mellor J, Tong G, Nicoll R. The actions of synaptically released zinc at hippocampal mossy fiber synapses. Neuron. 2000;26:187–196. doi: 10.1016/s0896-6273(00)81149-6. [DOI] [PubMed] [Google Scholar]
- Weisskopf MG, Nicoll RA. Presynaptic changes during mossy fibre LTP revealed by NMDA receptor-mediated synaptic responses. Nature. 1995;376:256–259. doi: 10.1038/376256a0. [DOI] [PubMed] [Google Scholar]
- Williams S, Johnston D. Muscarinic depression of long-term potentiation in CA3 hippocampal neurons. Science. 1988;242:84–87. doi: 10.1126/science.2845578. [DOI] [PubMed] [Google Scholar]
- Wittenberg GM, Wang SS. Malleability of spike-timing-dependent plasticity at the CA3-CA1 synapse. J Neurosci. 2006;26:6610–6617. doi: 10.1523/JNEUROSCI.5388-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witter MP, Amaral DG. Hippocampal formation. In: Paxinos G, editor. The Rat Nervous System. 3rd edn. Amsterdam: Elsevier; 2004. pp. 635–704. Chap 21, pp. [Google Scholar]
- Yeckel MF, Kapur A, Johnston D. Multiple forms of LTP in hippocampal CA3 neurons use a common postsynaptic mechanism. Nat Neurosci. 1999;2:625–633. doi: 10.1038/10180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalutsky RA, Nicoll RA. Comparison of two forms of long-term potentiation in single hippocampal neurons. Science. 1990;248:1619–1624. doi: 10.1126/science.2114039. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]