

Abstract
The molecular geometry of the title compound, C7H7N3O2S, does not differ much from that of the previously reported 4-toluenesulfonyl analogue. Unlike the latter compound, however, molecules of the title compound associate primarily via π–π stacking interactions of their benzene rings [centroid–centroid distance = 3.5865 (8) Å], forming columnar stacks along the crystallographic 21 axes. These stacks are interconnected via weak C—H⋯O and C—H⋯N hydrogen bonds.
Related literature
For crystal structure of 1-(p-toluenesulfonyl)-1H-1,2,3-benzotriazole, see: Rodríguez et al. (2005 ▶). For the preparation of the title compound and examples of its synthetic use, see: Katritzky et al. (1992 ▶, 2000 ▶).
Experimental
Crystal data
C7H7N3O2S
M r = 197.22
Monoclinic,
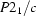
a = 9.3685 (3) Å
b = 7.0627 (2) Å
c = 12.4994 (3) Å
β = 92.984 (2)°
V = 825.93 (4) Å3
Z = 4
Mo Kα radiation
μ = 0.36 mm−1
T = 150 K
0.50 × 0.30 × 0.25 mm
Data collection
Nonius KappaCCD diffractometer
14989 measured reflections
1886 independent reflections
1674 reflections with I > 2σ(I)
R int = 0.023
Refinement
R[F 2 > 2σ(F 2)] = 0.030
wR(F 2) = 0.082
S = 1.09
1886 reflections
119 parameters
H-atom parameters constrained
Δρmax = 0.29 e Å−3
Δρmin = −0.42 e Å−3
Data collection: COLLECT (Nonius, 2000 ▶); cell refinement: SCALEPACK (Otwinowski & Minor, 1997 ▶); data reduction: DENZO (Otwinowski & Minor, 1997 ▶) and SCALEPACK; program(s) used to solve structure: SIR97 (Altomare et al., 1999) ▶; program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: PLATON (Spek, 2009 ▶); software used to prepare material for publication: PLATON.
Supplementary Material
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536810040778/im2237sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536810040778/im2237Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| C5—H5⋯O2i | 0.93 | 2.55 | 3.270 (2) | 135 |
| C6—H6⋯O1ii | 0.93 | 2.55 | 3.451 (2) | 164 |
| C8—H8B⋯N3iii | 0.96 | 2.61 | 3.446 (2) | 145 |
| C8—H8C⋯O2iv | 0.96 | 2.40 | 3.325 (2) | 161 |
Symmetry codes: (i) 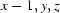 ; (ii)
; (ii) 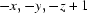 ; (iii)
; (iii) 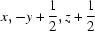 ; (iv)
; (iv) 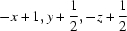 .
.
Acknowledgments
Financial support from the Ministry of Education of the Czech Republic (project No. MSM0021620857) is gratefully acknowledged.
supplementary crystallographic information
Comment
The title compound, 1-(methanesulfonyl)-1H-1,2,3-benzotriazole, is a useful and readily accessible organic reagent, acting as a convenient source of the benzotriazolyl anion. For instance, it reacts smoothly with carboxylic acids in the presence of a base to afford the corresponding 1-acyl-1H-1,2,3-benzotriazoles, which can be subsequently converted to amides in typically good yields (Katritzky et al., 2000).
The molecular structure of the title compound (Fig. 1) is rather unexceptional, particularly in view of the geometric data reported earlier for the related 1H-1,2,3-benzotriazole derivative, 1-(p-toluenesulfonyl)-1H-1,2,3-benzotriazole (Rodríguez et al., 2005). The N—N bonds within the triazole ring clearly maintain their localized character (cf. N1—N2 = 1.389 (2) Å, N3—N2 = 1.288 (2) Å), which is, however, not reflected in the adjacent bonds. The lengths of the remaining in-ring bonds, N1—C7A, C7A—C3A and C3A—N3, differ by less than ca 0.006 Å, while all in-ring angles span a range of 103.5 (1)–109.9 (1) °. On the other hand, the variation in the analogous parameters describing the geometry of the annelating benzene ring is less pronounced (cf. C—C = 1.371 (2)–1.408 (2) Å, C—C—C = 115.5 (1)–122.7 (1) °).
The methanesulfonyl group binds to the triazole ring somewhat unsymmetrically, which is best demonstrated by the difference in the S—N1—N2 (120.12 (9) °) and S—N1—C7A (129.8 (1) °) angles. Moreover, it is angularly distorted: The bond angles around sulfur span a range of 103.43 (6)–120.31 (7) ° with the N1—S—C8 and O1—S—O2 angles being the lower and upper limit, respectively. The remaining angles do not differ much in the pairs: N1—S—O(1/2) (ca 105 °, difference ca 0.4 °), C8—S1—O(1/2) (ca 110 °, difference ca 1.2 °). Indeed, such a variation in bond angles corresponds with distances to the sulfur atom (S—O1 1.425 (1), S—O2 1.419 (1), S—N1 1.692 (1), S—C8 1.744 (2) Å) as the most acute angle is associated with the shortest bonds.
In the crystal, molecules of the title compound assemble viaπ–π stacking interactions of their benzene rings (Fig. 2a). Since this interaction involves molecules related by the crystallographic 21 screw axes, it results in the formation of infinite columnar stacks in the direction of the crystallographic b axis. It is worth pointing out that the observed separation of the ring centroids [Cg···Cg (-x, 1/2 + y, 1/2-z; 3.5865 (8) Å] is slightly shorter than that reported for α-graphite (ca 3.65 Å), where, however, the rings are slipped by ca 1.42 Å. Finally, the neighboring columnar stacks are interlinked by soft C—H···O and C—H···N hydrogen bonds (Table 1) into a complicated three-dimensional array (Figs. 2b and 2c).
Experimental
The title compound was synthesized from 1H-1,2,3-benzotriazole and methanesulfonyl chloride as described in the literature with a yield of 91% (Katritzky et al., 2000). Crystals suitable for X-ray diffraction analysis were obtained by crystallization from warm benzene.
Refinement
H-atoms were included in calculated positions and refined as riding atoms with fixed C—H distances [C—H = 0.96 Å for CH3, and 0.93 Å for aromatic CH] and Uiso(H) assigned to 1.5Ueq(C) (CH3) or 1.2Ueq(C) (aromatic CH) of their bonding carbon atom.
Figures
Fig. 1.

Molecular structure of the title compound showing displacement ellipsoids for the non-H atoms at the 50% probability level. Hydrogen atoms are presented as spheres with arbitrary radii.
Fig. 2.

(a) Section of columnar stacks connected by π–π interactions of the benzene rings [Cg···Cg = 3.5865 (8) Å]. (b) H-bond interactions generated by the molecules of the title compounds. (c) View of the unit cell along the crystallographic a axis.
Crystal data
| C7H7N3O2S | F(000) = 408 |
| Mr = 197.22 | Dx = 1.586 Mg m−3 |
| Monoclinic, P21/c | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P 2ybc | Cell parameters from 2027 reflections |
| a = 9.3685 (3) Å | θ = 1.0–27.5° |
| b = 7.0627 (2) Å | µ = 0.36 mm−1 |
| c = 12.4994 (3) Å | T = 150 K |
| β = 92.984 (2)° | Block, colourless |
| V = 825.93 (4) Å3 | 0.50 × 0.30 × 0.25 mm |
| Z = 4 |
Data collection
| Nonius KappaCCD diffractometer | 1674 reflections with I > 2σ(I) |
| Radiation source: fine-focus sealed tube | Rint = 0.023 |
| horizontally mounted graphite crystal | θmax = 27.5°, θmin = 2.2° |
| Detector resolution: 9.091 pixels mm-1 | h = −12→12 |
| ω and π scans to fill the Ewald sphere | k = −9→9 |
| 14989 measured reflections | l = −16→16 |
| 1886 independent reflections |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.030 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.082 | H-atom parameters constrained |
| S = 1.09 | w = 1/[σ2(Fo2) + (0.0431P)2 + 0.2983P] where P = (Fo2 + 2Fc2)/3 |
| 1886 reflections | (Δ/σ)max < 0.001 |
| 119 parameters | Δρmax = 0.29 e Å−3 |
| 0 restraints | Δρmin = −0.42 e Å−3 |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two least-squares planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving least-squares planes. |
| Refinement. Refinement of F2 against all diffractions. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > 2σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on all data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| S | 0.39056 (3) | 0.07483 (5) | 0.36695 (2) | 0.02263 (12) | |
| O1 | 0.32920 (11) | −0.04099 (16) | 0.44586 (8) | 0.0318 (3) | |
| O2 | 0.51042 (11) | 0.01011 (18) | 0.31253 (8) | 0.0337 (3) | |
| N1 | 0.25880 (13) | 0.10614 (17) | 0.27080 (9) | 0.0236 (3) | |
| N2 | 0.29032 (13) | 0.10541 (18) | 0.16344 (9) | 0.0274 (3) | |
| N3 | 0.17185 (13) | 0.11648 (19) | 0.10673 (9) | 0.0289 (3) | |
| C3A | 0.05897 (15) | 0.1243 (2) | 0.17395 (11) | 0.0233 (3) | |
| C4 | −0.08866 (16) | 0.1336 (2) | 0.14889 (12) | 0.0290 (3) | |
| H4 | −0.1253 | 0.1381 | 0.0784 | 0.035* | |
| C5 | −0.17615 (16) | 0.1359 (2) | 0.23349 (12) | 0.0295 (3) | |
| H5 | −0.2746 | 0.1413 | 0.2200 | 0.035* | |
| C6 | −0.12050 (16) | 0.1302 (2) | 0.34029 (12) | 0.0290 (3) | |
| H6 | −0.1837 | 0.1318 | 0.3953 | 0.035* | |
| C7 | 0.02458 (16) | 0.1222 (2) | 0.36641 (11) | 0.0273 (3) | |
| H7 | 0.0611 | 0.1199 | 0.4370 | 0.033* | |
| C7A | 0.11237 (14) | 0.11801 (19) | 0.27980 (11) | 0.0218 (3) | |
| C8 | 0.41970 (16) | 0.3025 (2) | 0.41766 (11) | 0.0277 (3) | |
| H8A | 0.4901 | 0.2977 | 0.4762 | 0.042* | |
| H8B | 0.3319 | 0.3521 | 0.4424 | 0.042* | |
| H8C | 0.4531 | 0.3828 | 0.3623 | 0.042* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| S | 0.02097 (19) | 0.0276 (2) | 0.01921 (18) | 0.00087 (12) | 0.00002 (12) | 0.00117 (12) |
| O1 | 0.0327 (6) | 0.0361 (6) | 0.0263 (5) | −0.0063 (5) | −0.0026 (4) | 0.0100 (4) |
| O2 | 0.0245 (5) | 0.0461 (7) | 0.0306 (5) | 0.0088 (5) | 0.0010 (4) | −0.0071 (5) |
| N1 | 0.0209 (6) | 0.0317 (6) | 0.0183 (5) | 0.0002 (5) | 0.0016 (4) | 0.0007 (4) |
| N2 | 0.0275 (6) | 0.0369 (7) | 0.0180 (5) | −0.0017 (5) | 0.0030 (4) | 0.0007 (5) |
| N3 | 0.0264 (6) | 0.0402 (7) | 0.0201 (6) | −0.0023 (5) | 0.0002 (5) | 0.0017 (5) |
| C3A | 0.0252 (7) | 0.0222 (7) | 0.0223 (6) | −0.0016 (5) | −0.0002 (5) | 0.0008 (5) |
| C4 | 0.0266 (7) | 0.0293 (7) | 0.0304 (7) | −0.0015 (6) | −0.0052 (6) | 0.0009 (6) |
| C5 | 0.0220 (7) | 0.0251 (7) | 0.0413 (8) | −0.0012 (6) | 0.0005 (6) | 0.0008 (6) |
| C6 | 0.0263 (7) | 0.0262 (7) | 0.0352 (8) | 0.0000 (6) | 0.0100 (6) | 0.0008 (6) |
| C7 | 0.0290 (7) | 0.0294 (7) | 0.0238 (7) | 0.0010 (6) | 0.0038 (5) | 0.0010 (6) |
| C7A | 0.0212 (6) | 0.0217 (6) | 0.0225 (6) | 0.0001 (5) | 0.0005 (5) | 0.0006 (5) |
| C8 | 0.0301 (7) | 0.0290 (7) | 0.0239 (6) | −0.0017 (6) | 0.0001 (5) | −0.0003 (6) |
Geometric parameters (Å, °)
| S—O2 | 1.4185 (10) | C4—H4 | 0.9300 |
| S—O1 | 1.4254 (11) | C5—C6 | 1.408 (2) |
| S—N1 | 1.6919 (12) | C5—H5 | 0.9300 |
| S—C8 | 1.7444 (15) | C6—C7 | 1.382 (2) |
| N1—C7A | 1.3848 (17) | C6—H6 | 0.9300 |
| N1—N2 | 1.3890 (16) | C7—C7A | 1.3936 (19) |
| N2—N3 | 1.2878 (17) | C7—H7 | 0.9300 |
| N3—C3A | 1.3856 (18) | C8—H8A | 0.9600 |
| C3A—C7A | 1.3905 (18) | C8—H8B | 0.9600 |
| C3A—C4 | 1.4037 (19) | C8—H8C | 0.9600 |
| C4—C5 | 1.371 (2) | ||
| O2—S—O1 | 120.31 (7) | C4—C5—H5 | 119.2 |
| O2—S—N1 | 105.55 (6) | C6—C5—H5 | 119.2 |
| O1—S—N1 | 105.13 (6) | C7—C6—C5 | 122.41 (14) |
| O2—S—C8 | 111.01 (7) | C7—C6—H6 | 118.8 |
| O1—S—C8 | 109.79 (7) | C5—C6—H6 | 118.8 |
| N1—S—C8 | 103.43 (6) | C6—C7—C7A | 115.48 (13) |
| C7A—N1—N2 | 109.87 (11) | C6—C7—H7 | 122.3 |
| C7A—N1—S | 129.75 (9) | C7A—C7—H7 | 122.3 |
| N2—N1—S | 120.12 (9) | N1—C7A—C3A | 103.50 (11) |
| N3—N2—N1 | 108.12 (11) | N1—C7A—C7 | 133.75 (13) |
| N2—N3—C3A | 109.37 (11) | C3A—C7A—C7 | 122.74 (13) |
| N3—C3A—C7A | 109.12 (12) | S—C8—H8A | 109.5 |
| N3—C3A—C4 | 129.85 (13) | S—C8—H8B | 109.5 |
| C7A—C3A—C4 | 121.02 (13) | H8A—C8—H8B | 109.5 |
| C5—C4—C3A | 116.74 (13) | S—C8—H8C | 109.5 |
| C5—C4—H4 | 121.6 | H8A—C8—H8C | 109.5 |
| C3A—C4—H4 | 121.6 | H8B—C8—H8C | 109.5 |
| C4—C5—C6 | 121.60 (14) |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| C5—H5···O2i | 0.93 | 2.55 | 3.270 (2) | 135 |
| C6—H6···O1ii | 0.93 | 2.55 | 3.451 (2) | 164 |
| C8—H8B···N3iii | 0.96 | 2.61 | 3.446 (2) | 145 |
| C8—H8C···O2iv | 0.96 | 2.40 | 3.325 (2) | 161 |
Symmetry codes: (i) x−1, y, z; (ii) −x, −y, −z+1; (iii) x, −y+1/2, z+1/2; (iv) −x+1, y+1/2, −z+1/2.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: IM2237).
References
- Altomare, A., Burla, M. C., Camalli, M., Cascarano, G. L., Giacovazzo, C., Guagliardi, A., Moliterni, A. G. G., Polidori, G. & Spagna, R. (1999). J. Appl. Cryst.32, 115–119.
- Katritzky, A. R., He, H.-Y. & Suzuki, K. (2000). J. Org. Chem.65, 8210–8213. [DOI] [PubMed]
- Katritzky, A. R., Shobana, N., Pernak, J., Afridi, A. S. & Fan, W.-Q. (1992). Tetrahedron, 48, 7817–7822.
- Nonius (2000). COLLECT Nonius BV, Delft, The Netherlands.
- Otwinowski, Z. & Minor, W. (1997). Methods in Enzymology, Vol. 276, Macromolecular Crystallography, Part A, edited by C. W. Carter Jr & R. M. Sweet, pp. 307–326. New York: Academic Press.
- Rodríguez, R., Nogueras, M., Cobo, J., Low, J. N. & Glidewell, C. (2005). Acta Cryst. E61, o2795–o2797.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Spek, A. L. (2009). Acta Cryst. D65, 148–155. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536810040778/im2237sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536810040778/im2237Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report