

Abstract
Treatment of brain disease with recombinant proteins is difficult due to the blood-brain barrier. As an alternative to direct injections into the brain, we studied whether application of high concentrations of therapeutic enzymes via intrathecal (IT) injections could successfully drive uptake across the ependyma to treat brain disease. We studied IT enzyme replacement therapy with recombinant human iduronidase (rhIDU) in canine mucopolysaccharidosis I (MPS I, Hurler syndrome), a lysosomal storage disorder with brain and meningeal involvement. Monthly or quarterly IT treatment regimens with rhIDU achieved supranormal iduronidase enzyme levels in the brain, spinal cord, and spinal meninges. All regimens normalized total brain glycosaminoglycan (GAG) storage and reduced spinal meningeal GAG storage by 58–70%. The improvement in GAG storage levels persisted three months after the final IT dose. The successful use of enzyme therapy via the CSF represents a potentially useful approach for lysosomal storage disorders.
Keywords: Mucopolysaccharidosis I, lysosomal storage disorder, intrathecal, enzyme replacement therapy, central nervous system, cerebrospinal fluid, Hurler, Scheie, Hurler-Scheie, pachymeningitis
Introduction
Lysosomal storage diseases affecting the brain are difficult to treat due to the blood-brain barrier that blocks uptake of circulating therapeutic proteins. Many invasive strategies have been used or proposed for circumventing the blood-brain barrier, including intraparenchymal injections of therapeutic proteins, intraparenchymal gene therapy, chemical or physical agents to open the blood-brain barrier (such as mannitol), and hematopoietic stem cell transplantation [1–9]. Treatment via the cerebrospinal fluid has not been successful in the past due to the inability of the proteins to traverse the ependymal layer and diffuse through brain tissue, even though this route would be clinically easier for application to patients. Despite some success in animal models, many of these therapies have been difficult to translate to the clinical realm.
Rare lysosomal storage disorders, such as mucopolysaccharidosis I (MPS I, Hurler syndrome), MPS VI (Maroteaux-Lamy syndrome), Gaucher disease, and Fabry disease are currently treated by intravenous enzyme replacement therapy [10–16]. Enzyme replacement therapy has successfully treated some aspects of the physical disease in MPS patients and has been well tolerated. Its usefulness in MPS I and other lysosomal storage diseases with a neurodegenerative component has been hampered by its inability to cross the blood-brain barrier to a sufficient extent at usual doses.
The concept of IT enzyme replacement therapy is based on the rationale that even small amounts of these enzymes in the cerebrospinal fluid (CSF) represent very large concentration gradients relative to the affinity constants for the receptors for these enzymes on neurons and other cell types. For example, providing 1 mg of iduronidase to the approximately 20 mL of CSF in an MPS I dog provides a concentration that is about 1400 fold the uptake affinity constant for the mannose 6-phosphate receptor found on the surface of cells that binds and targets iduronidase to the lysosome [17]. This level is 1.4 million times the half-maximal concentration required for the correction of lysosomal storage in MPS I fibroblasts in culture [17]. Given this very large concentration gradient, the intrinsic diffusibility of iduronidase through brain tissue and a very efficient uptake process, we reasoned that tiny amounts of enzyme could cross the ependymal layer and still achieve adequate uptake at the end site around neurons and glia [18]. Our previous research in dogs has shown that weekly injections of IT rhIDU can drive rhIDU to penetrate several millimeters into brain tissue, normalize brain glycosaminoglycan (GAG, the material that accumulates in MPS disease) levels and substantially reduce meningeal storage [18]. Here, we show that this approach is sufficiently effective that successful treatment can be accomplished using an infrequent monthly or quarterly (every 3 month) regimen in a large animal model of MPS I.
Methods
The canine MPS I colony is derived from a beagle/Plott hound mix. The Los Angeles Biomedical Institute at Harbor-UCLA (formerly the Harbor-UCLA Research and Education Institute) is an AAALAC accredited facility. The study was approved by the institutional Animal Care and Use Review Committee.
Compound formulation
RhIDU was donated by BioMarin Pharmaceutical (Novato, California). It consists of 0.6 mg/mL rhIDU (117,000 to 150,000 units/mL, depending on formulation) in formulation buffer (150 mM NaCl; 100 mM sodium phosphate, 0.001% polysorbate 80, pH 5.8). The enzyme is stored at 2–8° and protected from excess heat and light.
Intrathecal injection experiments
Dogs affected with canine MPS I received IT administrations of 1.38 mg of rhIDU (270,000 units, in volumes of 2.3 mL) alone or diluted in the artificial CSF solution, Elliotts B (Ben Venue Laboratories, Bedford, Ohio) for a total volume of 6.9 mL. Dogs receiving low-dose IT rhIDU were given 0.46 mg rhIDU (90,000 units) diluted in Elliotts B for a total volume of 2.3 mL. The IT injections were administered to the cisterna magna of the anesthetized dogs as described below.
Animals were fasted overnight. Anesthesia was induced with Propofol (6 mg/kg), and maintained with 5% isoflurane and oxygen. Animals were intubated with a 6 or 7 mm endotracheal tube. Subcutaneous atropine (0.06 mg/kg) was used to reduce salivation. A 1½-inch, 22-gauge spinal needle was inserted into the cisterna magna under sterile conditions. Approximately 0.5–1.0 mL CSF was collected. RhIDU was injected intrathecally into the hub of the spinal needle manually with a syringe over 2 minutes. The spinal needle was removed and the animals were recovered. A sixteenth dog, originally on the monthly protocol, died prematurely from a brainstem hematoma during an IT procedure and was not included in the analysis.
Clinical Assessments
Vital signs (heart rate, body temperature, and respiratory rate) were monitored at each intrathecal injection. Growth was observed throughout the therapy by body weight measurements taken every 2 weeks. Neurological examinations were performed by a veterinarian prior to each dose of intrathecal rhIDU. The evaluations included general appearance, posture, gait, motor, sensory and reflexes. The evaluations were not blinded for reasons of practicality: the veterinarian examining the dogs also administered intrathecal injections.
Routine laboratory studies
Blood samples were taken at each IT procedure (once per month or once every three months) for complete blood counts with differential and chemistry panels. CSF samples were taken at each IT injection for glucose, protein, color, specific gravity, and cell counts. CSF specimens contaminated with blood were not used for analysis.
Tissue evaluations
Tissue preparation and analysis were performed as described by Kakkis, et al [19]. Forty-eight hours or three months after the last IT rhIDU injection, the animals were necropsied and their brains, spinal cords, meninges and caudae equinae removed. The left hemispheres of the brains were fixed in 4% paraformaldehyde/2% glutaraldehyde in phosphate buffer (pH 7.2) and then cut into approximately 1 cm coronal sections for histopathology. The right hemispheres were sectioned coronally and snap frozen at approximately −80°C to be assayed biochemically for α-L-iduronidase activity and GAG storage. One cervical, thoracic, and lumbar section of spinal cord and meninges were taken from each animal for iduronidase and GAG assays. Spinal meninges consisted of dura and arachnoid materes assayed together.
Tissue samples (100–500 mg) were thawed and homogenized in three volumes of PAD buffer (10 mM phosphate, pH 5.8, 0.02% sodium azide and 0.1 mM dithiothreitol) with 0.1% Triton X-100. The homogenates were then assayed for α-L-iduronidase using a flurometric assay as per Kakkis, et al [19]. Protein concentrations in the extracts were determined by the Bradford method using reagents from Bio-Rad Laboratories (Hercules, California). Results were expressed as units per mg protein. The 4-methylumbelliferyl-α-L-iduronide substrate (4-MUI) used in these studies was obtained from Toronto Research Chemicals (North York, Ontario, Canada). When 4-MUI is used at the very high concentration of 3 mM, the assay detects trace amounts of β-glucuronidase activity due to trace amounts of glucuronide contamination in the substrate. For the dogs studied after a 3-month hiatus, a 4-MUI substrate manufactured by Glycosynth (Cheshire, UK) was used. The Glycosynth substrate has a glucuronide contamination of <0.1%.
GAG levels were quantified by an Alcian blue dye binding method as described by Kakkis, et al. and quantified within the linear range with dermatan sulfate standards [20]. Iduronidase activity and GAG levels were determined from independent triplicate assays of 4 to 48 independent brain sections per animal.
Histopathologic evaluation was performed on samples from the parietal cortex, cerebellum, spinal cord and meninges. The tissues were postfixed in osmium tetroxide and embedded in Spurr’s resin. Brain sections for light microscopy were stained with toluidine blue. Thin sections of neocortex were selected for electron microscopy and evaluated using a JOEL 100 CX electron microscope. Samples of the cervical, thoracic and lumbar spinal cord, cauda equina, brain, and meninges were fixed in 10% neutral buffered formalin for evaluation by standard light microscopy with hematoxylin and eosin stain.
Statistics
Means and standard deviations were calculated in standard fashion for each animal. Means of the means and their standard deviation were then calculated for each treatment group and compared to normal or untreated MPS I dogs using an unpaired, two-tailed student’s t-test with p < 0.05 considered significant.
Results
IT rhIDU diffuses widely and penetrates brain tissue
Based on prior work that found an effective treatment dose, we initiated studies of clinical practical regimens to ascertain if successful reduction in lysosomal storage could be achieved [19]. A total of fifteen dogs with canine MPS I were given IT rhIDU at monthly or quarterly intervals for a total of 4 or 3 injections respectively via cisterna magna injections (Fig. 1). Nine dogs received a1.38 mg (270,000 units) dose of IT rhIDU. Six dogs received the monthly regimen with a lower ~1/3rd dose (90,000 units) of rhIDU to assess whether even lower levels of enzyme might also achieve significant iduronidase levels and storage reduction. Four dogs receiving the low-dose rhIDU were evaluated three months after the end of therapy, to determine if storage reduction could be sustained over this interval (low-dose hiatus regimen). All other animals were evaluated 48 hours after the end of therapy and the brains assessed for rhIDU, biochemical measure of GAG storage and pathology. To mimic the clinical situation more accurately, these canines were also provided IV enzyme replacement therapy at 0.58 mg/kg (125,000 units/kg) once weekly as is approved for human use. The IV enzyme replacement is not expected to result in CNS benefit at this dose based on prior data [19–21]. These dogs were compared with four MPS I dogs previously published that received four weekly doses of about 1 mg IT rhIDU and no IV treatment [19].
Fig. 1.
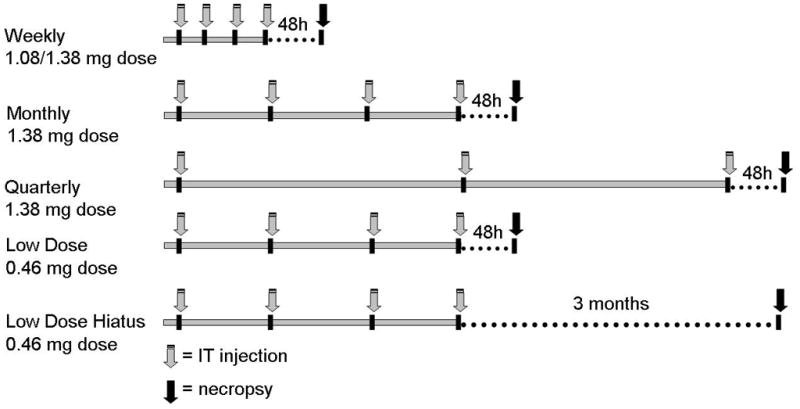
IT rhIDU regimens and doses received by the dogs. Dogs ranged in age from 13 to 40 months at the end of the study and consisted of 14 males and 5 females. Gray arrows indicate IT rhIDU injection. The weekly regimen (n=4) consisted of four ~ 1 mg (270,000 units) doses of IT rhIDU at weekly intervals. The monthly regimen (n=5) consisted of four 1.38 mg (270,000 units) doses of IT rhIDU at monthly intervals. Quarterly regimen (n=4) consisted of three 1.38 mg (270,000 units) doses IT rhIDU at three month intervals. Dogs were necropsied 48 hours after the last IT rhIDU injection (black arrows). Dogs receiving low-dose IT rhIDU received 0.46 mg (90,000 units) per dose at monthly intervals, and were necropsied 48 hours (n=2) or three months (n=4) after the final IT rhIDU dose (black arrows). Dogs on monthly, quarterly, and low dose regimens received concomitant weekly 0.58 mg/kg (125,000 units/kg) IV rhIDU. During the three-month hiatus, dogs continued to receive weekly IV rhIDU until necropsied.
At the end of the treatment regimen, the brains of the canines were collected, and divided in half, with one hemisphere for biochemical evaluations and the other hemisphere for histopathology. When multiple brain specimens from diverse areas in the brain were analyzed for iduronidase activity, very high levels of rhIDU were achieved with all treatment regimens in most treated CNS regions (Fig. 2). IT rhIDU distributed widely throughout brain tissue, penetrating into deeper tissue layers (≥3mm below the surface). At the 1.38 mg (270,000 units) dose, the monthly regimen resulted in 21-fold normal levels (248 ± 59.5 units per mg protein, p<0.0001, vs. normal 11.9 ± 1.95, n=5), and the quarterly regimen gave 20-fold normal levels (234 ± 117, p=0.0036 vs. normal). Similarly, weekly IT rhIDU provided iduronidase levels in the brain that reached 23-fold normal levels (277 ± 89.1, p= 0.0003 vs. normal) [19]. In the spinal cord, iduronidase levels were 6-fold with monthly, 5-fold with quarterly, and 13-fold normal with weekly IT rhIDU. Spinal meninges of treated dogs had rhIDU levels that were 285-fold normal with the monthly regimen, 264-fold normal with the quarterly regimen, and 300-fold normal on weekly IT rhIDU. A low-dose (0.48mg, 90,000 units) monthly regimen resulted in significant though lower iduronidase levels in all tissues. Brain iduronidase levels were roughly 3-fold normal (41.5 ± 19.2), spinal cord 2-fold, and spinal meninges 62-fold. All dogs on these treatment regimens had enzyme levels in each tissue type that were well above the few percent of normal iduronidase activity needed for correction of lysosomal storage.
Fig. 2.

Iduronidase levels in MPS I dogs treated with IT rhIDU. (a) Distribution of iduronidase activity in treated canine brains. Normal brain iduronidase activity is 11.9 ± 1.95 units/mg protein. In the brain, core samples in the temporoparietal regions were divided into surface and deep regions. Deep regions were at least three mm below the brain surface. All but one region had supranormal iduronidase activity (above 12 units/mg); the level in this region was 11.7 units/mg. Iduronidase levels are also shown in the spinal cord and spinal meninges. Spinal meninges sections consist of dura and arachnoid materes assayed together. Sections were taken from the cervical (C2-3), thoracic (T4-5), and lumbar (L4-5) regions. Normal spinal cord iduronidase is 11.7 ± 2.38; meninges, 15.4 ± 3.55. Values from 2 to 338 fold-normal were achieved with treatment. (b) Bar graph illustrating supranormal levels of iduronidase in total brain, spinal cord, and spinal meninges with monthly, quarterly, and low-dose IT rhIDU. As expected, lower iduronidase levels are achieved with the lower dose.
Initial results showed no detectable iduronidase in the CNS of low-dose hiatus dogs. With a more sensitive substrate, the assay detected 0.099 ± 0.033, which was 1.20% of the normal value 8.26 ± 0.746 for that substrate. Since the assay was performed on whole brain tissue, it is unclear whether this represents enzyme activity within the brain itself or solely within capillary endothelium.
IT rhIDU reduces GAG storage
The high levels of iduronidase resulted in substantial reduction of GAG levels in the brain and spinal meninges (Fig. 3). In the brain, weekly, monthly, quarterly, and low-dose monthly regimens all reduced GAG storage to normal levels. There was no significant variation in brain GAG levels among regimens. Brain GAG was normalized in all dogs regardless of age, which is important as older dogs have more brain GAG storage (Fig. 3b). In the spinal meninges, all three regimens achieved equivalent GAG storage reduction of up to 70% versus untreated MPS I dogs. Spinal meningeal GAG levels in weekly-treated dogs were reduced 57% and reached 15.3 ± 5.56 μg/mg dry weight, versus a level in untreated MPS I dogs of 35.9 ± 3.03 (n=2). The monthly regimen reduced spinal meningeal GAG levels 65% to 12.4 ± 4.96, the quarterly regimen 66% to 12.2 ± 2.69, and the low-dose regimen 70% to 10.6 ± 2.09. The reduction in spinal meningeal GAG, though large, did not quite reach normal levels (4.78 ± 0.818, n=4). In the spinal cord, where untreated storage levels are generally low (5.04 ± 0.933), there was no significant reduction of GAG storage. Spinal cord GAG levels with weekly therapy were 3.43 ± 0.718 μg/mg; with monthly, 4.23 ± 0.669, with quarterly, 4.62 ± 0.637, and with low-dose 5.03 ± 0.907. Normal spinal cord GAG was 3.02 ± 0.707 (n=4).
Fig. 3.

Quantitative glycosaminoglycan (GAG) storage in brain, spinal cord, and spinal meninges of normal dogs, untreated MPS I dogs, and MPS I dogs treated with IT rhIDU. (a) GAG levels in the brain were normal in all treatment groups. Mean GAG storage in the spinal meninges was reduced by more than half in all treatment groups versus untreated MPS I dogs. (b) Brain GAG levels in the IT-treated dogs reached normal levels regardless of age or treatment regimen.
Surprisingly, a three month hiatus following therapy did not lead to GAG reaccumulation. Low-dose hiatus dogs had normal brain GAG levels, 4.35 ± 0.783 μg/mg (p=0.0017 vs. untreated). Spinal meninges GAG levels were 9.74 ± 3.40, representing a 73% reduction from untreated values. Spinal cord levels were also similar to those in treated dogs evaluated 48 hours after therapy (3.98 ± 2.14). Storage levels in the brain and spinal meninges appear to be equally reduced with weekly, monthly, quarterly, and low-dose IT rhIDU, and a three month interval does not appear to allow for storage levels to rise.
Intrathecal rhIDU reduces histologic evidence of lysosomal storage
Having shown a biochemical improvement in lysosomal storage, we next evaluated storage histologically to determine the success of IT rhIDU in individual cell types. All IT-treated MPS I dogs showed lysosomal storage reduction in at least some cell types in the brain at 48 hours after the final IT dose. Lysosomal storage was reduced in neocortical leptomeninges and perivascular cells in 14 of the 15 MPS I dogs treated with weekly, monthly, and quarterly ~ 1 mg (270,000 units), and low-dose monthly 0.46 mg (90,000 units) IT rhIDU regimens. Neuronal storage was evaluated by electron microscopy (EM) in the treated MPS I dogs and compared to two untreated MPS I dogs. In neurons and glia where storage is relatively mild to moderate, a reduction of lysosomal storage was observed in 14 of the 15 treated dogs evaluated (Fig. 4). One dog treated with low-dose monthly rhIDU (Bd) appeared to have no reduction in neuronal storage, though the dog did have reduction in other cell types. Though storage reduction in neurons was moderate, the character of the material changed markedly with treatment. In untreated MPS I dogs, neuronal storage took the form of granular, flocculent, membranous and zebra body lysosomal storage, while in treated dogs, there were aggregates of electron dense complex material with and without stacks of membranous material. The change in the character of the neuronal storage most likely represents decreased GAG and secondary ganglioside storage associated with the membranous and zebra body storage. The residual electron-dense bodies may also be age related changes.
Fig. 4.

Pathological evaluation of IT-treated MPS I dogs shows improved GAG storage in perivascular cells (top panels) and neurons (bottom panels) compared with untreated MPS I dogs. LM, light microscopy; EM, electron microscopy. Toluidine blue stain of perivascular cells (centered in each upper panel) shows lysosomal storage (arrowhead) in an untreated dog that is not apparent in that of the treated dog. The blood vessel lumen is marked with an asterisk. Electron microscopy (lower panels) shows lipid and GAG storage vesicles (arrows) in the neuron of an untreated dog that is greatly reduced in a treated dog. Treated dogs did have some neuronal storage, though it was decreased from untreated levels.
Light microscopy of the spinal cord and spinal meninges of all treated MPS I dogs showed reduced lysosomal storage versus untreated MPS I dogs. There remained, however, some cytoplasmic vacuolation (storage accumulation) in scattered macrophages and meningothelial cells predominantly in the thoracic to lumbar segments of the spinal column. There was no apparent difference in morphology of spinal neurons of treated and untreated dogs at the light microscopic level.
Following a three-month hiatus, storage did not reaccumulate in neurons of 2 of 4 treated dogs. Two dogs (Cu, El) had reduced neuronal storage three months after the last treatment that was comparable to IT-treated dogs evaluated at 48 hours. The remaining 2 dogs had evidence of storage reaccumulation in neurons. We were unable to detect improvement in the neocortical menigeal cells, perivascular cells, or glia, despite the quantitative reduction in overall GAG storage measured biochemically in these dogs evaluated three months after the final IT rhIDU dose.
IT rhIDU results in clinical improvement
We performed neurologic evaluations on affected animals at treatment intervals to determine whether there may be clinical benefit to rhIDU. Two dogs treated with IT rhIDU (Um and Ye) had neurologic signs at the beginning of treatment. One symptomatic dog (Um) with gait disturbances attributed to cord compression was followed with neurologic assessments before and after four monthly IT treatments. Before treatment, the dog could not bear weight for more than a few seconds and would not move spontaneously. After treatment, the dog ran actively, though its gait on occasion was still wobbly (Supplementary Video). This dog had received prior intravenous enzyme replacement therapy without improvement in these symptoms, consistent with our prior experience. A second symptomatic dog with weak gait (Ye) did not improve with IT treatment. This dog was terminated after the third IT treatment and had severe extruded discs compressing the spinal cord. The remaining animals had no neurologic signs prior to, during, or following treatment with IT rhIDU.
Safety of rhIDU in MPS I dogs
Though IT rhIDU had been generally well-tolerated by normal dogs, some of the first IT-treated MPS I dogs experienced temporary side effects [19]. Ten of the nineteen treated MPS I dogs had hyperventilation and five had twitching during recovery from anesthesia. Five dogs had seizures in the post-procedure period that responded immediately to diazepam. In our prior research with normal dogs, two dogs receiving IT rhIDU had suffered seizures, which had responded to dialyzing the formulation buffer [19]. We attributed these immediate side effects to the low pH (5.5) of rhIDU. Diluting rhIDU 1/3 by volume in Elliotts B artificial CSF solution which contains bicarbonate buffer increased its pH to 6.1. Seventeen dogs, including three dogs that had side effects with undiluted IT rhIDU, experienced no seizures, hyperventilation, or twitching when treated with the diluted enzyme. One dog, initially on 1.38 mg (270,000 units) monthly IT rhIDU, died abruptly following the second IT administration from a large brainstem hematoma caused by a traumatic tap.
Reduction in inflammatory infiltrate with less frequent and lower dose IT rhIDU
In our previous research using weekly IT rhIDU, treated dogs had developed a moderate lymphoplasmocytic meningitis which did not impact them clinically [19]. Using less frequent and/or low-dose IT rhIDU appears to reduce the meningitis, in those dogs with evidence of an immune response (Fig. 5). There was absent to mild meningitis in dogs receiving quarterly IT rhIDU, and no meningitis in five of six dogs receiving low-dose rhIDU. The remaining low-dose dog (Bd) had a moderate, neutrophilic meningitis, which is not typical for an immune-mediated meningitis from IT rhIDU. Some dogs were made tolerant to rhIDU prior to study enrollment, which may limit our ability to assess their immune response [21]. CSF leukocyte counts at study end were ≤5 cells/μL in 6 dogs, 15–18 cells/μL in 3 dogs, and 74 cells/μL in one dog receiving monthly, quarterly, and low-dose IT regimens. There were modest elevations in the peripheral leukocyte count for seven dogs receiving IT rhIDU (16,900–24,100 cells per μL). There were no other clinically significant changes in serum chemistries or other cell counts during treatment.
Fig. 5.

Reduced meningitis with less frequent dosing and immune suppression in dogs treated with IT rhIDU. (a) A nontolerant dog treated with weekly IT rhIDU developed a moderate meningitis, as shown by hematoxylin and eosin stain of the spinal meninges. A dog receiving the quarterly regimen (b) and a dog receiving the low dose regimen (c) developed a much milder degree of meningitis. Both dogs had received immunosuppressive drugs, and the low dose dog (c) became tolerant to iduronidase. A dog treated with the low dose regimen, evaluated after a three month hiatus, also developed minimal meningitis (d). This dog was not tolerant to iduronidase. Cell differentials and pathological evaluation showed the meningitis to consist of lymphocytes, plasma cells, and monocytes/macrophages. This infiltrate was seen in the neocortical and spinal meninges, and was absent in the parenchyma of the brain and spinal cord.
Discussion
Treatment of brain disease via the CSF represents a potential strategy for lysosomal storage disorders which had not been previously thought to be viable as an effective approach to treatment, based on studies with growth factors [1, 2]. In this study, we successfully treated brain disease in MPS I dogs using an enzyme that can readily diffuse through tissue and efficiently enter cells. This was achieved at a dose and frequency that could be translated to clinical use. All IT-treated MPS I dogs had mean iduronidase levels in the brain, spinal cord, and spinal meninges that were many-fold above normal. GAG storage in the brain was normal in all MPS I dogs treated with IT rhIDU. Moreover, GAG levels remained low three months after the last IT rhIDU dose.
The intrathecal approach to treatment had been thought unlikely to succeed, due to the difficulty in traversing the ependymal lining of the brain and diffusing into the cells beneath. The drivers for success with rhIDU were the very high concentration gradient achievable in the spinal fluid with only a very small dose, the diffusability of the enzyme and its high-affinity, receptor-mediated uptake into cells. The ependymal layer in MPS I dogs may also be more permeable than in normal dogs. Higher levels of iduronidase were achieved in the brain in MPS I versus normal dogs receiving IT rhIDU, though both groups achieved high levels [19]. Conceptually, a large gradient of an enzyme with an efficient uptake mechanism at the final destination in the cell were enough to achieve adequate treatment of the brain in this lysosomal storage model of degenerative brain disease.
Improvement in neurologic symptoms in one affected dog treated with IT enzyme replacement therapy suggests that the therapy could have clinical benefit in patients. There is an early report case of a human patient receiving IT rhIDU using our method (R. Giugliani, et al., abstract, 2005). The patient had some improvements in strength, pain, ambulation (by twelve-minute walk test), and pulmonary function. There was no evidence of meningitis or other adverse effects from IT therapy.
In this study, we showed that IT rhIDU can not only diffuse widely throughout the CNS and treat disease there, but also that it can work effectively with a clinically applicable injection frequency and dose, even as infrequently as every 3 months. Brain GAG storage, measured biochemically, did not reaccumulate 3 months after IT rhIDU. A similar observation was made in MPS VII mice, in which cortical neurons maintained a slight GAG reduction one year after recombinant human beta-glucuronidase treatment [22]. The apparent slow reaccumulation of storage in cortical neurons of MPS I dogs receiving IT rhIDU suggests that, as in MPS VII mice, their enzyme requirement may be low. The very high initial iduronidase levels likely provide sufficient residual therapeutic enzyme for the three-month period, based on an estimated half-life of seven days in brain [18]. IT treatment is already in clinical use for chronic pain relief and CNS cancer, and it is less invasive and more clinically practical than other potential therapies using intraparenchymal injections. A low-dose, monthly or quarterly schedule makes IT treatment more feasible for use in patients as an outpatient procedure. The ability to treat brain disease via IT enzyme replacement therapy has important implications for the treatment of other lysosomal storage diseases, many of which affect the brain primarily or even exclusively.
Supplementary Material
Acknowledgments
Funding provided by grants from the Ryan Foundation and BioMarin Pharmaceutical, Inc. We thank Rita Esquivel, Hayden Manuel, Catherine Jabagat, Catalina Guerra, Sarah Snider and Dan Garner for their technical assistance. We thank Karen Russell, DVM, at Texas A&M Department of Veterinary Pathobiology for assistance with canine laboratory specimens.
References
- 1.Gill SS, Patel NK, Hotton GR, O’Sullivan K, McCarter R, Bunnage M, Brooks DJ, Svendsen CN, Heywood P. Direct brain infusion of glial cell line-derived neurotrophic factor in Parkinson disease. Nat Med. 2003;9:589–595. doi: 10.1038/nm850. [DOI] [PubMed] [Google Scholar]
- 2.Bobo RH, Laske DW, Akbasak A, Morrison PF, Dedrick RL, Oldfield EH. Convection-enhanced delivery of macromolecules in the brain. Proc Natl Acad Sci USA. 1994;91:2076–2080. doi: 10.1073/pnas.91.6.2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Consiglio A, Quattrini A, Martino S, Bensadoun JC, Dolcetta D, Trojani A, Benaglia G, Marchesini S, Cestari V, Oliverio A, Bordignon C, Naldini L. In vivo gene therapy of metachromatic leukodystrophy by lentiviral vectors: correction of neuropathology and protection against learning impairments in affected mice. Nat Med. 2001;7:310–316. doi: 10.1038/85454. [DOI] [PubMed] [Google Scholar]
- 4.Frisella WA, O’Connor LH, Vogler CA, Roberts M, Walkley S, Levy B, Daly TM, Sands MS. Intracranial injection of recombinant adeno-associated virus improves cognitive function in a murine model of mucopolysaccharidosis type VII. Mol Ther. 2001;3:351–358. doi: 10.1006/mthe.2001.0274. [DOI] [PubMed] [Google Scholar]
- 5.Kroll RA, Neuwelt EA. Outwitting the blood-brain barrier for therapeutic purposes: osmotic opening and other means. Neurosurg. 1998;42:1083–1099. doi: 10.1097/00006123-199805000-00082. [DOI] [PubMed] [Google Scholar]
- 6.Hobbs JR, Hugh-Jones K, Barrett AJ, Byrom N, Chambers D, Henry K, James DC, Lucas CF, Rogers TR, Benson PF, Tansley LR, Patrick AD, Mossman J, Young EP. Reversal of clinical features of Hurler’s disease and biochemical improvement after treatment by bone marrow transplantation. Lancet. 1981;2:709–712. doi: 10.1016/s0140-6736(81)91046-1. [DOI] [PubMed] [Google Scholar]
- 7.Whitley CB, Belani KG, Chang PN, Summers CG, Blazar BR, Tsai MY, Latchaw RE, Ramsay NKC, Kersey JH. Long-term outcome of Hurler syndrome following bone marrow transplantation. Am J Med Genet. 1993;46:209–218. doi: 10.1002/ajmg.1320460222. [DOI] [PubMed] [Google Scholar]
- 8.Peters C, Shapiro EG, Anderson J, Henslee-Downey PJ, Klemperer MR, Cowan MJ, Saunders EF, deAlarcon PA, Twist C, Nachman JB, Hale GA, Harris RE, Rozans MK, Kurtzberg J, Grayson GH, Williams TE, Lenarsky C, Wagner JE, Krivit W, the members of The Storage Disease Collaborative Study Group Hurler syndrome: II. Outcome of HLA-genotypically identical sibling and HLA-haploidentical related donor bone marrow transplantation in fifty-four children. Blood. 1998;91:2601–2608. [PubMed] [Google Scholar]
- 9.Staba SL, Escolar ML, Poe M, Kim Y, Martin PL, Szabolcs P, Allison-Thacker J, Wood S, Wenger DA, Rubinstein P, Hopwood JJ, Krivit W, Kurtzberg J. Cord-blood transplants form unrelated donors in patients with Hurler’s syndrome. N Engl J Med. 2004;350:1960–1969. doi: 10.1056/NEJMoa032613. [DOI] [PubMed] [Google Scholar]
- 10.Kakkis ED, Muenzer J, Tiller GE, Waber L, Belmont J, Passage M, Izykowski B, Phillips J, Doroshow R, Walot I, Hoft R, Neufeld E. Enzyme-replacement therapy in mucopolysaccharisosis I. N Engl J Med. 2001;344:182–188. doi: 10.1056/NEJM200101183440304. [DOI] [PubMed] [Google Scholar]
- 11.Wraith JE, Clarke LA, Beck M, Kolodny EH, Pastores GM, Muenzer J, Rapoport DM, Berger KI, Swiedler SJ, Kakkis ED, Braakman T, Chadbourne E, Walton-Bowen K, Cox GF. Enzyme replacement therapy for mucopolysaccharidosis I: a randomized, double-blinded, placebo-controlled, multinational study of recombinant human α-L-iduronidase (laronidase) J Pediatr. 2004;144:581–588. doi: 10.1016/j.jpeds.2004.01.046. [DOI] [PubMed] [Google Scholar]
- 12.Harmatz P, Kramer WG, Hopwood JJ, Simon J, Butensky E, Swiedler SJ on behalf of the Mucopolysaccharidosis VI Study Group. Pharmacokinetic profile of recombinant human N-acetylgalactosamine 4-sulphatase enzyme replacement therapy in patients with mucopolysaccharidosis VI (Maroteaux–Lamy syndrome): a phase I/II study. Acta Padiatrica. 2005;94(Suppl 447):61–68. doi: 10.1111/j.1651-2227.2005.tb02115.x. [DOI] [PubMed] [Google Scholar]
- 13.Barton NW, Brady RO, Dambrosia JM, Di Bisceglie AM, Doppelt SH, Hill SC, Mankin HJ, Murray GJ, Parker RI, Argoff CE, Grewal RP, Yu KT, et al. Replacement therapy for inherited enzyme deficiency: macrophage-targeted glucocerebrosidase for Gaucher’s disease. N Engl J Med. 1991;324:1464–1470. doi: 10.1056/NEJM199105233242104. [DOI] [PubMed] [Google Scholar]
- 14.Eng CM, Guffon N, Wilcox WR, Germain DP, Lee P, Waldek S, Caplan L, Linthorst GE, Desnick RJ the International Fabry Disease Study Group. Safety and efficacy of recombinant human a-galactosidase A replacement therapy in Fabry’s disease. N Engl J Med. 2001;345:9–16. doi: 10.1056/NEJM200107053450102. [DOI] [PubMed] [Google Scholar]
- 15.Schiffmann R, Murray GJ, Treco D, Daniel P, Sellos-Moura M, Myers M, Quirk JM, Zirzow GC, Borowski M, Loveday K, Anderson T, Gillespie F, Oliver KL, Jeffries NO, Doo E, Liang TJ, Kreps C, Gunter K, Frei K, Crutchfield K, Selden RF, Brady RO. Infusion of α-galactosidase A reduces tissue globotriaosylceramide storage in patients with Fabry disease. Proc Natl Acad Sci USA. 2000;97:365–370. doi: 10.1073/pnas.97.1.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sifuentes M, Doroshow R, Hoft R, Mason G, Walot I, Diament M, Okazaki S, Huff K, Cox GF, Swiedler SJ, Kakkis ED. A follow-up study of MPS I patients treated with laronidase enzyme replacement therapy for 6 years. Mol Genet Metab. 2006 doi: 10.1016/j.ymgme.2006.08.007. in press. [DOI] [PubMed] [Google Scholar]
- 17.Kakkis ED, Matynia A, Jonas AJ, Neufeld EF. Overexpression of the human lysosomal enzyme α-L-iduronidase in Chinese hamster ovary cells. Prot Expr Purif. 1994;5:225–232. doi: 10.1006/prep.1994.1035. [DOI] [PubMed] [Google Scholar]
- 18.Belichenko PV, Dickson PI, Passage M, Jungles S, Mobley WC, Kakkis ED. Penetration, diffusion, and uptake of recombinant human α-L-iduronidase after intraventricular injection into the rat brain. Mol Genet Metab. 2005;86:141–149. doi: 10.1016/j.ymgme.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 19.Kakkis E, McEntee M, Vogler C, Le S, Levy B, Belichenko P, Mobley W, Dickson P, Hanson S, Passage M. Intrathecal enzyme replacement therapy reduces lysosomal storage in the brain and meninges of the canine model of MPS I. Mol Genet Metab. 2004;83:163–174. doi: 10.1016/j.ymgme.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 20.Kakkis ED, McEntee MF, Schmidtchen A, Neufeld EF, Ward DA, Gompf RE, Kania S, Bedolla C, Chien SL, Shull RM. Long-term and high-dose trials of enzyme replacement therapy in the canine model of mucopolysaccharidosis I. Biochem Mol Med. 1996;58:156–167. doi: 10.1006/bmme.1996.0044. [DOI] [PubMed] [Google Scholar]
- 21.Kakkis E, Lester T, Yang R, Tanaka C, Anand V, Lemontt J, Peinovich M, Passage M. Successful induction of immune tolerance to enzyme replacement therapy in canine mucopolysaccharidosis I. Proc Natl Acad Sci USA. 2004;101:829–834. doi: 10.1073/pnas.0305480101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vogler C, Sands MS, Levy B, Galvin N, Birkenmeier EH, Sly WS. Enzyme replacement with recombinant beta-glucuronidase in murine mucopolysaccharidosis type VII: impact of therapy during the first six weeks of life on subsequent lysosomal storage, growth, and survival. Pediatr Res. 1996;39:1050–1054. doi: 10.1203/00006450-199606000-00019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.