

Abstract
Cerebral vascular smooth muscle contractility plays a crucial role in controlling arterial diameter and, thereby, blood flow regulation in the brain. A number of K+ channels have been suggested to contribute to the regulation of diameter by controlling smooth muscle membrane potential (Em) and Ca2+ influx. Previous studies indicate that stromatoxin (ScTx1)-sensitive, Kv2-containing channels contribute to the control of cerebral arterial diameter at 80 mmHg, but their precise role and molecular composition were not determined. Here, we tested if Kv2 subunits associate with ‘silent’ subunits from the Kv5, Kv6, Kv8 or Kv9 subfamilies to form heterotetrameric channels that contribute to control of diameter of rat middle cerebral arteries (RMCAs) over a range of intraluminal pressure from 10 to 100 mmHg. The predominant mRNAs expressed by RMCAs encode Kv2.1 and Kv9.3 subunits. Co-localization of Kv2.1 and Kv9.3 proteins at the plasma membrane of dissociated single RMCA myocytes was detected by proximity ligation assay. ScTx1-sensitive native current of RMCA myocytes and Kv2.1/Kv9.3 currents exhibited functional identity based on the similarity of their deactivation kinetics and voltage dependence of activation that were distinct from those of homomultimeric Kv2.1 channels. ScTx1 treatment enhanced the myogenic response of pressurized RMCAs between 40 and 100 mmHg, but this toxin also caused constriction between 10 and 40 mmHg that was not previously observed following inhibition of large conductance Ca2+-activated K+ (BKCa) and Kv1 channels. Taken together, this study defines the molecular basis of Kv2-containing channels and contributes to our understanding of the functional significance of their expression in cerebral vasculature. Specifically, our findings provide the first evidence of heteromultimeric Kv2.1/Kv9.3 channel expression in RMCA myocytes and their distinct contribution to control of cerebral arterial diameter over a wider range of Em and transmural pressure than Kv1 or BKCa channels owing to their negative range of voltage-dependent activation.
Introduction
Potassium (K+) channels play a crucial role in the physiological control of membrane potential (Em) of vascular smooth muscle cells (VSMCs) and, thereby, contraction and arterial diameter (Nelson & Quayle, 1995; Davis & Hill, 1999; Jackson, 2005). Contraction of VSMCs is dependent in part on the influx of Ca2+ through voltage-gated Ca2+ channels (VGCCs). Cytosolic Ca2+ concentration ([Ca2+]i) is elevated following Em depolarization leading to the activation of myosin light chain kinase that phosphorylates the 20 kDa myosin light chain subunits to initiate cross-bridge cycling. In small resistance arteries, increased intravascular pressure causes depolarization, Ca2+ influx and constriction, a phenomenon referred to as the myogenic response (see Davis & Hill, 1999). The myogenic response is the fundamental mechanism underlying blood flow autoregulation, a critical determinant of capillary hydrostatic pressure, and essential to prevent pressure-induced damage of delicate downstream structures (Davis & Hill, 1999). There is now considerable evidence indicating that the level of K+ channel activity influences the extent of myogenic constriction in response to pressure elevation, as well as dilatation evoked by pressure reduction. Specifically, K+ channel activity is postulated to play a critical negative-feedback role, controlling the level of Em depolarization and, thereby, the extent of VGCC activation, Ca2+ influx and force generation owing to pressure elevation. Although progress has been made in identifying the K+ channels involved in the arterial myogenic response, there are gaps in our understanding that remain to be addressed. This lack of knowledge limits the development of novel therapies for the treatment of dysfunctional myogenic control of arterial diameter.
Previous studies have provided substantial evidence that Ca2+-activated BKCa and voltage-gated Kv1 channels contribute to the negative-feedback control of myogenic depolarization in cerebral resistance arteries (Brayden & Nelson, 1992; Nelson et al. 1995; Albarwani et al. 2003; Chen et al. 2006; Yang et al. 2009). These channels exhibit an increasing contribution to the control of Em and diameter owing to pressure-dependent activation; specifically, Kv1 channel gating is increased in response to the change in Em with pressure elevation, and BKCa channel activity is principally enhanced by Ca2+ release from intracellular Ca2+ stores (i.e. Ca2+ sparks) and Ca2+ influx owing to VGCC activation (Brayden & Nelson, 1992; Nelson et al. 1995; Knot & Nelson, 1995; Guia et al. 1999; Albarwani et al. 2003; Chen et al. 2006; Yang et al. 2009). Cerebral arterial BKCa channels are composed of KCa1.1 pore-forming α-subunits (i.e. Slo1 protein KCNMA1 gene product; Wei et al. 2005), and β1 modulatory subunits that stabilize the channel open state and increase Ca2+ sensitivity (Brenner et al. 2000; Cox & Aldrich, 2000). Selective inhibition of BKCa enhances the myogenic response of cerebral arteries (Brayden & Nelson, 1992; Nelson et al. 1995). Targeted deletion of the β1-subunit (Brenner et al. 2000) or reduced β1 subunit expression in hypertensive rats (Amberg & Santana, 2006) was shown to be associated with increased myogenic constriction and elevated blood pressure. Moreover, differences in β1-subunit expression level also appear to contribute to the distinct functional properties (e.g. varied Em–myogenic constriction relation) of skeletal versus cerebral resistance arteries (Yang et al. 2009).
Kv1 channels of VSMCs also possess a heteromultimeric subunit composition that may vary in a vessel-specific manner (Thorneloe et al. 2001; Albarwani et al. 2003; Plane et al. 2005). Rat cerebral arteries express heteromultimeric Kv1 channels containing at a minimum Kv1.2 and Kv1.5 (Albarwani et al. 2003; Chen et al. 2006). These α-subunits are likely to associate with accessory Kvβ subunits (Kvβ1–2), as was demonstrated for VSMCs of other vessels (Thorneloe et al. 2001; Plane et al. 2005). Selective pharmacological suppression of Kv1 channels with 4-aminopyridine (4-AP) (at ≤300 μm) or correolide (1–10 μm) causes depolarization and enhances the cerebral myogenic response (Knot & Nelson, 1995; Albarwani et al. 2003; Chen et al. 2006). Additionally, over-expression of a dominant-negative Kv1.5 subunit was found to enhance, but wild-type Kv1.5 suppressed, myogenic constriction of rat middle cerebral arteries (RMCAs; Chen et al. 2006).
Several recent observations indicate, however, that Kv1 and BKCa are not the only K+ channels that may contribute to the negative-feedback control of myogenic depolarization and constriction. Kv2 and Kv7 subunit message and/or protein expression has been detected in some arteries (Cox et al. 2008; Greenwood & Ohya, 2009) and inhibition of Kv2- and Kv7-containing channels also modulates myogenic control of arterial diameter (Amberg & Santana, 2006; Zhong et al. 2010). For example, Kv2.1 and Kv2.2 α-subunits are expressed by several vascular and non-vascular smooth muscle cell types (e.g. cerebral arteries, Amberg & Santana, 2006; pulmonary arteries, Patel et al. 1997; Coppock & Tamkum, 2001; Platoshyn et al. 2001; Smirnov et al. 2002; mesenteric arteries, Moreno-Dominguez et al. 2009; ductus arteriosis, Wu et al. 2007; aorta, Belevych et al. 2002; coronary artery, Thorne et al. 2002; placental vasculature, Wareing et al. 2006; corpus cavernosum, Malysz et al. 2002; urinary bladder, Thorneloe & Nelson, 2003; Chen et al. 2010; and gastrointestinal tract, Schmalz et al. 1998; Frey et al. 2000). Amberg & Santana (2006) showed that stromatoxin (ScTx1) suppression of Kv2 channels (Escoubas et al. 2002; Swartz, 2007) produced constriction in RMCAs at a transmural pressure of 80 mmHg. Also, abnormal regulation of arterial diameter in hypertension and in a canine model of subarachnoid haemorrhagic stroke was shown to be associated with a reduction in whole-cell Kv2 current and/or subunit expression (Yuan et al. 1998; Amberg & Santana, 2006; Jahromi et al. 2008a,b,c; Moreno-Dominguez et al. 2009). These data imply that Kv2-containing channels may also contribute to the negative-feedback regulation of the cerebral myogenic response.
An additional, important consideration that has not been addressed in the cerebral vasculature is that Kv2 subunits are known to co-assemble with Kv5, Kv6, Kv8 or Kv9 so-called γ-subunits to form hetermultimeric channels with a subunit stoichiometry of 3:1 (Kerschensteiner et al. 2005; Coetzee et al. 2006). Transcripts encoding members of the Kv5, Kv6 and Kv9 subfamilies are known to be expressed in VSMCs of pulmonary and placental arteries (Kv9.2 or Kv9.3, Patel et al. 1997; Platoshyn et al. 2001; Davies & Kozlowski, 2001; Wareing et al. 2006), and urinary bladder (Kv5.1 and Kv6.1–6.3, Thorneloe & Nelson, 2003; Kv9.3, Chen et al. 2010). The possibility that Kv2 channels of cerebral arterial myocytes might be heteromultimers was considered by Amberg & Santana (2006), but the molecular composition of the channels was not determined.
Kv5, Kv6, Kv8 and Kv9 subunits are commonly called ‘silent’ subunits because they do not form functional channels when expressed as homomultimers (Drewe et al. 1992; Hugnot et al. 1996; Salinas et al. 1997a,b; Shepard & Rae, 1999; Ottschytsch et al. 2002). However, when they combine with Kv2.1 or Kv2.2 to form heteromultimeric channels, the presence of the ‘silent’ subunits affects the biophysical properties of the channel complex. Specifically, the kinetics of activation and deactivation, voltage dependence of activation and inactivation, and/or current amplitude are modified by the presence of the ‘silent’ subunits (Patel et al. 1997; Salinas et al. 1997a,b; Kerschensteiner & Stocker, 1999; Shepard & Rae, 1999; Ottschytsch et al. 2002; Kerschensteiner et al. 2003, 2005). These alterations in function and expression have the potential to affect the contribution of the channels to the control of Em and arterial diameter. For example, genetic hypertension in the BPH mouse model was found to be associated with de novo expression of Kv6.3 subunits leading to a reduction in net Kv current in mesenteric arterial VSMCs that possibly contributes to the elevated blood pressure observed in this model (Moreno-Dominguez et al. 2009).
Whether ‘silent’ subunits associate with Kv2 subunits in arterial myocytes to modify the range of transmural pressure over which the channels contribute to control of Em and the myogenic response is an important question that must be addressed to understand the cerebral myogenic response and blood flow autoregulation. Additionally, this knowledge may provide insights concerning the expression of multiple types of Kv channels by resistance arterial VSMCs and the pathophysiological significance of reduced Kv2-containing channel expression as a cause of abnormal control of diameter. Here, we tested the hypothesis that ScTx1-sensitive Kv channels of RMCAs are heteromultimeric channels owing to the co-assembly of Kv2.1 or Kv2.2 with a Kv5, Kv6 or Kv9 subunit. We employed: (1) RT-PCR and quantitative, real-time PCR to define the extent of Kv2 and ‘silent’ subunit expression in RMCAs, (2) patch clamp analysis of native RMCA ScTx1-senstive and recombinant Kv2.1 and Kv2.1/Kv9.3 channels to permit a comparison of their functional properties, (3) pressurized arterial myography to evaluate the contribution of ScTx1-sensitive channels to control of RMCA diameter over a range of transmural pressure between 10 and 100 mmHg, and (4) proximity ligation assay (PLA; Fredriksson et al. 2002; Söderberg et al. 2006), to detect Kv2.1 and Kv9.3 protein expression at the plasma membrane of freshly isolated RMCA myocytes. Our data indicate that the ScTx1-sensitive Kv channels of RMCA myocytes are Kv2.1/Kv9.3 heteromultimeric channels that contribute to the control of myogenic constriction over a range of transmural pressure that is distinct from that previously identified for cerebral arterial Kv1, Kv7 and BKCa channels.
Methods
Ethical approval
Male Sprague–Dawley rats (250–275 g; Charles River, Montréal, Quebec, Canada) were maintained and killed by halothane inhalation and exsanguination according to the standards of the Canadian Council on Animal Care and The Journal of Physiology's ethical policies and regulations as indicated in Drummond (2009), and reviewed by the Animal Care Committee of the Faculty of Medicine, University of Calgary. A total of 180 rats were employed.
Intact cerebral arterial pressure myography
Rat brains were carefully removed and placed in ice-cold Krebs solution containing (in mm): NaCl 120, NaHCO3 25, KCl 4.8, NaH2PO4 1.2, MgSO4 1.2, glucose 11, CaCl2 2.5 (pH 7.4 when aerated with 95% air–5% CO2). Left and right RMCAs were removed from each brain, dissected free of the surrounding tissue and cut into 2–3 mm segments for arterial pressure myography as previously described (Chen et al. 2006; Johnson et al. 2009b). Briefly, RMCAs were mounted in an arteriograph chamber attached to a pressure myograph (Living Systems, Burlington, VT, USA) for measurement of outer diameter with an automated edge detection system (IonOptix, Milton, MA, USA). Endothelium intact or denuded vessels were used with no differences in behaviour noted. In the latter case, endothelial cells were removed from all arteries by briefly passing a stream of air through the vessel lumen and confirmed by loss of vasodilatation to 10 μm bradykinin. Arteries were warmed at 37°C for 10–15 min in Krebs saline solution, pressurized to 60 mmHg and allowed to develop active myogenic tone over 20–30 min. All arteries were subjected to two 5 min pressure steps from 20 to 80 mmHg to ensure development of stable myogenic constriction. Vessels (∼10%) that exhibited leaks and/or did not exhibit stable constriction at 80 mmHg were discarded.
RMCA myocyte isolation
Rat brains were carefully removed and placed in an ice-cold smooth muscle dissection solution (SMDS) containing (in mm): NaCl 60, sodium glutamate 80, KCl 5, MgCl2 2, glucose 10, and Hepes 10 (pH 7.4). RMCAs were dissected and single myocytes were enzymatically isolated using a method modified from Plane et al. (2005). Briefly, arteries were equilibrated in SMDS containing bovine serum albumin (BSA; 1 mg ml−1) at 37°C for 10 min, exposed to the same solution supplemented with papain (0.5 mg ml−1) and dithiothreitol (1.5 mg ml−1) at 37°C for 8–10 min, washed in ice-cold SMDS, then incubated in SMDS containing 100 μm Ca2+, BSA (1 mg ml−1) and collagenase (0.7 mg ml−1 type F and 0.4 mg ml−1 type H) at 37°C for 8–10 min and again washed in ice-cold SMDS. Isolated myocytes were liberated from the digested vessels by gentle trituration and kept in cold SMDS containing 1 mg ml−1 BSA until use (within 12 h).
Cell culture and transfection
HEK 293 cells were maintained on 10 cm plastic culture dishes in high glucose Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and 5% ampicillin–streptomycin at 37°C in 5% CO2 as previously described (Clément-Chomienne et al. 1999; Chen et al. 2006; Johnson et al. 2009a) Transfections were performed using FuGENE 6 Transfection Reagent as per the vendor's instructions. cDNAs encoding green fluorescent protein (GFP), Kv2.1, Kv9.2, Kv9.3, Kv1.2 and Kv1.5 were subcloned into pcDNA3.1. Cells were transfected with: (1) 4 μg plasmid containing GFP, (2) 4 μg GFP and 2 μg Kv2.1, (3) 4 μg GFP and 6 μg Kv9.3, (4) 4 μg GFP and 4 μg Kv1.2, (5) 4 μg GFP and 4 μg Kv1.5, (6) 4 μg GFP, 4 μg Kv1.2 and 4 μg Kv1.5, (7) 4 μg GFP, 2 μg Kv2.1 and 6 μg Kv9.3, (8) 4 μg GFP, 2 μg Kv2.1 and 6 μg Kv9.2, (9) 4 μg GFP, 2 μg Kv2.1, 6 μg Kv9.3 and 4 μg Kv1.5, (10) 4 μg GFP, 6 μg Kv9.3 and 4 μg Kv1.5, (11) 4 μg GFP, 4 μg Kv2.1 and 4 μg Kv1.5, (12) 4 μg GFP, 4 μg Kv1.2 and 4 μg Kv2.1. Transfected cells were re-plated and employed for patch clamp analysis after 36–48 h or fixed for PLA at 70% confluence.
Patch clamp electrophysiology
Whole-cell currents due to native Kv current and heterologous expression of Kv2.1, Kv2.1/Kv9.3 and Kv2.1/Kv9.2 channels in HEK 293 cells were recorded and analysed as previously described (Chen et al. 2006; Johnson et al. 2009a). Briefly, cells were superfused with a bath solution containing (in mm): NaCl 120, NaHCO3 3, KCl 4.2, KH2PO4 1.2, MgCl2 2, CaCl2 0.1, glucose 10, and Hepes 10 (pH 7.4). The patch pipette solution contained (in mm): potassium gluconate 110, KCl 30, MgCl2 0.5, Hepes 5, EGTA 10, Na2ATP 5, and GTP 1 (pH 7.2). The junction potential was determined to be 15 mV for the recording conditions employed and this value was used to correct all voltage protocols. Current–voltage (I–V) relations were determined using 325 ms step pulses to between −95 and +25 or +45 mV in increments of 10 mV from a holding potential of −75 mV. End-pulse current amplitudes were normalized to cell capacitance to permit determination of current density (pA pF−1). Cell capacitance was determined by integration of the capacity transient recorded for a step from −75 to −70 mV. Peak tail current amplitudes of native and recombinant channel currents were determined during 625 ms repolarizing steps to −45 and −55 mV, respectively. To determine the time constants for tail current decay, native and recombinant currents were assessed at a similar voltage of −45 mV following steps to +25 mV. Mean changes in I–V relation, current density and fractional changes in current amplitude at +15 mV owing to toxin or drug treatment were compared by Student's t test or ANOVA followed by Bonferoni's post hoc test.
RT-PCR and real-time QPCR
RT-PCR and real-time quantitative PCR (QPCR) were carried out as previously described (Thorneloe et al. 2001; Plane et al. 2005). Briefly, total RNA was extracted from rat brain, endothelium-denuded, intact RMCAs and isolated RMCA myocytes using an RNeasy Mini kit with DNase treatment (Qiagen, Mississauga, Canada) and first strand cDNA synthesized using the Sensiscript RT kit (Qiagen) with oligo d(T) primer. Primer pairs to detect Kv2.1, Kv2.2, Kv5.1, Kv6.1–Kv6.3 and Kv9.1–Kv9.3 were designed or purchased from Qiagen (Mississauga, Canada) to permit quantification of transcript abundance in mRNA samples of RMCAs by real-time QPCR. The sequence of subunit-specific primers (5′–3′) that were designed in-house were: Kv2.2 (91 bp): forward TTGATAACACCTGCTGC, reverse GATGGCCAGGATCTTTG; Kv6.3 (119 bp): forward TGTCTATGGTGGTGCTGTG, reverse AGCTTCAATTATCCCGGA; β-actin (98 bp): forward TATGAGGGTTACGCGCTCCC, reverse ACGCTCGGTCAGGATCTTCA. All other primers were purchased from Qiagen. Each primer set used: (1) had an efficiency of >90% that did not differ by >5% at an annealing temperature of ∼58°C, (2) produced a single peak with no evidence of additional amplicons or dimer formation during melt curve analysis, and (3) yielded amplicons of an expected size. Real-time PCR was performed with SYBR-Green and a reaction with a hot start at 95°C for 15 min, followed by 40 cycles of 94°C for 15 s, 58°C for 30 s and 72°C for 30 s. Threshold cycle was determined using a Bio-Rad iCycler and vendor-supplied software, and transcript abundance was calculated by the 2−ΔΔCt method using β-actin as the reference for normalization (Livak & Schmittgen, 2001). Kv2.1 and Kv9.3 subunit expression in RMCA myocytes was confirmed by RT-PCR (40 cycles; primer pairs Kv2.1 (267 bp): forward ACACCATCACCATCTCTCAAGG, reverse CTAAATTGTCAGCTCACCCCGA; Kv9.3 (569 bp): forward TCCCATCACCATCATCTTCAA, reverse GCCGTAGCAATAAATCCTTC) using mRNA samples derived from populations of 200–300 freshly isolated, individually selected RMCA myocytes; amplicons of appropriate size and sequence were detected for both subunits. Primers for the neuronal K+ channel, Erg-3, and the endothelial factor, endothelin-1, were used as markers of contamination of the myocyte mRNA samples by neuronal and endothelial message, respectively (Plane et al. 2005).
Proximity ligation assay (PLA)
Freshly dissociated RMCA myocytes or transfected HEK 293 cells were studied using the Duolink in situ PLA detection kit 563 (Olink, Uppsala, Sweden) as per the vendor's instructions. Briefly, cells were fixed in phosphate-buffered saline (PBS) containing 3% paraformaldehyde for 15 min, permeabilized in PBS containing 0.1% Triton X-100 for 15 min and quenched in PBS containing 100 mm glycine for 5 min. Cells were then washed with PBS, blocked for 30 min at 37°C in Duolink blocking solution, and incubated overnight at 4°C with pairs of primary antibodies in Duolink antibody diluent solution; i.e. rabbit anti-Kv2.1 (Alomone Labs Ltd, Jerusalem, Israel) and goat anti-Kv9.3 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) or mouse anti-Kv1.2 (UC Davis/NIH NeuroMab, Davis, CA, USA) and rabbit anti-Kv1.5 (Alomone). Control experiments employed only one primary antibody or cells transfected with cDNAs encoding only one type of Kv subunit. Cells were labelled with Duolink PLA PLUS and MINUS probes for 2 h at 37°C. Anti-goat PLUS and anti-rabbit MINUS were employed to detect Kv2.1 and Kv9.3 co-assembly, whereas anti-mouse PLUS and anti-rabbit MINUS were used for Kv1.2 and Kv1.5. The secondary antibodies of PLA PLUS and MINUS probes are attached to synthetic oligonucleotides that hybridize when in close proximity (i.e. <40 nm separation). The hybridized oligonucleotides are then ligated prior to rolling circle amplification. The concatemeric amplification products extending from the oligonucleotide arm of the PLA probes were then detected using red fluorescent fluorophore-tagged, complementary oligonucleotide sequences and a Zeiss ApoTome epifluorescence microscope (Carl Zeiss, NY, USA) using 0.3–0.5 μm optical sections.
Materials
All chemicals were purchased from Sigma-Aldrich (Oakville, ON, Canada) unless indicated otherwise. FuGENE 6 Transfection Reagent was from Roche (IN, USA). Papain was from Worthington Biochemical Corp. (Lakewood, NJ, USA). Proximity ligation assays (PLAs) used the Duolink in situ PLA kit (Olink, Uppsala, Sweden). Stromatoxin (ScTx1), Kv1.5 and Kv2.1 antibodies were from Alomone, Kv1.2 antibody was from UC Davis/NIH NeuroMab, Kv9.2 and Kv9.3 clones were from Thermo Scientific Open Biosystem (Huntsville, AL, USA), goat Kv9.3 antibody was from Santa Cruz Biotechnology, and chicken Kv9.3 antibody was from Abcam Inc. (Cambridge, MA, USA).
Statistical analysis
Where applicable values are presented as the means ± s.e.m., with n indicating the number of experiments for a given treatment. Statistical differences were determined using Student's t test or repeated-measures ANOVA followed by Bonferroni's post hoc test. A level of P < 0.05 was considered to be statistically significant.
Results
ScTx1 enhances the myogenic response of RMCAs
Figure 1A and B shows the concentration-dependent vasoconstriction evoked by 5–30 nm ScTx1 of RMCAs pressurized to 80 mmHg. A near peak constriction was obtained at 80 mmHg with 30 nm ScTx1 (n = 4), as described previously (Amberg & Santana, 2006). ScTx1 always evoked a stable, sustained vasoconstriction that was of similar magnitude during repeated applications of the toxin and unaffected by endothelial removal (data not shown). Figure 1C and D shows representative recordings and mean ± s.e.m. values (n = 7) of RMCA diameter for transmural pressures between 10 and 100 mmHg in control Krebs saline solution (containing 2.5 mm Ca2+), during exposure to 30 nm ScTx1, and then zero Ca2+-containing (2 mm EGTA) Krebs solution. RMCA diameter was maintained or decreased with increasing transmural pressure in normal Krebs solution. ScTx1 treatment caused a significant decrease in arterial diameter and an increase in active constriction (i.e. the difference between the passive diameter recorded in zero Ca2+-containing solution and in control or ScTx1-containing Krebs solution) at all pressures tested, including the range of 10–40 mmHg (Fig. 1C–E). Figure 1F shows a representative example of the constriction evoked by 30 nm ScTx1 at 10 mmHg that was 27 ± 4.5 μm in 17 vessels; there was no change in diameter in four untreated, time control vessels (data not shown).
Figure 1. Enhanced RMCA myogenic response by ScTx1.
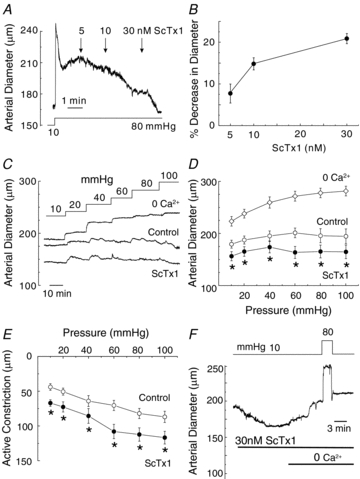
A, representative recording of arterial diameter at 80 mmHg showing concentration-dependent constriction of RMCA treated with 5–30 nm ScTx1. B, mean values ± s.e.m. (n = 4) for the percentage decrease in RMCA diameter in the presence of ScTx1 at 80 mmHg. C and D, representative recordings and mean values ± s.e.m. (n = 7) of RMCA diameter between 10 and 100 mmHg under control conditions, and following 30 nm ScTx1 prior to exposure to zero Ca2+ solution to determine passive diameter at each pressure. E, mean values ± s.e.m. (n = 7) for active constriction in micrometres under control conditions and after treatment with ScTx1 (value for active constriction is the difference between control or ScTx1 diameter and passive diameter). F, representative recording of vasoconstriction evoked by ScTx1 at 10 mmHg and block of constriction at 10 and 80 mmHg in ScTx1 and zero Ca2+ solution. *Significantly different (P < 0.05) from value in control solution.
Zero Ca2+-containing solution blocked the constriction evoked by ScTx1 indicating that Ca2+ influx from the extracellular space was required at 10 and 80 mmHg (Fig. 1F). Given that Em between 10 and 40 mmHg was reported to be <−50 mV in rat posterior cerebral arteries (Knot & Nelson, 1998), the nature of the channel(s) responsible for the influx of Ca2+ warranted examination. Figure 2A–D shows representative recordings and mean ± s.e.m. values (n = 4–6 vessels in each group) for RMCA diameter at 10 mmHg in the presence of ScTx1 followed by treatment with diltiazem (10 μm), nifedipine (1 μm), mibefradil (1 μm) or SKF96365 (10–30 μm), prior to exposure to zero extracellular Ca2+ solution. Diltiazem, nifedipine and mibefradil reversed the ScTx1-induced constriction, but SKF96365 had no effect, although it was able to suppress the myogenic response evoked by a subsequent pressure step from 10 to 80 mmHg (Fig. 2A–D). A similar lack of reversal of ScTx1-evoked constriction was observed in five additional vessels.
Figure 2. Inhibition of VGCCs but not NSCCs reversed ScTx1-evoked vasoconstriction.
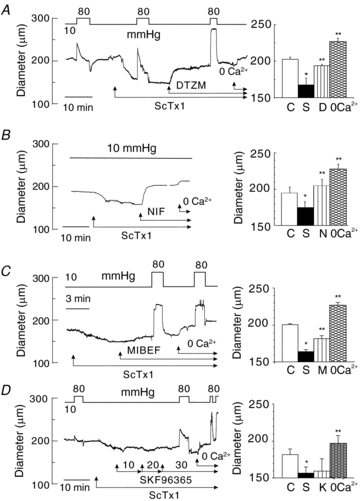
A, representative recording and mean values ± s.e.m. (n = 4) of RMCA diameter at 10 mmHg under control conditions and following sequential treatment with ScTx1 (30 nm), diltiazem (10 μm) and zero Ca2+ solution. Brief steps to 80 mmHg show that the extent of myogenic response was enhanced by ScTx1 and inhibited by diltiazem. B, representative recording and mean values ± s.e.m. (n = 6) of RMCA diameter at 10 mmHg under control conditions and following sequential treatment with ScTx1 (30 nm), nifedipine (1 μm) and zero Ca2+ solution. C, representative recording and mean values ± s.e.m. (n = 4) of RMCA diameter at 10 mmHg under control conditions and following sequential treatment with ScTx1 (30 nm), mibefradil (1 μm) and zero Ca2+ solution. Brief steps to 80 mmHg show that the myogenic response was inhibited by mibefradil to level observed in zero Ca2+. D, representative recording and mean values ± s.e.m. (n = 4) of RMCA diameter at 10 mmHg under control conditions and following sequential treatment with ScTx1 (30 nm), 10, 20 and 30 μm SKF96365(K) and zero Ca2+ solution. SKF96365 did not affect ScTx1-evoked constriction, and brief steps to 80 mmHg indicated that the extent of myogenic response was reduced by SKF96365 confirming effective block of NSCCs was achieved. * and ** are significantly different (P < 0.05) from value in control solution and ScTx1, respectively.
Kv2.1 and Kv9.3 subunits are expressed in RMCAs
The voltage range reported for the activation of homomeric Kv2.1 channels heterologously expressed in Xenopus oocytes or mammalian cells is positive to −40 mV (Patel et al. 1997; Salinas et al. 1997a,b; Kerschensteiner & Stocker, 1999; Shepard & Rae, 1999; Ottschytsch et al. 2002; Kerschensteiner et al. 2003, 2005). Based on the ability of ScTx1 to evoke constriction of RMCAs between 10 and 40 mmHg at which Em is <–50 mV (Knot & Nelson, 1998), it seemed likely that the Kv2-containing channels contained a ‘silent’ subunit to account for ScTx1 sensitivity at low pressures. We therefore examined the expression of transcripts encoding members of the Kv5, Kv6 and Kv9 subfamilies previously demonstrated to be expressed in smooth muscle cells (Patel et al. 1997; Platoshyn et al. 2001; Davies & Kozlowski, 2001; Wareing et al. 2006; Thorneloe & Nelson, 2003; Chen et al. 2010). QPCR primer pairs were designed or purchased (Qiagen) to quantify the respective levels of message for each subunit. Each of the QPCR primer pairs employed generated amplicons of appropriate size using mRNA derived from rat brain (Fig. 3A). Transcript abundance relative to β-actin was quantified by real-time QPCR using the 2−ΔΔCt method and mRNA derived from endothelium-denuded RMCAs of 12 rats. Substantial expression of Kv2.1 and Kv9.3 was consistently detected in all experiments, but transcripts for Kv2.2, Kv5.1, Kv6.1–6.3, Kv9.1 and Kv9.2 were expressed only at very low or undetectable levels (n = 4–10; Fig. 3B). We subsequently confirmed the VSMC-specific expression of Kv2.1 and Kv9.3 by RT-PCR using populations of 250–300 individually selected RMCA myocytes from cell isolations from vessels of three rats and a second set of primer pairs. Contamination of the myocyte mRNA samples by neuronal or endothelial message was excluded by the absence of any evidence of transcripts encoding the neuronal K+ channel, Erg3, or the endothelial factor, endothelin-1 (data not shown).
Figure 3. Kv2.1 and Kv9.3 are predominant transcripts expressed in RMCA.
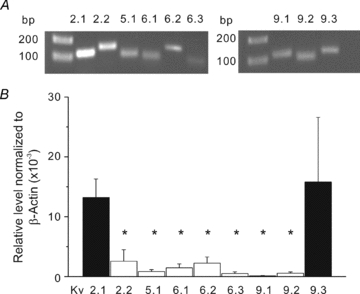
A, representative gels indicating generation of amplicons of appropriate sizes using QPCR pimer pairs for Kv2.1, Kv2.2, Kv5.1, Kv6.1–Kv6.3, Kv9.1–9.3 and mRNA extracted from rat brain. Similar results were obtained from 7 additional experiments. B, mean values ± s.e.m. (n = 4–10) for level of Kv2.1, Kv2.2, Kv5.1, Kv6.1–Kv6.3, Kv9.1–9.3 transcript expression relative to β-actin determined by QPCR and mRNA derived from RMCAs from different rats. Relative transcript levels were determined using the 2−ΔΔCt method. Specfic expression of Kv2.1 and Kv9.3 was confirmed using mRNA from isolated RMCA myocytes. *Significantly different (P < 0.05) from value for Kv2.1.
Properties of ScTx1-sensitive Kv current of RMCA myocytes
Based on the abundance of Kv2.1 and Kv9.3 message expression in RMCAs, we employed standard whole-cell voltage clamp recording methods to test for functional identity of the native ScTx1-sensitive current of freshly isolated myocytes with currents owing to heterologous expression of Kv2.1 or Kv2.1/Kv9.3 channels in HEK 293 cells at 22 and 35°C. The voltage-clamp protocol employed applied 300 ms voltage steps from a holding potential of −75 mV to test potentials between −95 and +25 mV in 10 mV increments followed by repolarization to −45 mV to detect tail current in the absence and presence of 100 nm ScTx1 (Fig. 4A). This concentration of ScTx1 was employed to permit ∼90% inhibition of Kv2.1/Kv9.3 current based on experiments using expressed channels (Fig. 1 in online Supplemental Material; Escoubas et al. 2002). Digital subtraction of residual current in ScTx1 from that in control conditions yielded the ScTx1-sensitive current component at each voltage (right of Fig. 4A). Current density (pA pF−1) versus voltage relations for RMCA Kv current in the absence and presence of ScTx1 (100 nm), as well as the ScTx1-sensitive component, were determined from the end-pulse current amplitude normalized to cell capacitance and plotted as a function of the step voltage (Fig. 4B and C). Slowly deactivating tail currents were apparent on repolarization to −45 mV, and these were significantly reduced by ScTx1 (Fig. 4A). Both the net and residual components of Kv current recorded ± ScTx1 were outwardly rectifying, as was the ScTx1-sensitive component that accounted for ∼60% of total Kv current recorded at +25 mV. Increasing the temperature of the bathing solution to 35°C caused a marked increase in amplitude, as well as the kinetics of activation and deactivation (Fig. 4D). The level of steady-state activation of native current as a function of voltage was determined at 22 and 35°C by normalization of tail current amplitude at −45 mV following steps to between −95 and +45 mV (Fig. 4E). Increasing temperature from 22 to 35°C caused a significant leftward shift in the activation curve from ∼–9 mV to −25 mV (Table 1) and an increase in current in the range of −55 to −25 mV (Fig. 4E).
Figure 4. Native ScTx1-sensitive Kv current of RMCA myocytes.
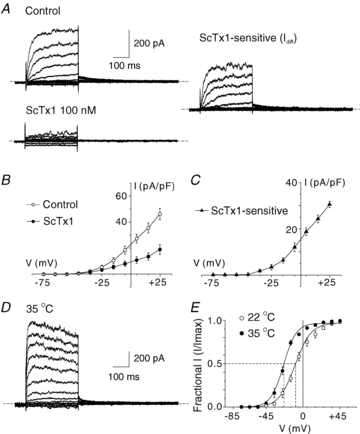
A, representative recordings of whole-cell Kv current of an RMCA myocyte in the absence (Control) and presence of 100 nm ScTx1 (left) and the ScTx1-sensitive current (right) determined by digital subtraction of ScTx1 from Control current at 22°C. Voltage steps of 325 ms duration between −95 and +45 mV in increments of 10 mV prior to repolarization to −45 mV were applied from a holding potential of −75 mV. Similar recordings were obtained from 7 additional myocytes from cell isolations of RMCAs of 3 rats. B and C, mean values ± s.e.m. (n = 6) for net whole-cell and ScTx1-sensitive Kv current I–V relations. D, representative recording of RMCA ScTx1-sensitive Kv current at 35°C. E, mean values ± s.e.m. for normalized tail current amplitude versus command step voltage for native current at 22 and 35°C (n = 6 and 3, respectively) that exhibited complete suppression of tail currents following treatment with ScTx1. Continuous lines represent best fits to the data points using a standard Boltzmann function.
Table 1.
Biophysical properties of native, Kv2.1, Kv2.1/Kv9.2 and Kv2.1/Kv9.3 currents
| Native Kv | Kv2.1/Kv9.3 | Kv2.1 | Kv2.1/Kv9.2 | |
|---|---|---|---|---|
| V0.5 act at 22°C | −8.5 ± 1.9 mV | −12.8 ± 4.6 mV | −4.8 ± 1.1 mV* | −13.3 ± 1.3 mV |
| (n = 6) | (n = 8) | (n = 6) | (n = 6) | |
| V0.5 act at 35°C | −25.2 ± 3 mV | −28.4 ± 1.3 mV | −14.6 ± 3 mV* | n.a. |
| (n = 3) | (n = 5) | (n = 4) | ||
| Deact τfast at +25 mV and 22°C | 82 ± 22 ms | 66 ± 11 ms | 16 ± 1 ms* | 14 ± 0.5 ms* |
| (n = 6) | (n = 8) | (n = 6) | (n = 6) | |
| Deact τslow at +25 mV and 22°C | 442 ± 60 ms | 298 ± 60 ms | 69 ± 18 ms* | 53 ± 3 ms* |
| (n = 6) | (n = 8) | (n = 6) | (n = 6) |
V0.5 act, V0.5 value for half-maximal activation. Deact τ, deactivation time constant.
Significantly different (P ≤ 0.05) from value for native current. All values are based on n cells obtained from three or more rats or cell transfections.
Comparison of recombinant and native ScTx1-sensitive current of RMCA myocytes
Whole-cell currents owing to homomultimeric Kv2.1 and heteromultimeric Kv2.1 and Kv9.3 were recorded under identical conditions as native RMCA myocyte current. Figure 5A and B shows representative families (at 22 and 35°C) and mean current density versus voltage relations for Kv2.1 and Kv2.1/Kv9.3 currents (at 22°C), respectively. Both recombinant channel types were inhibited by 100 nm ScTx1 and 3 mm 4-AP (see Figs 1 and 2 in the online Supplemental Material), as previously reported (Patel et al. 1997; Escoubas et al. 2002). However, the amplitude of Kv2.1/Kv9.3 current within the physiological range of Em was significantly greater than that of Kv2.1 at both temperatures. This is evident in Fig. 5C, which shows that at 35°C, current due to Kv2.1/Kv9.3, but not Kv2.1 was activated during steps from −75 to −55 and −45 mV. Also, the decay of the tail current on repolarization to −55 mV was slower for Kv2.1/Kv9.3 compared to Kv2.1 at both temperatures (Fig. 5A); this is readily apparent in the expanded representative recordings at 22°C shown in Fig. 5D.
Figure 5. Kv2.1 and Kv2.1/Kv9.3 currents in HEK293 cells.
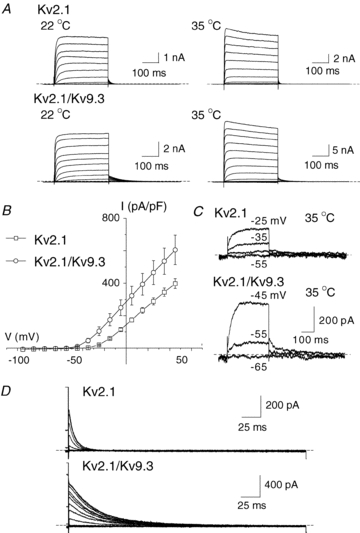
A, representative families of whole-cell currents due to expression of Kv2.1 (upper) and Kv2.1/Kv9.3 (lower) channels in HEK 293 cells recorded at 22 and 35°C (left and right, respectively). Voltage steps of 325 ms duration between −95 and +45 mV in increments of 10 mV prior to repolarization to −55 mV were applied from a holding potential of −75 mV. B, mean values ± s.e.m. (n = 6 and 8, respectively) for current density (pA pF−1) versus voltage relation for Kv2.1 and Kv2.1/Kv9.3 channels in HEK 293 cells at 22°C. C, representative Kv2.1 and Kv2.1/Kv9.3 currents recorded in response to command steps to between −55 and −25 mV and between −65 and −45 mV, respectively. Note the activation of current due to heteromultimeric, but not homomultimeric, channels at −55 mV. D, representative families of expanded Kv2.1 and Kv2.1/Kv9.3 tail currents recorded at −50 mV at 22°C illustrating the significantly slower deactivation kinetics of the heteromultimeric channels.
Figure 6 shows expanded tail currents for ScTx1-sensitive current of RMCA myocytes and recombinant Kv2.1, Kv2.1/Kv9.2 and Kv2.1/Kv9.3 channels recorded at −45 mV following steps to +25 mV (Fig. 6A) and superimposed recordings of each normalized to peak amplitude of native tail current (Fig. 6B). Each recording was best fitted by a bi-exponential function (continuous lines). The decay of the native and Kv2.1/Kv9.3 tail currents were identical and slower than that of Kv2.1. The similarity in rate of deactivation of the native and Kv2.1/Kv9.3 currents versus Kv2.1 was also apparent from the values determined for the fast and slow time constants of the decay (Table 1). For comparison, representative and mean data for deactivation of Kv2.1/Kv9.2 currents are also presented in Fig. 6 and Table 1. Although the voltage dependence of activation of Kv2.1/Kv9.2 current was similar to that of Kv2.1/Kv9.3, the fast and slow time constants of deactivation were significantly smaller and not different from those of Kv2.1 homomultimeric channels, as was previously reported for Kv2.1/Kv9.2 and Kv2.1/Kv9.1 channels expressed in Xenopus oocytes (Salinas et al. 1997b).
Figure 6. Decay of native ScTx1-sensitive Kv current mimics that of Kv2.1/Kv9.3, but not Kv2.1 or Kv2.1/Kv9.2, channels.
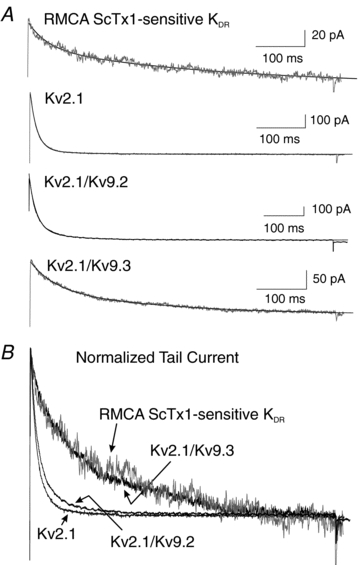
A, expanded representative recordings of tail currents of native RMCA ScTx1-sensitive Kv, Kv2.1, Kv2.1/Kv9.2 and Kv2.1/Kv9.3 channels at −45 mV following steps to +25 mV at 22°C. Continuous lines through each recording represent the best fit using a two-exponential function. B, superimposed tail current recordings from panel A with currents for Kv2.1, Kv2.1/Kv9.2 and Kv2.1/Kv9.3 normalized to value of peak tail current of native channels.
A further comparison between the native and recombinant currents was made through determination of the voltage dependence of activation of Kv2.1 and Kv2.1/Kv9.3 currents at 22 and 35°C. For clarity, Fig. 7A shows that steady-state activation of Kv2.1/9.3 occurred over a more negative voltage range compared to Kv2.1 at 22°C. Mean values ± s.e.m. for half-maximal activation of the native and recombinant currents of several cells/myocytes are indicated in Table 1. Figure 7B and C shows that increasing the temperature to 35°C caused a negative shift in the voltage dependence of activation of Kv2.1 and Kv2.1/Kv9.3 current. When compared to the activation of native current at the same temperatures (indicated by the dashed lines; data are from Fig. 4E), the activation curves for Kv2.1/Kv9.3 at 22 and 35°C, but not those for Kv2.1 homomeric channels, closely mimicked the activation of the native current (Fig. 7 and Table 1).
Figure 7. Voltage dependence of activation of Kv2.1/Kv9.3, but not Kv2.1, channels mimics that of native Kv current.
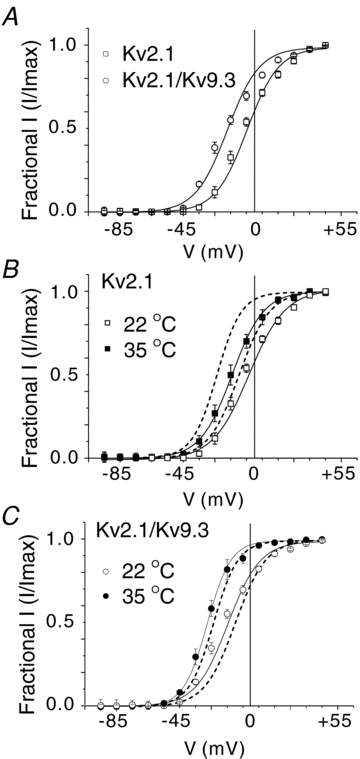
A, mean values ± s.e.m. for normalized tail current amplitude versus command step voltage for Kv2.1 and Kv2.1/Kv9.3 current at 22°C (n = 6 and 8, respectively). Note the negative shift in voltage dependence of Kv2.1/Kv9.3. B, mean values ± s.e.m. for normalized tail current amplitude versus command step voltage for Kv2.1 current at 22 and 35°C (n = 6 and 3, respectively). Dashed lines indicate the relation for activation of native current from Fig. 4E for comparison; note lack of similarity in voltage dependence of native and recombinant channel current activation at both recording temperatures. C, mean values ± s.e.m. for normalized tail current amplitude versus command step voltage for Kv2.1/Kv9.3 current at 22 and 35°C (n = 8 and 5, respectively). Dashed lines indicate the relation for activation of native current from Fig. 4E for comparison; note the similarity in voltage dependence of native and recombinant channel current activation at both recording temperatures.
PLA detection of Kv2.1/Kv9.3 co-localization in RMCA myocytes
We attempted to detect the co-assembly of Kv2.1 and Kv9.3 subunits using a co-immunoprecipitation approach that was previously used for Kv1.2/Kv1.5 channels of rabbit portal vein and rat cerebral arteries (Thorneloe et al. 2001; Albarwani et al. 2003). We detected Kv2.1 in anti-Kv2.1 immunoprecipitates of RMCAs and HEK 293 cells transfected with Kv2.1/Kv9.3 (see Fig. 3 in online Supplemental Material), but the commercially available Kv9.3 antibodies were not of sufficient quality to detect the presence of Kv9.3, even when a high sensitivity three-step immunoblotting technique was used (Johnson et al. 2009b). For this reason, we employed PLA to detect the presence of Kv2.1 and Kv9.3 at the plasma membrane of RMCAs.
To validate the PLA method, we first determined whether the technique could detect the presence of Kv1.2/Kv1.5 and Kv2.1/Kv9.3 channels expressed in HEK 293 cells. Figure 8 shows representative images of Hoechst 33342-stained cells transfected with cDNAs encoding GFP and Kv1.2/Kv1.5 (Fig. 8A) or Kv2.1/Kv9.3 (Fig. 8C) treated with Kv1.2 and Kv1.5 or Kv2.1 and Kv9.3 primary antibodies, respectively, and appropriately matched PLA PLUS and MINUS probes. PLA signals were consistently detected at the plasma membrane of green fluorescent, GFP-positive cells, but not in untransfected GFP-negative cells (100–200 cells for each). In contrast, PLA signals were not detected in 100–200 GFP-positive cells from three transfections using cDNAs for only one subunit of each pair; Fig. 8B and D show cells expressing only Kv1.5 or Kv2.1 and probed for Kv1.2/Kv1.5 and Kv2.1/Kv9.3, respectively (similar results were obtained for Kv1.2 and Kv9.3 cells; data not shown).
Figure 8. PLA detection of plasma membrane expression of recombinant Kv channel protein in HEK 293 cells.
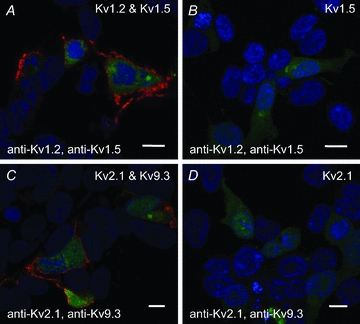
A, PLA reaction product indicated by red fluorescent dots was detected at the periphery of GFP-positive HEK 293 cells transfected with cDNAs encoding GFP, Kv1.2 and Kv1.5, but not in non-transfected GFP-negative cells probed with Kv1.2 and Kv1.5 primary antibodies. Here and in subsequent panels, the nuclei of GFP-positive and -negative cells are indicated by the blue Hoechst 33342 stain, the Kv channel cDNAs and primary antibodies used are indicated in the upper right and lower left corners, respectively, the scale bars are 10 μm in length and each image is an optical section of 0.3–0.5 μm thickness at a mid-cell depth. B, lack of PLA reaction product at cell periphery of GFP-positive cells transfected with Kv1.5 only (i.e. no Kv1.2) and probed with Kv1.2 and Kv1.5 primary antibodies. C, PLA signals were detected at the periphery of GFP-positive cells transfected with Kv2.1 and Kv9.3, but not in GFP-negative cells probed with Kv2.1 and Kv9.3 primary antibodies. D, lack of PLA signals at cell periphery of GFP-positive cells transfected with Kv2.1 (i.e. no Kv9.3) and probed with Kv2.1 and Kv9.3 primary antibodies.
PLA was then employed to detect the presence of Kv1.2/Kv1.5 or Kv2.1/Kv9.3 at the plasma membrane of freshly isolated RMCA myocytes. Figure 9A shows differential interference contrast (DIC) and fluorescence micrographs of a representative myocyte probed for Kv1.2 and Kv1.5 co-localization. PLA signals were detected at the cell periphery of this myocyte and an additional 60 myocytes from three cell isolations using RMCAs of different rats. No PLA signals were detected when the Kv1.5 primary antibody was omitted while applying identical PLA PLUS and MINUS secondary antibodies (Fig. 9B). Figure 9C–F shows representative myocytes from four cell isolations that were probed for Kv2.1 and Kv9.3 co-localization. An identical detection of PLA signals was obtained for 80 additional myocytes, but no reaction product was detected when the Kv9.3 primary antibody was omitted (Fig. 9G and H). These observations are consistent with previous data showing co-assembly of Kv1.2/Kv1.5 and show, for the first time, the co-localization of Kv9.3 and Kv2.1 at the plasma membrane of RMCA myocytes.
Figure 9. PLA detection of plasma membrane expression of Kv channel protein in RMCA myocytes.
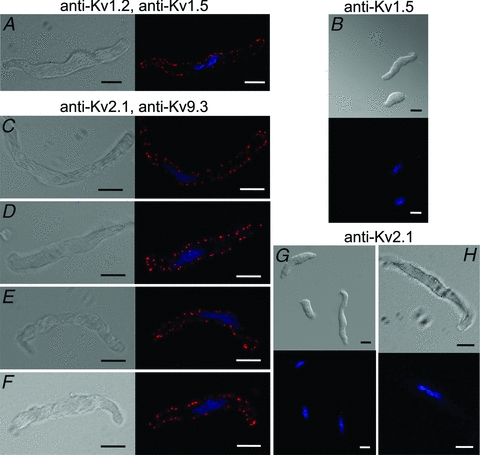
A, DIC (left) and fluorescence (right) micrographs of an RMCA myocyte probed for co-localization of Kv1.2 and Kv1.5 with PLA signals at cell periphery when probed with Kv1.2 and Kv1.5 primary antibodies. Here and in subsequent panels, the nuclei of GFP-positive and -negative cells are indicated by the blue Hoechst 33342 stain, the primary antibodies used are indicated above the panels, the scale bars are 10 μm in length and each image is an optical section of 0.3–0.5 μm thickness at a mid-cell depth. B, lack of PLA signals in two myocytes probed for Kv1.2 and Kv1.5 co-localization when Kv1.2 primary antibody was omitted. C–F, four representative RMCA myocytes exhibiting PLA signals when probed with Kv2.1 and Kv9.3 primary antibodies. G, lack of PLA signals in three myocytes probed for Kv2.1 and Kv9.3 co-localization when Kv9.3 primary antibody was omitted. H, single RMCA myocyte at high magnification showing lack of PLA signals for Kv2.1 and Kv9.3 co-localization when Kv9.3 primary antibody was omitted.
The presence of localized ‘hot spots’ of PLA signal detection at the plasma membrane of RMCA myocytes suggested the possibility of Kv channel targeting to microdomains. It is well-known that Kv1 subunits do not co-assemble with Kv2 or Kv9 (Coetzee et al. 2006); however, the separation of adjacent channels may be reduced by over-expression in a heterologous cell type, or due to targeting to microdomains, such as the reported targeting of Kv channels to lipid rafts (Martens et al. 2001; Xia et al. 2004). If the Kv channels of RMCAs are targeted to microdomains and at a higher local density, the channel-to-channel separation may fall within the range of <40 nm required for hybridization of the PLA probes. In this case, PLA signals would be detected in the absence of co-assembly within a single channel complex. We examined the possibility that Kv1 and Kv2/Kv9.3 channels target to similar microdomains in RMCA myocytes and whether the expression of Kv9.3 at the plasma membrane could be detected with anti-Kv1.5 in addition to anti-Kv2.1. We first studied HEK 293 cells transfected with cDNAs encoding GFP and (1) Kv1.2 and Kv2.1, (2) Kv1.5 and Kv9.3, (3) Kv1.5, Kv9.3 and Kv2.1 and (4) Kv1.5 and Kv2.1. Figure 10A shows that PLA signals were detected in cells expressing Kv1.2 and Kv2.1 using Kv1.2 and Kv2.1 antibodies. This indicates that the commercial PLA probes can detect the co-localization of subunits in adjacent Kv1 and Kv2 channels. In contrast, PLA signals were not detected with anti-Kv1.5 and anti-Kv9.3 using cells expressing Kv1.5 and Kv9.3 alone, a result that is consistent with previous studies showing the lack of membrane expression of homomultimeric Kv9.3 channels (Fig. 10B). However, product was detected in cells with co-incident expression of Kv2.1 with Kv1.5 and Kv9.3 indicating the rescue of Kv9.3 trafficking in the presence of Kv2.1 (Fig. 10C). That the positive PLA reaction in Fig. 10C required the presence of Kv9.3 at the plasma membrane was confirmed by the absence of product in cells expressing homomultimeric Kv1.5 and Kv2.1 channels and probed with the same pair of Kv1.5 and Kv9.3 antibodies (Fig. 10D). Finally, Fig. 10E and F shows that PLA signals were detected in RMCA myocytes probed with Kv1.5 and Kv9.3 or Kv1.2 and Kv2.1 primary antibodies (similar data were obtained for >40 myocytes from three cell isolations in each case). This suggests that Kv1.2/Kv1.5 and Kv2.1/Kv9.3 channels may be trafficked to similar regions of the plasma membrane of RMCA myocytes resulting in a channel–channel separation of <40 nm.
Figure 10. PLA detection of Kv subunit proteins in adjacent channel complexes in HEK 293 cells and RMCA myocytes.
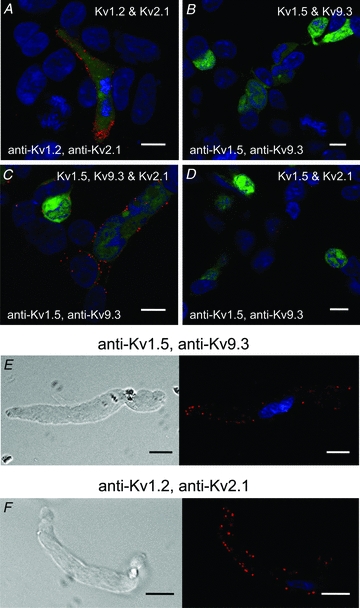
A, PLA signals were detected at the periphery of GFP-positive cells transfected with Kv1.2 and Kv2.1, but not in GFP-negative cells probed with Kv1.2 and Kv2.1 primary antibodies. B, PLA signals were not detected at the periphery of GFP-positive cells transfected with Kv1.5 and Kv9.3 and probed with Kv1.5 and Kv9.3 primary antibodies. C, PLA signals were detected at the periphery of GFP-positive cells transfected with Kv1.5, Kv9.3 and Kv2.1, but not in GFP-negative cells probed with Kv1.5 and Kv9.3 primary antibodies. D, PLA signals were not detected at cell periphery of GFP-positive cells transfected with Kv1.5 and Kv2.1 and probed with Kv1.5 and Kv9.3 primary antibodies. E, representative RMCA myocyte exhibiting PLA signals at cell periphery when probed with Kv1.5 and Kv9.3 primary antibodies. F, representative RMCA myocyte exhibiting PLA signals at cell periphery when probed with Kv1.2 and Kv2.1 primary antibodies.
Discussion
This is the first study to identify the presence of heteromultimeric Kv2.1/Kv9.3 channels in RMCA myocytes and provide evidence of a distinct functional contribution to control of cerebral resistance arterial diameter. Previous studies provided evidence that ScTx1-sensitive, Kv2-containing channels oppose myogenic constriction of cerebral arteries at 80 mmHg and that modulation of their expression via changes in calcineurin and NFATc3 signalling is associated with angiotensin-induced hypertension (Amberg et al. 2004; Amberg & Santana, 2006). Here we show that: (1) transcripts encoding Kv2.1 and Kv9.3 are the predominant mRNAs encoding Kv2 and ‘silent’ subunits expressed in RMCAs, (2) co-localized Kv2.1 and Kv9.3 are present at the plasma membrane of RMCA myocytes, (3) ScTx1-sensitive native Kv and Kv2.1/Kv9.3 channels exhibit functional identity based on deactivation kinetics and voltage dependence of activation that are distinct from those of homomultimeric Kv2.1 channels, and (4) Kv2.1/Kv9.3 currents were detected at all voltages ≥−55 mV at 35°C and ScTx1 induced constriction of RMCAs over the entire range of intra-luminal pressure from 10 to 100 mmHg studied. Taken together, our findings indicate that heteromultimeric Kv2.1/Kv9.3 channels are expressed by RMCA myocytes and contribute to the control of Em and the myogenic response over a wider range of intra-luminal pressure than that previously reported for Kv1, Kv7 and BKCa channels (Brayden & Nelson, 1992; Nelson et al. 1995; Knot & Nelson, 1995; Chen et al. 2006; Yang et al. 2009; Zhong et al. 2010).
Our findings indicate that the predominant Kv2-containing channel type expressed by RMCA myocytes is a heteromultimeric channel containing Kv2.1 and Kv9.3. Homomultimeric Kv2.1 channels may be present in RMCAs, but at a level of expression that does not have a significant influence on the functional properties of the native ScTx1-sensitive Kv current. We attribute the ScTx1-sensitive current of RMCA myocytes to heteromultimeric Kv2.1/Kv9.3 channels for the following reasons.
First, expression of message encoding Kv2.2 and the ‘silent’ subunits, Kv5.1, Kv6.1–6.3, and Kv9.1–9.2 was very low or undetectable in RMCAs and more than 10-fold less than the level of Kv2.1 and Kv9.3 transcripts. Kv5.1 expression was previously reported for RMCAs (Amberg & Santana, 2006), and we also detected Kv5.1 transcripts by RT-PCR. However, the level of expression was very low compared to Kv9.3 when assessed by quantitative real-time PCR.
Second, in this study, we employed the PLA technique to detect co-localization of Kv2.1 and Kv9.3 subunit proteins at the plasma membrane of RMCA myocytes. The commercially available Duolink PLA PLUS and MINUS probes employed in this study consist of synthetic oligonucleotides attached to species-specific secondary antibodies that interact with primary antibodies bound to proteins of interest. If the proteins are separated by <40 nm, the oligonucleotides will hybridize and following ligation are capable of generating a unique concatameric oligonucleotide product extending from one oligonucleotide by rolling circle amplification (Fredriksson et al. 2002). The amplification products are then detected with complementary oligonucleotides conjugated to a fluorophore as individual fluorescent dots at sites of protein co-localization. The necessity for dual recognition of the co-localized proteins in the PLA method has the advantage of minimizing detection errors in immunocytochemical analysis owing to possible non-specific binding of the primary antibodies employed, and the lack of precision in assessing co-localization by comparing the extent of overlap of two fluorescence signals. To validate the method, we employed antibody pairs against Kv1.2 and Kv1.5 or Kv2.1 and Kv9.3, and detected PLA signals in HEK cells expressing Kv1.2/Kv1.5 or Kv2.1/Kv9.3 heteromultimeric, but not homomultimeric, Kv1.5, Kv1.2, Kv2.1 or Kv9.3 channels. Moreover, we also detected co-localization of Kv1.2 and Kv1.5 in RMCA myocytes consistent with the findings of Albarwani et al. (2003) showing co-assembly of Kv1.2 and Kv1.5 by co-immunoprecipitation from protein extracts of rat cerebral arteries.
It is important to recognize that the PLA probes employed here do not exclusively detect subunits co-assembled within single channel complexes. Rather, the Duolink PLUS and MINUS oligonucleotides were of sufficient length to detect proteins within adjacent channel complexes. Specifically, Kv1.5 co-localization with Kv9.3 and Kv1.2 with Kv2.1 was detected at the periphery of HEK 293 cells and RMCA myocytes although these subunit pairs do not co-assemble (Coetzee et al. 2006). Nevertheless, this caveat is not a limitation in the present study, as Kv9.3 is not expressed at the membrane unless assembled in a heteromultimeric complex with Kv2 subunits (Salinas et al. 1997a,b; Shepard & Rae, 1999; Ottschytsch et al. 2002; and confirmed here by the lack of PLA signals for Kv1.5 and Kv9.3 in the absence of Kv2.1 expression). The PLA signals detected at the periphery of RMCA myocytes using Kv9.3 and Kv2.1 or Kv9.3 and Kv1.5 antibody pairs indicates the presence of Kv9.3 trafficking to the plasma membrane and, therefore, the expression of heteromultimeric Kv2.1/Kv9.3 channels.
Third, the ScTx1-sensitive current of RMCA myocytes is a delayed rectifier K+ current that exhibits functional identity with currents owing to heteromultimeric Kv2.1/Kv9.3, but not homomultimeric Kv2.1 channels expressed in HEK 293 cells. ScTx1 is known to suppress Kv currents due to Kv4 channels, as well as homomultimeric and heteromultimeric Kv2 channels (Escoubas et al. 2002). We do not attribute the effect of ScTx1 to an inhibition of Kv4 channels based on the absence of a rapid, transient component of outward Kv current (here and previous studies: e.g. Albarwani et al. 2003; Amberg & Santana, 2006; Luykenaar et al. 2009). On the other hand, the fast and slow time constants for decay of tail current amplitude were similar for native ScTx1-sensitive and Kv2.1/Kv9.3 channels, and significantly greater than the values for Kv2.1 channels under identical conditions. Moreover, the voltage dependence of native current activation, as indicated by the V0.5 value for half-maximal activation at 22 and 35°C, was similar to that of Kv2.1/Kv9.3, but not Kv2.1 channels. The similarities in rate of tail current decay and voltage dependence of activation are consistent with the view that the native ScTx1-sensitive current of RMCA myocytes is due to the expression of heteromultimeric Kv2.1/Kv9.3, but not homomultimeric Kv2.1 channels. Moreover, these characteristics also exclude any contribution of heteromultimeric channels containing Kv2.1 with Kv9.1, Kv9.2, Kv5 or Kv6 subunits. Salinas et al. (1997b) previously showed that voltage dependence of activation was left-shifted by ∼–10 mV, but there was little/no difference in rate of deactivation of Kv2.1/Kv9.1 and Kv2.1/9.2 compared to Kv2.1 channels expressed in Xenopus oocytes and confirmed here for Kv2.1/Kv9.2 expressed in mammalian HEK293 cells. It is also unlikely that Kv5 or Kv6 subunits are involved, as the voltage dependence of activation of Kv2.1/Kv5.1 channels is positive to that of Kv2.1 channels (Kramer et al. 1998) and Kv6 subunits do not exhibit as large a shift in activation or slowing of deactivation as Kv9.3, and Kv6.3 reduces Kv2.1 current amplitude (Post et al. 1996; Zhu et al. 1999; Sano et al. 2002; Thorneloe & Nelson, 2003; Bocksteins et al. 2009; Moreno-Dominguez et al. 2009).
Fourth, the vasoconstriction induced by ScTx1 between 10 and 40 mmHg is consistent with a role for Kv2.1/Kv9.3, but not Kv2.1 channels in control of RMCA diameter. We do not attribute the actions of ScTx1 to the block of channels in the endothelium, as denudation did not affect the response to ScTx1. Also, it is unlikely that an ScTx1-induced release of vasoactive neurotransmitter(s) was involved, as sustained constrictions of identical amplitude were observed during repeated applications of the toxin. Rather, we attribute the vasoconstriction evoked by ScTx1 at <40 mmHg to the inhibition of Kv2.1/Kv9.3 channels leading to depolarization and Ca2+ influx via VGCCs (T-type and/or L-type VGCCs; Kuo et al. 2010). This view is based on (1) the demonstrated activity of Kv2.1/Kv9.3 channels at voltages consistent with the level of Em between 10 and 40 mmHg (Knot & Nelson, 1995, 1998), and (2) the reversal of the ScTx1-induced constriction by diltiazem, nifedipine or mebifradil, but not SKF96365. Although we did not determine the effect of ScTx1 on membrane potential of RMCAs between 10 and 40 mmHg, previous studies of rat and rabbit cerebral arteries indicate that Em in this range of pressure is ≤−50 mV (Knot & Nelson, 1995, 1998). This range of Em is negative to the activation voltage for homomultimeric Kv2 channels, but within the range associated with steady-state activity of Kv2.1/Kv9.3 channels. As shown here and in previous studies (e.g. Patel et al. 1997), Kv2.1/Kv9.3 activate over a more negative range of voltage than Kv2.1 channels; substantial Kv2.1/Kv9.3, but not Kv2.1, current activation was detected at −55 mV at a near physiological temperature of 35°C. Also, the membrane potential of COS cells co-expressing Kv2.1 and Kv9.3 was −50.6 ± 0.9 versus−30.7 ± 1.3 mV when transfected with Kv2.1 alone (Patel et al. 1997). For this reason, inhibition of Kv2.1/Kv9.3, but not Kv2.1, channels can account for ScTx1-evoked constriction of RMCAs at 10–40 mmHg. In the absence of pharmacological compounds that selectively interact with Kv9.3, future experiments will need to employ a molecular approach involving subunit-specific knock-down with siRNAs or antisense oligonucleotides to provide the necessary specific suppression of Kv9.3 required to determine the role of this ‘silent’ subunit in control of RMCA diameter.
The mechanisms responsible for the arterial myogenic response are not fully understood. The current view holds that increased pressure elicits Em depolarization attributable to the activation of NSCCs, chloride channels and/or VGCCs, with the amplitude of current increasing proportionally with greater pressure elevation (Nelson & Quayle, 1995; Davis & Hill, 1999). Very precise control over the extent of myogenic depolarization is necessary to permit the low amplitude, steady-state changes in Em and diameter that are essential for physiological regulation of blood pressure and blood flow (Nelson & Quayle, 1995; Nelson et al. 1995; Davis & Hill, 1999). There is considerable evidence that pressure-dependent activation of VSMC K+ channels permits negative-feedback regulation of myogenic depolarization and diameter. For example, previous studies provide pharmacological and molecular evidence that Kv1 and BKCa channels are activated by myogenic depolarization and increased Ca2+ sparks/Ca2+-influx, respectively, to oppose the change in Em, and prevent action potential initiation and oscillatory vasomotion (Brayden & Nelson, 1992; Nelson et al. 1995; Knot & Nelson, 1995; Guia et al. 1999; Brenner et al. 2000; Amberg & Santana, 2006; Albarwani et al. 2003; Plane et al. 2005; Chen et al. 2006; Yang et al. 2009). More recent data indicate that Kv7 channels also contribute to the control of the myogenic response of RMCAs (Zhong et al. 2010). These observations combined with the present findings and those of Amberg & Santana (2006) concerning Kv2-containing channels indicate that at least four different K+ channels expressed in cerebral arteries contribute to control of the myogenic response. The specific reasons why four different channels are required are not evident. However, it is reasonable to propose that multiple channels with different functional properties are required to integrate the complex array of intrinsic (Em, cytosolic Ca2+ concentration, and intracellular modulators such as kinases) and extrinsic (endothelial, neuronal, parechymal and metabolic) signals that determine the extent of myogenic depolarization and constriction under varied physiological conditions. For example, the different mechanisms of activation of Kv1 and BKCa are clearly able to provide for distinct modes of control of Em via changes in Em or cytosolic Ca2+, respectively. Our findings suggest that multiple Kv channel subtypes with differences in voltage dependence of activation may be required to permit graded, voltage-dependent control of Em over the entire pressure range. Previous studies indicated that suppression of Kv1 current enhanced myogenic constriction of cerebral arteries in a pressure-dependent manner above 40 mmHg and an Em positive to ∼−50 mV (Knot & Nelson, 1995, 1998; Chen et al. 2006). Here, we show that inhibition of Kv2.1/Kv9.3 channels with ScTx1 enhanced the myogenic response at ≥40 mmHg, but the toxin also caused vasoconstriction between 10 to 40 mmHg. This suggests that Kv2.1/Kv9.3 channels contribute to the control of RMCA diameter over a wider range of Em and pressure than Kv1 and, possibly, play a predominant role in determining the level of Em over the lower physiological range of 40–60 mmHg. Kv2.1/Kv9.3 current may also be required to maintain voltage-dependent negative-feedback control of myogenic depolarization when Em is hyperpolarized below the activation range of Kv1 and BKCa channels by vasodilators that increase voltage-independent ATP-sensitive or inwardly rectifying K+ current (Nelson & Quayle, 1995). Future studies are necessary to address these important issues and to clarify the specific roles and importance of each K+ conductance in the myogenic regulation of arterial diameter in health and disease.
Acknowledgments
This work was supported by a grant from the Canadian Institutes of Health Research (CIHR; MOP-13505). W.C.C. is the Andrew Family Professor in Cardiovascular Research and M.P.W. is an Alberta Heritage Foundation for Medical Research (AHFMR) Scientist and recipient of a Canada Research Chair (Tier 1) in Vascular Smooth Muscle Research. X.Z.Z. and K.A.R. were supported by salary funds from the Andrew Family Professorship held by W.C.C. and the Kertland Family Fund in support of The Smooth Muscle Research Group, respectively. C.H.L. and A.F.E. were supported by AHFMR and NSERC Studentships and AHFMR and CIHR Fellowships, respectively. The authors thank Dr. R. Turner for access to the Zeiss ApoTome epifluorescence microscope (funded by AHFMR and CIHR) and Mirna Kruskic for her expert technical assistance.
Glossary
Abbreviations
- NSCC
non-selective cation channel
- PLA
proximity ligation assay
- RMCA
rat middle cerebral artery
- ScTx1
stromatoxin
- VGCC
voltage-gated Ca2+ channel
- VSMC
vascular smooth muscle cell
Author contributions
X.Z.Z. and W.C.C. were responsible for the conception and design of all experiments and wrote the manuscript. X.Z.Z. conducted all electrophysiological analyses with the exception of the 4-AP and Kv2.1/Kv9.2 experiments conducted by K.A.R. X.Z.Z. and E.J.W. performed pressure myograph experiments, X.Z.Z. and C.H.L. conducted the PLA experiments, X.Z.Z. and A.F.Y. completed immunoblotting and co-immunoprecipitation experiments, E.J.W. maintained all cell cultures, performed cell transfections, and provided all plasmids. X.Z.Z. and E.J.W. completed the RT-PCR and real-time PCR experiments, and M.P.W. contributed to the design and interpretation of the PLA experiments. All authors have approved the submitted version of this manuscript. The work was and conducted in the laboratory of W.C.C. in The Smooth Muscle Research Group at the University of Calgary. The authors have no disclosures.
References
- Albarwani S, Nemetz LT, Madden JA, Tobin AA, England SK, Pratt PF, Rusch NJ. Voltage-gated K+ channels in rat small cerebral arteries: molecular identity of the functional channels. J Physiol. 2003;551:751–763. doi: 10.1113/jphysiol.2003.040014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amberg GC, Rossow CF, Navedo MF, Santana LF. NFATc3 regulates Kv2.1 expression in arterial smooth muscle. J Biol Chem. 2004;279:47326–47334. doi: 10.1074/jbc.M408789200. [DOI] [PubMed] [Google Scholar]
- Amberg GC, Santana LF. Kv2 channels oppose myogenic constriction of rat cerebral arteries. Am J Physiol Cell Physiol. 2006;291:C348–C356. doi: 10.1152/ajpcell.00086.2006. [DOI] [PubMed] [Google Scholar]
- Belevych AE, Beck R, Tammaro P, Poston L, Smirnov SV. Developmental changes in the functional characteristics and expression of voltage-gated K+ channel currents in rat aortic myocytes. Cardiovasc Res. 2002;54:152–161. doi: 10.1016/s0008-6363(02)00227-4. [DOI] [PubMed] [Google Scholar]
- Bocksteins E, Labro AJ, Mayeur E, Bruyns T, Timmermans JP, Adriaensen D, Snyders DJ. Conserved negative charges in the N-terminal tetramerization domain mediate efficient assembly of Kv2.1 and Kv2.1/Kv6.4 channels. J Biol Chem. 2009;284:31625–31634. doi: 10.1074/jbc.M109.039479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brayden JE, Nelson MT. Regulation of arterial tone by activation of calcium-dependent potassium channels. Science. 1992;256:532–535. doi: 10.1126/science.1373909. [DOI] [PubMed] [Google Scholar]
- Brenner R, Perez GJ, Bonev AD, Eckman DM, Kosek JC, Wiler SW, Patterson AJ, Nelson MT, Aldrich RW. Vasoregulation by the β1 subunit of the calcium-activated potassium channel. Nature. 2000;407:870–876. doi: 10.1038/35038011. [DOI] [PubMed] [Google Scholar]
- Chen M, Kellett WF, Petkov GV. Voltage-gated K+ channels sensitive to stromatoxin-1 regulate myogenic and neurogenic contractions of rat urinary bladder smooth muscle. Am J Physiol Regul Integr Comp Physiol. 2010;299:R177–R184. doi: 10.1152/ajpregu.00036.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen TT, Luykenaar KD, Walsh EJ, Walsh MP, Cole WC. Key role of Kv1 channels in vasoregulation. Circ Res. 2006;99:53–60. doi: 10.1161/01.RES.0000229654.45090.57. [DOI] [PubMed] [Google Scholar]
- Clément-Chomienne O, Ishii K, Walsh MP, Cole WC. Identification, cloning and expression of rabbit vascular smooth muscle Kv1.5 and comparison with native delayed rectifier K+ current. J Physiol. 1999;515:653–667. doi: 10.1111/j.1469-7793.1999.653ab.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppock EA, Tamkun MM. Differential expression of KV channel α- and β-subunits in the bovine pulmonary arterial circulation. Am J Physiol Lung Cell Mol Physiol. 2001;281:L1350–L1360. doi: 10.1152/ajplung.2001.281.6.L1350. [DOI] [PubMed] [Google Scholar]
- Coetzee WA, Amarillo Y, Chiu J, Chow A, Lau D, McCormack T, Moreno H, Nadal MS, Ozaita A, Pountney D, Saganich M, Vega-Saenz de Miera E, Rudy B. Molecular diversity of K+ channels. Ann NY Acad Sci. 2006;868:233–255. doi: 10.1111/j.1749-6632.1999.tb11293.x. [DOI] [PubMed] [Google Scholar]
- Cox DH, Aldrich RW. Role of the β1 subunit in large-conductance Ca2+-activated K+ channel gating energetics. Mechanisms of enhanced Ca2+ sensitivity. J Gen Physiol. 2000;116:411–432. doi: 10.1085/jgp.116.3.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox RH, Fromme SJ, Folander KL, Swanson RJ. Voltage gated K+ channel expression in arteries of Wistar-Kyoto and spontaneously hypertensive rats. Am J Hypertens. 2008;21:213–218. doi: 10.1038/ajh.2007.44. [DOI] [PubMed] [Google Scholar]
- Davies AR, Kozlowski RZ. Kv channel subunit expression in rat pulmonary arteries. Lung. 2001;179:147–161. doi: 10.1007/s004080000057. [DOI] [PubMed] [Google Scholar]
- Davis MJ, Hill MA. Signaling mechanisms underlying the vascular myogenic response. Physiol Rev. 1999;79:387–423. doi: 10.1152/physrev.1999.79.2.387. [DOI] [PubMed] [Google Scholar]
- Drewe JA, Verma S, Frech G, Joho RH. Distinct spatial and temporal expression patterns of K+ channel mRNAs from different subfamilies. J Neurosci. 1992;12:538–548. doi: 10.1523/JNEUROSCI.12-02-00538.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escoubas P, Diochot S, Celerier ML, Nakajima T, Lazdunski M. Novel tarantula toxins for subtypes of voltage-dependent potassium channels in the Kv2 and Kv4 subfamilies. Mol Pharmacol. 2002;62:48–57. doi: 10.1124/mol.62.1.48. [DOI] [PubMed] [Google Scholar]
- Fredriksson S, Gullberg M, Jarvius J, Olsson C, Pietras K, Gustafsdottir SM, Ostman A, Landegren U. Protein detection using proximity-dependent DNA ligation assays. Nat Biotechnol. 2002;20:473–477. doi: 10.1038/nbt0502-473. [DOI] [PubMed] [Google Scholar]
- Frey BW, Lynch FT, Kinsella JM, Horowitz B, Sanders KM, Carl A. Blocking of cloned and native delayed rectifier K channels from visceral smooth muscles by phencyclidine. Neurogastroenterol Motil. 2000;12:509–516. doi: 10.1046/j.1365-2982.2000.00225.x. [DOI] [PubMed] [Google Scholar]
- Greenwood IA, Ohya S. New tricks for old dogs: KCNQ expression and role in smooth muscle. Br J Pharmacol. 2009;156:1196–1203. doi: 10.1111/j.1476-5381.2009.00131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guia A, Wan X, Courtemanche M, Leblanc N. Local Ca2+ entry through L-type Ca2+ channels activates Ca2+-dependent K+ channels in rabbit coronary myocytes. Circ Res. 1999;84:1032–1042. doi: 10.1161/01.res.84.9.1032. [DOI] [PubMed] [Google Scholar]
- Hugnot JP, Salinas M, Lesage F, Guillemare E, de Weille J, Heurteaux C, Mattei MG, Lazdunski M. Kv8.1, a new neuronal potassium channel subunit with specific inhibitory properties towards Shab and Shaw channels. EMBO J. 1996;15:3322–3331. [PMC free article] [PubMed] [Google Scholar]
- Jackson WF. Potassium channels in the peripheral microcirculation. Microcirculation. 2005;12:113–127. doi: 10.1080/10739680590896072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahromi BS, Aihara Y, Ai J, Zhang ZD, Nikitina E, Macdonald RL. Voltage-gated K+ channel dysfunction in myocytes from a dog model of subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2008a;28:797–811. doi: 10.1038/sj.jcbfm.9600577. [DOI] [PubMed] [Google Scholar]
- Jahromi BS, Aihara Y, Ai J, Zhang ZD, Weyer G, Nikitina E, Yassari R, Houamed KM, Macdonald RL. Preserved BK channel function in vasospastic myocytes from a dog model of subarachnoid hemorrhage. J Vasc Res. 2008b;45:402–415. doi: 10.1159/000124864. [DOI] [PubMed] [Google Scholar]
- Jahromi BS, Aihara Y, Ai J, Zhang ZD, Weyer G, Nikitina E, Yassari R, Houamed KM, Macdonald RL. Temporal profile of potassium channel dysfunction in cerebrovascular smooth muscle after experimental subarachnoid haemorrhage. Neurosci Lett. 2008c;440:81–86. doi: 10.1016/j.neulet.2008.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RP, El-Yazbi AF, Hughes MF, Schriemer DC, Walsh EJ, Walsh MP, Cole WC. Identification and functional characterization of protein kinase A-catalyzed phosphorylation of potassium channel Kv1.2 at serine 449. J Biol Chem. 2009a;284:16562–16574. doi: 10.1074/jbc.M109.010918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RP, El-Yazbi AF, Takeya K, Walsh EJ, Walsh MP, Cole WC. Ca2+ sensitization via phosphorylation of myosin phosphatase targeting subunit at threonine-855 by Rho kinase contributes to the arterial myogenic response. J Physiol. 2009b;587:2537–2553. doi: 10.1113/jphysiol.2008.168252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerschensteiner D, Stocker M. Heteromeric assembly of Kv2.1 with Kv9.3: effect on the state dependence of inactivation. Biophys J. 1999;77:248–257. doi: 10.1016/S0006-3495(99)76886-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerschensteiner D, Monje F, Stocker M. Structural determinants of the regulation of the voltage-gated potassium channel Kv2.1 by the modulatory α-subunit Kv9.3. J Biol Chem. 2003;278:18154–18161. doi: 10.1074/jbc.M213117200. [DOI] [PubMed] [Google Scholar]
- Kerschensteiner D, Soto F, Stocker M. Fluorescence measurements reveal stoichiometry of K+ channels formed by modulatory and delayed rectifier α-subunits. Proc Natl Acad Sci U S A. 2005;102:6160–6165. doi: 10.1073/pnas.0500468102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knot HJ, Nelson MT. Regulation of membrane potential and diameter by voltage-dependent K+ channels in rabbit myogenic cerebral arteries. Am J Physiol Heart Circ Physiol. 1995;269:H348–H355. doi: 10.1152/ajpheart.1995.269.1.H348. [DOI] [PubMed] [Google Scholar]
- Knot HJ, Nelson MT. Regulation of arterial diameter and wall [Ca2+] in cerebral arteries of rat by membrane potential and intravascular pressure. J Physiol. 1998;508:199–209. doi: 10.1111/j.1469-7793.1998.199br.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer JW, Post MA, Brown AM, Kirsch GE. Modulation of potassium channel gating by coexpression of Kv2.1 with regulatory Kv5.1 or Kv6.1 α-subunits. Am J Physiol Cell Physiol. 1998;274:C1501–C1510. doi: 10.1152/ajpcell.1998.274.6.C1501. [DOI] [PubMed] [Google Scholar]
- Kuo IY, Ellis A, Seymour VA, Sandow SL, Hill CE. Dihydropyridine-insensitive calcium currents contribute to function of small cerebral arteries. J Cereb Blood Flow Metab. 2010;30:1226–1239. doi: 10.1038/jcbfm.2010.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔC(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Luykenaar KD, El-Rahman RA, Walsh MP, Welsh DG. Rho-kinase-mediated suppression of KDR current in cerebral arteries requires an intact actin cytoskeleton. Am J Physiol Heart Circ Physiol. 2009;296:H917–H926. doi: 10.1152/ajpheart.01206.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malysz J, Farrugia G, Ou Y, Szurszewski JH, Nehra A, Gibbons SJ. The Kv2.2 α subunit contributes to delayed rectifier K+ currents in myocytes from rabbit corpus cavernosum. J Androl. 2002;23:899–910. [PubMed] [Google Scholar]
- Martens JR, Sakamoto N, Sullivan SA, Grobaski TD, Tamkun MM. Isoform-specific localization of voltage-gated K+ channels to distinct lipid raft populations. Targeting of Kv1.5 to caveolae. J Biol Chem. 2001;276:8409–8414. doi: 10.1074/jbc.M009948200. [DOI] [PubMed] [Google Scholar]
- Moreno-Dominguez A, Cidad P, Miguel-Velado E, Lopez-Lopez JR, Perez-Garcia MT. De novo expression of Kv6.3 contributes to changes in vascular smooth muscle cell excitability in a hypertensive mice strain. J Physiol. 2009;587:625–640. doi: 10.1113/jphysiol.2008.165217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson MT, Cheng H, Rubart M, Santana LF, Bonev AD, Knot HJ, Lederer WJ. Relaxation of arterial smooth muscle by calcium sparks. Science. 1995;270:633–637. doi: 10.1126/science.270.5236.633. [DOI] [PubMed] [Google Scholar]
- Nelson MT, Quayle JM. Physiological roles and properties of potassium channels in arterial smooth muscle. Am J Physiol Cell Physiol. 1995;268:C799–C822. doi: 10.1152/ajpcell.1995.268.4.C799. [DOI] [PubMed] [Google Scholar]
- Ottschytsch N, Raes A, Van Hoorick D, Snyders DJ. Obligatory heterotetramerization of three previously uncharacterized Kv channel α-subunits identified in the human genome. Proc Natl Acad Sci U S A. 2002;99:7986–7991. doi: 10.1073/pnas.122617999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AJ, Lazdunski M, Honore E. Kv2.1/Kv9.3, a novel ATP-dependent delayed-rectifier K+ channel in oxygen-sensitive pulmonary artery myocytes. EMBO J. 1997;16:6615–6625. doi: 10.1093/emboj/16.22.6615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plane F, Johnson R, Kerr P, Wiehler W, Thorneloe K, Ishii K, Chen T, Cole W. Heteromultimeric Kv1 channels contribute to myogenic control of arterial diameter. Circ Res. 2005;96:216–224. doi: 10.1161/01.RES.0000154070.06421.25. [DOI] [PubMed] [Google Scholar]
- Platoshyn O, Yu Y, Golovina VA, McDaniel SS, Krick S, Li L, Wang JY, Rubin LJ, Yuan JX. Chronic hypoxia decreases KV channel expression and function in pulmonary artery myocytes. Am J Physiol Lung Cell Mol Physiol. 2001;280:L801–L812. doi: 10.1152/ajplung.2001.280.4.L801. [DOI] [PubMed] [Google Scholar]
- Post MA, Kirsch GE, Brown AM. Kv2.1 and electrically silent Kv6.1 potassium channel subunits combine and express a novel current. FEBS Lett. 1996;399:177–182. doi: 10.1016/s0014-5793(96)01316-6. [DOI] [PubMed] [Google Scholar]
- Salinas M, de Weille J, Guillemare E, Lazdunski M, Hugnot JP. Modes of regulation of shab K+ channel activity by the Kv8.1 subunit. J Biol Chem. 1997a;272:8774–8780. doi: 10.1074/jbc.272.13.8774. [DOI] [PubMed] [Google Scholar]
- Salinas M, Duprat F, Heurteaux C, Hugnot JP, Lazdunski M. New modulatory α subunits for mammalian Shab K+ channels. J Biol Chem. 1997b;272:24371–24379. doi: 10.1074/jbc.272.39.24371. [DOI] [PubMed] [Google Scholar]
- Sano Y, Mochizuki S, Miyake A, Kitada C, Inamura K, Yokoi H, Nozawa K, Matsushime H, Furuichi K. Molecular cloning and characterization of Kv6.3, a novel modulatory subunit for voltage-gated K+ channel Kv2.1. FEBS Lett. 2002;512:230–234. doi: 10.1016/s0014-5793(02)02267-6. [DOI] [PubMed] [Google Scholar]
- Schmalz F, Kinsella J, Koh SD, Vogalis F, Schneider A, Flynn ER, Kenyon JL, Horowitz B. Molecular identification of a component of delayed rectifier current in gastrointestinal smooth muscles. Am J Physiol Gastrointest Liver Physiol. 1998;274:G901–G911. doi: 10.1152/ajpgi.1998.274.5.G901. [DOI] [PubMed] [Google Scholar]
- Shepard AR, Rae JL. Electrically silent potassium channel subunits from human lens epithelium. Am J Physiol Cell Physiol. 1999;277:C412–C424. doi: 10.1152/ajpcell.1999.277.3.C412. [DOI] [PubMed] [Google Scholar]
- Smirnov SV, Beck R, Tammaro P, Ishii T, Aaronson PI. Electrophysiologically distinct smooth muscle cell subtypes in rat conduit and resistance pulmonary arteries. J Physiol. 2002;538:867–878. doi: 10.1113/jphysiol.2001.013003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söderberg O, Gullberg M, Jarvius M, Ridderstrale K, Leuchowius KJ, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson LG, Landegren U. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods. 2006;3:995–1000. doi: 10.1038/nmeth947. [DOI] [PubMed] [Google Scholar]
- Swartz KJ. Tarantula toxins interacting with voltage sensors in potassium channels. Toxicon. 2007;49:213–230. doi: 10.1016/j.toxicon.2006.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorne GD, Conforti L, Paul RJ. Hypoxic vasorelaxation inhibition by organ culture correlates with loss of Kv channels but not Ca2+ channels. Am J Physiol Heart Circ Physiol. 2002;283:H247–H253. doi: 10.1152/ajpheart.00569.2001. [DOI] [PubMed] [Google Scholar]
- Thorneloe KS, Chen TT, Kerr PM, Grier EF, Horowitz B, Cole WC, Walsh MP. Molecular composition of 4-aminopyridine-sensitive voltage-gated K+ channels of vascular smooth muscle. Circ Res. 2001;89:1030–1037. doi: 10.1161/hh2301.100817. [DOI] [PubMed] [Google Scholar]
- Thorneloe KS, Nelson MT. Properties and molecular basis of the mouse urinary bladder voltage-gated K+ current. J Physiol. 2003;549:65–74. doi: 10.1113/jphysiol.2003.039859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wareing M, Bai X, Seghier F, Turner CM, Greenwood SL, Baker PN, Taggart MJ, Fyfe GK. Expression and function of potassium channels in the human placental vasculature. Am J Physiol Regul Integr Comp Physiol. 2006;291:R437–R446. doi: 10.1152/ajpregu.00040.2006. [DOI] [PubMed] [Google Scholar]
- Wei AD, Gutman GA, Aldrich R, Chandy KG, Grissmer S, Wulff H. International Union of Pharmacology. LII. Nomenclature and molecular relationships of calcium-activated potassium channels. Pharmacol Rev. 2005;57:463–472. doi: 10.1124/pr.57.4.9. [DOI] [PubMed] [Google Scholar]
- Wu C, Hayama E, Imamura S, Matsuoka R, Nakanishi T. Developmental changes in the expression of voltage-gated potassium channels in the ductus arteriosus of the fetal rat. Heart Vessels. 2007;22:34–40. doi: 10.1007/s00380-006-0926-0. [DOI] [PubMed] [Google Scholar]
- Xia F, Gao X, Kwan E, Lam PP, Chan L, Sy K, Sheu L, Wheeler MB, Gaisano HY, Tsushima RG. Disruption of pancreatic β-cell lipid rafts modifies Kv2.1 channel gating and insulin exocytosis. J Biol Chem. 2004;279:24685–24691. doi: 10.1074/jbc.M314314200. [DOI] [PubMed] [Google Scholar]
- Yang Y, Murphy TV, Ella SR, Grayson TH, Haddock R, Hwang YT, Braun AP, Peichun G, Korthuis RJ, Davis MJ, Hill MA. Heterogeneity in function of small artery smooth muscle BKCa: involvement of the beta1-subunit. J Physiol. 2009;587:3025–3044. doi: 10.1113/jphysiol.2009.169920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan JX, Aldinger AM, Juhaszova M, Wang J, Conte JV, Jr, Gaine SP, Orens JB, Rubin LJ. Dysfunctional voltage-gated K+ channels in pulmonary artery smooth muscle cells of patients with primary pulmonary hypertension. Circulation. 1998;98:1400–1406. doi: 10.1161/01.cir.98.14.1400. [DOI] [PubMed] [Google Scholar]
- Zhong XZ, Harhun MI, Olesen SP, Ohya S, Moffatt JD, Cole WC, Greenwood IA. Participation of KCNQ (Kv7) potassium channels in myogenic control of cerebral arterial diameter. J Physiol. 2010;588:3277–3293. doi: 10.1113/jphysiol.2010.192823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu XR, Netzer R, Bohlke K, Liu Q, Pongs O. Structural and functional characterization of Kv6.2 a new γ-subunit of voltage-gated potassium channel. Receptors Channels. 1999;6:337–350. [PubMed] [Google Scholar]