

Abstract
The title molecule, C14H11N3O4, is approximately planar, with an interplanar angle between the two benzene rings of 5.8 (2)°. In the crystal, four molecules are linked by an R
4
4(12) motif with pairs of strong O—H⋯O and N—H⋯O hydrogen bonds. The motif is situated about the crystallographic centres of symmetry and it is composed of two pairs of parallel molecules. This quadruplet of molecules is further extended by symmetry-equivalent hydrogen bonds to form layers parallel to the (10 ) plane. In addition to the hydrogen bonds, there is also a weak π–π interaction between the benzene rings.
) plane. In addition to the hydrogen bonds, there is also a weak π–π interaction between the benzene rings.
Related literature
For medical applications of hydrazones, see: Ajani et al. (2010 ▶); Angelusiu et al. (2010 ▶); Zhang et al. (2010 ▶). For related structures, see: Ahmad et al. (2010 ▶); Huang & Wu (2010 ▶); Ji & Lu (2010 ▶); Khaledi et al. (2010 ▶); Singh & Singh (2010 ▶); Zhou & Yang (2010 ▶). For background to hydrogen bonds, see: Desiraju & Steiner (1999 ▶). For hydrogen-bonding motifs, see: Etter et al. (1990 ▶). PLATON (Spek, 2009 ▶) was used to analyse the π–π interactions.
Experimental
Crystal data
C14H11N3O4
M r = 285.26
Monoclinic,
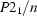
a = 9.987 (3) Å
b = 8.967 (3) Å
c = 15.108 (4) Å
β = 106.560 (3)°
V = 1296.8 (6) Å3
Z = 4
Mo Kα radiation
μ = 0.11 mm−1
T = 298 K
0.13 × 0.10 × 0.10 mm
Data collection
Bruker SMART 1000 CCD diffractometer
Absorption correction: multi-scan (SADABS; Bruker, 2001 ▶) T min = 0.986, T max = 0.989
6579 measured reflections
2768 independent reflections
1787 reflections with I > 2σ(I)
R int = 0.028
Refinement
R[F 2 > 2σ(F 2)] = 0.044
wR(F 2) = 0.119
S = 1.03
2768 reflections
197 parameters
H atoms treated by a mixture of independent and constrained refinement
Δρmax = 0.21 e Å−3
Δρmin = −0.17 e Å−3
Data collection: SMART (Bruker, 2007 ▶); cell refinement: SAINT (Bruker, 2007 ▶); data reduction: SAINT; program(s) used to solve structure: SHELXTL (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXTL; molecular graphics: SHELXTL; software used to prepare material for publication: SHELXTL.
Supplementary Material
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536810043564/fb2226sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536810043564/fb2226Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N2—H2⋯O1i | 0.91 (2) | 2.11 (2) | 2.931 (2) | 149.3 (16) |
| O1—H1⋯O2ii | 0.85 (2) | 1.82 (2) | 2.6573 (17) | 167 (2) |
Symmetry codes: (i)  ; (ii)
; (ii)  .
.
Table 2. Overview of π–π ring interactions in the structure.
Cg1 and Cg2 are the centroids of the C1–C6 and C9–C14 benzene rings, respectively.
| Centroid–centroid | Distance (Å) | Symmetry code |
|---|---|---|
| Cg1–Cg2 | 3.6803 (16) | 1 − x, −y, 1 − z |
Acknowledgments
The authors acknowledge the Jiangsu Provincial Key Laboratory of Coastal Wetland Bioresources and Environmental Protection for open financial support (project No. JLCBE07026).
supplementary crystallographic information
Comment
In the last few months, a number of hydrazone compounds have been reported for their medical applications (Ajani et al., 2010; Angelusiu et al., 2010; Zhang et al., 2010). Recent structure analyses of some members of this family of compounds have also been reported (Ahmad et al., 2010; Huang & Wu, 2010; Ji & Lu, 2010; Khaledi et al., 2010; Singh & Singh, 2010; Zhou & Yang, 2010). In this paper, we report the structure of the new hydrazone compound, N'-(3-hydroxybenzylidene)-4-nitrobenzohydrazide.
The title molecule is shown in Fig. 1. The molecule is approximately planar, with the interplanar angle between the two benzene rings equal to 5.8 (2)°. The bond lengths and angles are comparable with the hydrazone compounds cited above.
Four title molecules are linked by the motif R44(12) (Etter et al., 1990) with pairs of strong O—H···O and strong N—H···O hydrogen bonds (Desiraju & Steiner, 1999). For the hydrogen bonds, see Table 1. The motif R44(12) is situated about the crystallographic centres of symmetry with the Wyckoff position 2c for the present setting. This motif is composed of two pairs of parallel molecules. This quadruplet of the title molecules is further extended by the symmetry equivalent H-bonds into the layers parallel to the planes (101). In addition to the hydrogen bonds there is also a weak π-electron ring–π-electron ring interaction (Table 2) between the benzene rings in the structure (Spek, 2009).
Experimental
4-Nitrobenzohydrazide (0.181 g, 1 mmol) and 3-hydroxybenzaldehyde (0.122 g, 1 mmol) were mixed in 50 ml of methanol at room temperature. The mixture was stirred at room temperature for 30 min to give a yellow solution of the product. After keeping the above solution in air for 5 d, yellow block-shaped crystals with average size of 0.1 mm × 0.2 mm × 0.2 mm developed.
Refinement
All the H atoms were discernible in the difference electron density maps. However, the aryl H atoms were positioned into idealized positions and refined in riding atom approximation. The used constraints: C—H = 0.93 Å; Uiso(H) = 1.2Ueq(C). The positional parameters of the H atoms H1 and H2 involved in the strong hydrogen bonds were refined freely, however, with the constraints of the displacement parameters Uiso(H) = 1.5Ueq(O or N).
Figures
Fig. 1.

The title molecule showing 30% probability displacement ellipsoids and the atomic numbering scheme.
Fig. 2.

A quadruplet of the title molecules forming the motif R44(12). Intermolecular interactions are drawn as dashed lines. N (blue), O (red), C (grey), H (green).
Crystal data
| C14H11N3O4 | F(000) = 592 |
| Mr = 285.26 | Dx = 1.461 Mg m−3 |
| Monoclinic, P21/n | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P 2yn | Cell parameters from 1436 reflections |
| a = 9.987 (3) Å | θ = 2.7–26.3° |
| b = 8.967 (3) Å | µ = 0.11 mm−1 |
| c = 15.108 (4) Å | T = 298 K |
| β = 106.560 (3)° | Block, yellow |
| V = 1296.8 (6) Å3 | 0.13 × 0.10 × 0.10 mm |
| Z = 4 |
Data collection
| Bruker SMART 1000 CCD diffractometer | 2768 independent reflections |
| Radiation source: fine-focus sealed tube | 1787 reflections with I > 2σ(I) |
| graphite | Rint = 0.028 |
| ω scans | θmax = 27.0°, θmin = 2.2° |
| Absorption correction: multi-scan (SADABS; Bruker, 2001) | h = −11→12 |
| Tmin = 0.986, Tmax = 0.989 | k = −11→10 |
| 6579 measured reflections | l = −18→16 |
Refinement
| Refinement on F2 | Secondary atom site location: difference Fourier map |
| Least-squares matrix: full | Hydrogen site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.044 | H atoms treated by a mixture of independent and constrained refinement |
| wR(F2) = 0.119 | w = 1/[σ2(Fo2) + (0.0537P)2 + 0.0632P] where P = (Fo2 + 2Fc2)/3 |
| S = 1.03 | (Δ/σ)max < 0.001 |
| 2768 reflections | Δρmax = 0.21 e Å−3 |
| 197 parameters | Δρmin = −0.17 e Å−3 |
| 0 restraints | Extinction correction: SHELXTL (Sheldrick, 2008), Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 |
| 38 constraints | Extinction coefficient: 0.0088 (17) |
| Primary atom site location: structure-invariant direct methods |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| N1 | 0.00619 (14) | 0.30350 (16) | 0.94081 (9) | 0.0406 (4) | |
| N2 | 0.12358 (14) | 0.36267 (16) | 1.00310 (10) | 0.0398 (4) | |
| N3 | 0.69398 (17) | 0.6801 (2) | 1.24192 (14) | 0.0589 (5) | |
| O1 | −0.42069 (13) | 0.13866 (16) | 0.68671 (8) | 0.0555 (4) | |
| H1 | −0.493 (2) | 0.089 (2) | 0.6608 (16) | 0.083* | |
| O2 | 0.16825 (12) | 0.52109 (15) | 0.89819 (8) | 0.0533 (4) | |
| O3 | 0.76141 (18) | 0.7733 (2) | 1.21577 (13) | 0.0993 (6) | |
| O4 | 0.72752 (16) | 0.62850 (19) | 1.31937 (12) | 0.0842 (6) | |
| C1 | −0.18157 (16) | 0.12789 (18) | 0.91821 (11) | 0.0359 (4) | |
| C2 | −0.24225 (16) | 0.16628 (19) | 0.82649 (11) | 0.0372 (4) | |
| H2A | −0.2025 | 0.2405 | 0.7992 | 0.045* | |
| C3 | −0.36203 (17) | 0.0939 (2) | 0.77569 (11) | 0.0385 (4) | |
| C4 | −0.42103 (19) | −0.0178 (2) | 0.81556 (13) | 0.0452 (5) | |
| H4 | −0.5005 | −0.0675 | 0.7810 | 0.054* | |
| C5 | −0.3613 (2) | −0.0546 (2) | 0.90643 (14) | 0.0502 (5) | |
| H5 | −0.4015 | −0.1290 | 0.9334 | 0.060* | |
| C6 | −0.24222 (18) | 0.0171 (2) | 0.95848 (13) | 0.0449 (5) | |
| H6 | −0.2029 | −0.0086 | 1.0201 | 0.054* | |
| C7 | −0.05675 (17) | 0.20413 (19) | 0.97439 (12) | 0.0393 (4) | |
| H7 | −0.0222 | 0.1794 | 1.0365 | 0.047* | |
| C8 | 0.19947 (16) | 0.4687 (2) | 0.97659 (12) | 0.0369 (4) | |
| C9 | 0.32783 (16) | 0.52089 (18) | 1.04872 (11) | 0.0341 (4) | |
| C10 | 0.39976 (17) | 0.64059 (19) | 1.02656 (12) | 0.0400 (4) | |
| H10 | 0.3676 | 0.6855 | 0.9689 | 0.048* | |
| C11 | 0.51947 (18) | 0.6937 (2) | 1.09002 (13) | 0.0438 (5) | |
| H11 | 0.5681 | 0.7742 | 1.0756 | 0.053* | |
| C12 | 0.56488 (16) | 0.62500 (19) | 1.17461 (12) | 0.0410 (5) | |
| C13 | 0.49633 (17) | 0.50675 (19) | 1.19882 (12) | 0.0437 (5) | |
| H13 | 0.5292 | 0.4624 | 1.2566 | 0.052* | |
| C14 | 0.37696 (16) | 0.45478 (19) | 1.13512 (11) | 0.0407 (5) | |
| H14 | 0.3289 | 0.3745 | 1.1503 | 0.049* | |
| H2 | 0.1392 (19) | 0.341 (2) | 1.0640 (14) | 0.061* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| N1 | 0.0314 (7) | 0.0502 (9) | 0.0331 (8) | 0.0010 (7) | −0.0021 (6) | −0.0075 (7) |
| N2 | 0.0340 (7) | 0.0487 (9) | 0.0295 (8) | −0.0019 (7) | −0.0027 (6) | −0.0017 (7) |
| N3 | 0.0419 (9) | 0.0546 (11) | 0.0707 (13) | −0.0056 (8) | 0.0008 (9) | −0.0167 (10) |
| O1 | 0.0436 (8) | 0.0827 (11) | 0.0308 (7) | −0.0161 (7) | −0.0046 (6) | 0.0000 (7) |
| O2 | 0.0411 (7) | 0.0761 (10) | 0.0353 (8) | 0.0012 (6) | −0.0006 (6) | 0.0098 (7) |
| O3 | 0.0719 (11) | 0.1000 (14) | 0.1113 (15) | −0.0481 (10) | 0.0023 (10) | −0.0048 (11) |
| O4 | 0.0703 (10) | 0.0935 (13) | 0.0634 (11) | −0.0126 (9) | −0.0219 (8) | −0.0066 (10) |
| C1 | 0.0327 (9) | 0.0371 (10) | 0.0347 (10) | 0.0047 (7) | 0.0042 (7) | −0.0060 (8) |
| C2 | 0.0333 (9) | 0.0449 (10) | 0.0321 (10) | −0.0018 (7) | 0.0074 (7) | −0.0039 (8) |
| C3 | 0.0331 (9) | 0.0494 (11) | 0.0302 (10) | −0.0001 (8) | 0.0047 (7) | −0.0067 (8) |
| C4 | 0.0407 (10) | 0.0486 (11) | 0.0429 (11) | −0.0097 (8) | 0.0064 (8) | −0.0084 (9) |
| C5 | 0.0558 (12) | 0.0425 (11) | 0.0509 (12) | −0.0084 (9) | 0.0127 (10) | 0.0050 (9) |
| C6 | 0.0484 (11) | 0.0450 (11) | 0.0354 (11) | 0.0057 (8) | 0.0022 (8) | 0.0026 (8) |
| C7 | 0.0366 (9) | 0.0434 (10) | 0.0307 (10) | 0.0063 (8) | −0.0021 (7) | −0.0041 (8) |
| C8 | 0.0303 (9) | 0.0443 (10) | 0.0332 (10) | 0.0097 (8) | 0.0041 (7) | −0.0018 (8) |
| C9 | 0.0273 (8) | 0.0381 (9) | 0.0347 (10) | 0.0087 (7) | 0.0053 (7) | −0.0034 (7) |
| C10 | 0.0402 (9) | 0.0441 (11) | 0.0367 (10) | 0.0056 (8) | 0.0126 (8) | 0.0024 (8) |
| C11 | 0.0412 (10) | 0.0405 (10) | 0.0523 (12) | −0.0033 (8) | 0.0174 (9) | −0.0041 (9) |
| C12 | 0.0285 (9) | 0.0428 (10) | 0.0464 (11) | 0.0023 (8) | 0.0021 (8) | −0.0098 (9) |
| C13 | 0.0379 (10) | 0.0454 (11) | 0.0395 (11) | 0.0037 (8) | −0.0027 (8) | 0.0022 (8) |
| C14 | 0.0342 (9) | 0.0404 (10) | 0.0406 (11) | −0.0016 (7) | −0.0002 (8) | 0.0027 (8) |
Geometric parameters (Å, °)
| N1—C7 | 1.276 (2) | C4—H4 | 0.9300 |
| N1—N2 | 1.3830 (18) | C5—C6 | 1.383 (3) |
| N2—C8 | 1.346 (2) | C5—H5 | 0.9300 |
| N2—H2 | 0.91 (2) | C6—H6 | 0.9300 |
| N3—O3 | 1.208 (2) | C7—H7 | 0.9300 |
| N3—O4 | 1.213 (2) | C8—C9 | 1.501 (2) |
| N3—C12 | 1.481 (2) | C9—C10 | 1.385 (2) |
| O1—C3 | 1.365 (2) | C9—C14 | 1.390 (2) |
| O1—H1 | 0.85 (2) | C10—C11 | 1.386 (2) |
| O2—C8 | 1.229 (2) | C10—H10 | 0.9300 |
| C1—C2 | 1.388 (2) | C11—C12 | 1.374 (2) |
| C1—C6 | 1.391 (2) | C11—H11 | 0.9300 |
| C1—C7 | 1.462 (2) | C12—C13 | 1.367 (2) |
| C2—C3 | 1.385 (2) | C13—C14 | 1.382 (2) |
| C2—H2A | 0.9300 | C13—H13 | 0.9300 |
| C3—C4 | 1.384 (2) | C14—H14 | 0.9300 |
| C4—C5 | 1.372 (3) | ||
| C7—N1—N2 | 114.55 (14) | C1—C6—H6 | 120.2 |
| C8—N2—N1 | 120.57 (14) | N1—C7—C1 | 122.05 (16) |
| C8—N2—H2 | 120.2 (12) | N1—C7—H7 | 119.0 |
| N1—N2—H2 | 118.3 (12) | C1—C7—H7 | 119.0 |
| O3—N3—O4 | 123.56 (18) | O2—C8—N2 | 123.16 (15) |
| O3—N3—C12 | 117.62 (19) | O2—C8—C9 | 120.68 (16) |
| O4—N3—C12 | 118.81 (18) | N2—C8—C9 | 116.16 (15) |
| C3—O1—H1 | 111.7 (16) | C10—C9—C14 | 119.29 (15) |
| C2—C1—C6 | 119.55 (15) | C10—C9—C8 | 117.42 (15) |
| C2—C1—C7 | 121.39 (16) | C14—C9—C8 | 123.29 (16) |
| C6—C1—C7 | 119.04 (15) | C9—C10—C11 | 120.24 (16) |
| C3—C2—C1 | 119.96 (17) | C9—C10—H10 | 119.9 |
| C3—C2—H2A | 120.0 | C11—C10—H10 | 119.9 |
| C1—C2—H2A | 120.0 | C12—C11—C10 | 118.70 (17) |
| O1—C3—C4 | 121.66 (15) | C12—C11—H11 | 120.7 |
| O1—C3—C2 | 117.95 (16) | C10—C11—H11 | 120.7 |
| C4—C3—C2 | 120.37 (16) | C13—C12—C11 | 122.60 (15) |
| C5—C4—C3 | 119.48 (16) | C13—C12—N3 | 118.64 (17) |
| C5—C4—H4 | 120.3 | C11—C12—N3 | 118.75 (17) |
| C3—C4—H4 | 120.3 | C12—C13—C14 | 118.26 (16) |
| C4—C5—C6 | 120.98 (18) | C12—C13—H13 | 120.9 |
| C4—C5—H5 | 119.5 | C14—C13—H13 | 120.9 |
| C6—C5—H5 | 119.5 | C13—C14—C9 | 120.91 (17) |
| C5—C6—C1 | 119.64 (17) | C13—C14—H14 | 119.5 |
| C5—C6—H6 | 120.2 | C9—C14—H14 | 119.5 |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N2—H2···O1i | 0.91 (2) | 2.11 (2) | 2.931 (2) | 149.3 (16) |
| O1—H1···O2ii | 0.85 (2) | 1.82 (2) | 2.6573 (17) | 167 (2) |
Symmetry codes: (i) x+1/2, −y+1/2, z+1/2; (ii) −x−1/2, y−1/2, −z+3/2.
Table 2 Overview of π–π ring interactions in the structure
| Centroid–centroid* | Distance (Å) | Symmetry code |
| Cg1–Cg2 | 3.6803 (16) | 1 - x, -y, 1 - z |
* Cg1 and Cg2 are the centroids of the C1–C6 and C9–C14 benzene rings, respectively.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: FB2226).
References
- Ahmad, T., Zia-ur-Rehman, M., Siddiqui, H. L., Mahmud, S. & Parvez, M. (2010). Acta Cryst. E66, o1022. [DOI] [PMC free article] [PubMed]
- Ajani, O. O., Obafemi, C. A., Nwinyi, O. C. & Akinpelu, D. A. (2010). Bioorg. Med. Chem.18, 214–221. [DOI] [PubMed]
- Angelusiu, M. V., Barbuceanu, S. F., Draghici, C. & Almajan, G. L. (2010). Eur. J. Med. Chem.45, 2055–2062. [DOI] [PubMed]
- Bruker (2001). SADABS Bruker AXS Inc., Madison, Wisconsin, USA.
- Bruker (2007). SMART and SAINT Bruker AXS Inc., Madison, Wisconsin, USA.
- Desiraju, G. R. & Steiner, T. (1999). The Weak Hydrogen Bond in Structural Chemistry and Biology, p. 13. New York: Oxford University Press Inc.
- Etter, M. C., MacDonald, J. C. & Bernstein, J. (1990). Acta Cryst. B46, 256–262. [DOI] [PubMed]
- Huang, H.-T. & Wu, H.-Y. (2010). Acta Cryst. E66, o2729–o2730. [DOI] [PMC free article] [PubMed]
- Ji, X.-H. & Lu, J.-F. (2010). Acta Cryst. E66, o1514. [DOI] [PMC free article] [PubMed]
- Khaledi, H., Alhadi, A. A., Mohd Ali, H., Robinson, W. T. & Abdulla, M. A. (2010). Acta Cryst. E66, o105–o106. [DOI] [PMC free article] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Singh, V. P. & Singh, S. (2010). Acta Cryst. E66, o1172. [DOI] [PMC free article] [PubMed]
- Spek, A. L. (2009). Acta Cryst. D65, 148–155. [DOI] [PMC free article] [PubMed]
- Zhang, Y.-H., Zhang, L., Liu, L., Guo, J.-X., Wu, D.-L., Xu, G.-C., Wang, X.-H. & Jia, D.-Z. (2010). Inorg. Chim. Acta, 363, 289–293.
- Zhou, C.-S. & Yang, T. (2010). Acta Cryst. E66, o290. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536810043564/fb2226sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536810043564/fb2226Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report