

Abstract
The solution dynamics of an enzyme acid-β-glucocerebrosidase (GCase) probed at a physiologically relevant (lysosomal) pH by hydrogen/deuterium exchange mass spectrometry (HDX-MS) reveals very uneven distribution of backbone amide protection across the polypeptide chain. Highly mobile segments are observed even within the catalytic cavity alongside highly protective segments, highlighting the importance of the balance between conformational stability and flexibility for enzymatic activity. Forced oxidation of GCase that resulted in a 40–60% reduction in in vitro biological activity affects the stability of some key structural elements within the catalytic site. These changes in dynamics occur on a longer time scale that is irrelevant for catalysis, effectively ruling out loss of structure in the catalytic site as a major factor contributing to the reduction of the catalytic activity. Oxidation also leads to noticeable destabilization of conformation in remote protein segments on a much larger scale, which is likely to increase the aggregation propensity of GCase and affect its bioavailability. Therefore, it appears that oxidation exerts its negative impact on the biological activity of GCase indirectly, primarily through accelerated aggregation and impaired trafficking.
Keywords: hydrogen/deuterium exchange, electrospray ionization, protein ion charge-state distribution, mass spectrometry, lysosomal storage disorder, Gaucher's disease, biologics
Introduction
Acid-β-glucocerebrosidase (GCase) is an essential metabolic enzyme, which catalyzes the hydrolysis of glucocerebroside in the acidic environment of lysosomes.1 Mutations leading to the partial or complete impairment of GCase activity cause Type 1 Gaucher's disease, the most prevalent lysosomal storage disorder worldwide.2,3 The absence of functional GCase prevents glucosylceramide (derived from catabolism of glycosphingolipids by lysosomal hydrolases) from being degraded further, resulting in an accumulation of lipids that leads to symptoms such as hepatosplenomegaly, growth retardation, bone pain, and liver and heart failure.4
Enzyme replacement therapy initially using GCase purified from human placentae, and later produced as a recombinant protein, has revolutionized the care of patients with Type 1 Gaucher's disease, reversing many pathological consequences and preventing its progression.4 The therapy is administered through intravenous infusion of the therapeutic protein on a periodic schedule. Although gene therapy and use of small molecule “pharmacological chaperones” for stabilizing defective GCase have recently shown some promise,5,6 enzyme replacement therapy currently remains the standard of care for Type 1 Gaucher's disease.
As is the case with other biopharmaceuticals, protein conformation and stability of GCase are key elements of efficacy and safety. The crystal structure of GCase has provided insight into the molecular mechanism of its action as well as provided limited evidence of how localized conformational dynamics are important for the enzymatic activity of the protein.1 Segments with increased backbone flexibility were also revealed in recent hydrogen/deuterium exchange mass spectrometry (HDX-MS) studies of GCase dynamics7 at neutral pH, where several hot spots were identified against the backdrop of the predominantly stable protein. This fine balance between the overall stability of GCase and the highly localized flexibility is very important: out of many mutations implicated in diminishing the biological activity of GCase,8 several are known to compromise the protein stability (with likely negative repercussions for bioavailability), even though the catalytic activity is not affected directly.9 In fact, mutations in the GCase gene exert a large effect on susceptibility for Lewy body disorders, such as Parkinson's disease,10,11 with facile protein aggregation being one of the likely mechanisms.10
Although the wild-type and gene-activated GCase12 are capable of maintaining the delicate balance between the overall conformational stability (to enhance bioavailability) and localized flexibility (to optimize the enzymatic activity), subtle modifications of the protein structure due to nonenzymatic post-translational modifications (PTMs) may alter conformational dynamics of the protein in a very significant way. Protein oxidation is one of the most frequent PTM events13 that may occur both in vivo and during the storage of the protein substance. Extensive oxidation of GCase is known to impair its activity (data not shown), although the specific mechanism remains unknown.
In this work, we compared the conformational stability of intact and extensively oxidized GCase (GCase-ox) at a mildly acidic pH that would be typical of the lysosomal environment and where GCase exhibits optimal enzymatic activity. Although the uneven distribution of flexibility across the protein backbone at acidic pH is strikingly similar to that observed in neutral solutions,7 our findings also reveal a noticeable increase of local dynamics in GCase-ox. The regions with diminished conformational stability are localized in a few protein segments positioned at the entrance to the catalytic cavity, as well as the protein periphery. Taken together, the results of this work suggest that the greatly diminished activity of GCase caused by extensive oxidation is likely to result from the decrease of its conformational stability, rather than direct impairment of enzymatic function caused by the loss of structure around the catalytic site.
Results
Charge-state distribution of GCase ions in nanoelectrospray ionization mass spectra acquired at lysosomal pH
The electrospray ionization (ESI-MS) of intact GCase acquired at physiological pH (blue trace in Fig. 1) displays a narrow distribution of protein ion peaks with low charge density (charge states +13 to +16), consistent with a highly compact protein conformation in solution14; the reference spectrum of acid-unfolded GCase (presented in Fig. 1 with a gray trace) displays significantly higher extent of multiple charging and broader charge state distribution. The convoluted shape of the protein ion peaks at any given charge state reflects the presence of several glycoforms (the blue trace in the inset in Fig. 1 presents a deconvoluted mass distribution of intact GCase monomers). The only other ionic species present in the mass spectrum of intact GCase at pH 4.5 is represented by low-abundance signal in the high m/z region (above 5000). Estimation of the mass of this species based on the available ESI-MS data provides a value that exceeds the mass of the monomeric GCase species by a factor of two, which is likely to be a dimer; the low signal-to-noise ratio and large amount of adducts prevent accurate mass calculation for this species.
Figure 1.

Nano-ESI mass spectra (inset contains the deconvoluted spectra) of 6 μM GCase (blue) and GCase-ox (red) solutions in 50 mM ammonium acetate at pH 4.5. The gray trace shows a reference mass spectrum of GCase under denaturing conditions (50% acetonitrile and 5% acetic acid).
The most abundant ionic species in the ESI-MS of GCase-ox also populate the m/z region 3500–5000 (red trace in Fig. 1). However, the charge-state distribution is noticeably skewed toward higher charge states, making ion peaks at +17 to +19 clearly visible. Another distinct feature of this spectrum is the presence of moderate charge density ionic species in m/z region 2000–3000 (up to charge state +30), whose masses also correspond to monomeric GCase-ox. The ionic species appearing in the high m/z region of the spectrum (above 5000) correspond to the GCase-ox dimers (deconvoluted mass 125.8 ± 0.5 kDa), but their charge density is noticeably higher than that of the intact GCase dimers.
HDX-MS of intact GCase at lysosomal pH
HDX-MS data for intact GCase at pH 4.5 were collected at five time points (0.5, 5, 15, 60, and 120 min), chosen empirically based on preliminary data to best represent the dynamic range of the exchange reactions for various protein segments. An acid-unfolded GCase sample that was exchanged for 120 min was used to represent the exchange reaction endpoint as well as to determine the amount of back exchange for each peptide in HDX-MS measurements. According to calculations based on intrinsic exchange rates,15 completely unfolded GCase would undergo more than 99% exchange in these conditions. Figure 2(A) shows the evolution of isotopic distributions for several peptic fragments representing a highly protected region encompassing the catalytic site (232–240), and a less protected flexible segment (341–347), which is part of a flexible loop flanking one of the glutamic acid residues (E340) involved in catalysis. The rest of this loop (segment 348–351), which is further away from the catalytic site, displays even greater flexibility as evidenced by the rapid exchange of labile hydrogen atoms for deuterium atoms during the early stages of the exchange reactions [Fig. 2(A)]. These raw data, normalized with the end points of the exchange reaction for each peptide can be used to calculate the kinetics of isotope exchange in solution [Fig. 2(B)].
Figure 2.

The isotopic distributions of peptide ions representing peptic fragments (232–240, 341–347, and 348–351) of GCase following 0.5, 5, 15, 60, and 120 min of HDX in solution (A) and the kinetic plots showing exchange of backbone amide hydrogen atoms within these peptides (B).
The extent of protein backbone coverage and the spatial resolution achieved in HDX-MS measurements are dictated by the number and size of peptide fragments generated by the acidic protease (pepsin) following the quench of the exchange reaction in solution. GCase is composed of 497 amino acids (of which 34 are proline residues) and four high-mannose oligosaccharides. We identified 37 nonredundant peptic fragments of GCase under the conditions compatible with the HDX-MS protocol, which cover 87% of the protein sequence. As the information on the amide occupancy is lost for N-terminal residues in each peptic fragment (due to their conversion to amines upon peptide bond hydrolysis), the observed peptic fragments actually cover 378 backbone amides or 82% of the total 463 hydrogen-bearing backbone amide groups in the protein. The GCase segments covered in HDX-MS experiments include one of the two glutamic acid residues essential for catalysis and immediately flank the second residue. The largest gap in GCase sequence not covered by peptic fragments spans residues 91–105. It is possible that this leucine-rich segment is actually proteolyzed into smaller fragments that are not efficiently retained by the peptide trap, and therefore, elude MS detection. The number of nonproline backbone amide groups in observed peptic fragments ranged from 2 to 25. In some instances, spatial resolution was improved by subtracting data for two overlapping peptides that share an identical N- or C-terminus (producing segments 143–153 and 283–285).
Figure 3 represents mapping of HDX-MS data to the primary structure (A) and tertiary structure (B and C) of GCase. Although the protein appears to be highly protected overall (evident by the very modest 15% deuterium incorporation averaged across all measured segments at the earliest time point and only 35% incorporation at 120 min), several regions stand out against a backdrop of overall stability by displaying a highly dynamic character. The most dynamic region was covered by two peptic fragments (341–347 and 348–351) that are located immediately adjacent to the catalytically essential E340, which also maps to Loop 1 in the crystal structure (Fig. 3). Protein segments flanking Loop 1 on each side also displayed higher than average rates of exchange, which also included partial coverage of Loop 3. Loop 2 appears to be more stable in comparison with Loops 1 and 3 (Fig. 3). Other highly dynamic segments consist of coils between secondary structural elements as well as the N-terminal domain (Domain I).
Figure 3.

Protection of peptide fragments of GCase at pH 4.5 revealed by HDX-MS data plotted to the protein sequence (A) and HDX protection data for 0.5 (B) and 60 min (C) mapped to the crystal structure of the protein. Peptic fragments (excluding their respective N-terminal amino acids) are shown as horizontal bars below the sequence in Panel A and are colored according to the fraction of exchanged backbone amide hydrogen atoms. Each row of bars represents data collected at a designated time point (0.5, 5, 15, 60, and 120 min from top to bottom). The two active sites of glutamic acid residues are indicated in Panel B. Black-colored protein segments in Panels B and C indicate regions for which the HDX-MS data are not available.
Comparison of our HDX-MS data obtained under acidic conditions to the flexibility map of the wild-type GCase obtained earlier at pH 7.8 (Ref. 77) reveals similar protection patterns and highlights the stability of this protein over a wide pH range. Although the direct quantitative comparison of the data acquired at two different pH levels is made difficult by the slight incongruence of the two peptic maps and by the fact that intrinsic exchange rates are highly dependent on solution pH,16 there is obvious qualitative similarity between the two sets in terms of distribution of protection across the entire protein backbone. Two noticeable differences are observed, both favoring higher local protection under mildly acidic (lysosomal) conditions over neutral pH. Peptic fragment (185–197), which spans a loop in Domain III, displays a moderate level of exchange at pH 4.5, retaining nearly 50% of labile hydrogen atoms after 2 hours of exchange (slightly higher than the average, vide supra). In contrast, the similar peptide 187–197 at pH 7.8 represents an obvious dynamic hot spot in the sequence, displaying a staggering 70% exchange at the earliest time point, which dramatically exceeds the average protection level.7 Similarly, an increase in protection at lysosomal pH is also observed within Loop 2 [e.g., peptide (384–396) in Figure 3(A)], which is relatively stable at pH 4.5 compared to peptic fragment (386–398) that exhibits higher than average flexibility at pH 7.8.7
HDX-MS of oxidized GCase at lysosomal pH
There are 113 amino acid residues in GCase, which can be considered potential oxidation sites17 (nine methionine, seven cysteine, 22 arginine, 18 histidine, 26 phenylalanine, 12 tryptophan, and 19 tyrosine residues); forced oxidation of GCase produces a distribution of species with different patterns of oxidation. According to the liquid chromatography (LC)-MS analysis of a proteolyzed sample of the oxidized protein, the sites most affected by oxidation are M53, M335, and M450, although noticeable oxidation was also observed at two other sites, that is, oxidation of C126 and C342 to sulfinic acid (Fig. 4). The total number of GCase species exhibiting all possible oxidation patterns within these five sites is 32 (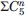 , where
, where 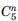 is a number of combinations of n elements out of 5, and summation is made from 0 to 5), and physical separation of these species is practically impossible. Therefore, the entire oxidized sample is treated as a single species (GCase-ox). Analysis of conformational dynamics of GCase-ox carried out with HDX-MS reveals a slight decrease of the overall degree of the backbone protection when averaged across the entire length of the polypeptide chain (32% HDX at 100 min vs. 30% for the intact GCase sample), although the affected segments are localized within only a few protein regions, as changes were detected in seven peptic fragments of GCase-ox (out of a total of 35). Three exhibited a moderate increase in HDX (ΔHDX values between 5 and 6%) and the other four were more significant (ΔHDX values between 10 and 20%).
is a number of combinations of n elements out of 5, and summation is made from 0 to 5), and physical separation of these species is practically impossible. Therefore, the entire oxidized sample is treated as a single species (GCase-ox). Analysis of conformational dynamics of GCase-ox carried out with HDX-MS reveals a slight decrease of the overall degree of the backbone protection when averaged across the entire length of the polypeptide chain (32% HDX at 100 min vs. 30% for the intact GCase sample), although the affected segments are localized within only a few protein regions, as changes were detected in seven peptic fragments of GCase-ox (out of a total of 35). Three exhibited a moderate increase in HDX (ΔHDX values between 5 and 6%) and the other four were more significant (ΔHDX values between 10 and 20%).
Figure 4.
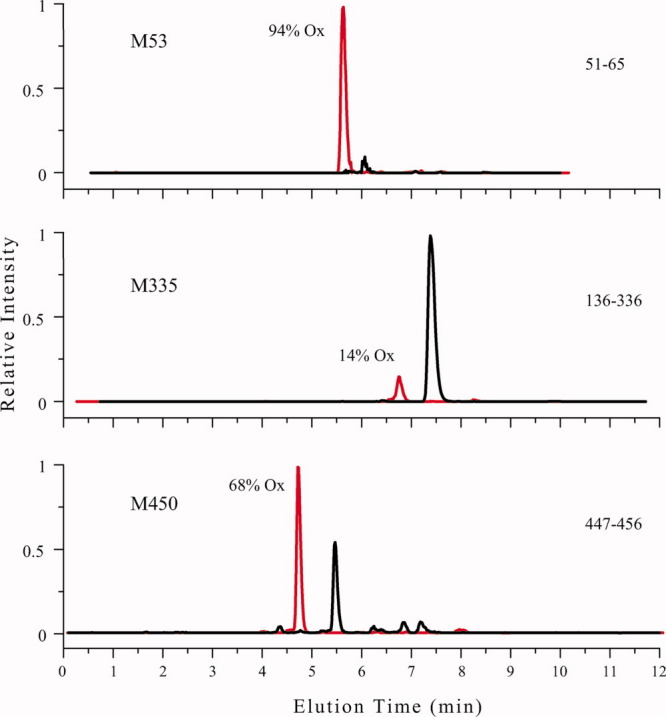
Extracted ion chromatograms for a peptic digest of GCase-ox used to quantitate the extent of oxidation for the indicated residues. Data are plotted red for the oxidized species and black for nonoxidized species. Each panel is labeled by the peptic fragment used, the site of oxidation, and the percentage oxidation calculated for each residue.
Of the peptides whose backbone protection was affected by protein oxidation, three exhibited bimodal isotopic distributions, with two examples shown in Figure 5. This phenomenon is usually a hallmark of the so-called EX1 exchange regime16,18 and reveals the existence of at least two different conformations in solution with markedly different levels of backbone protection. Each of the bimodal isotopic distributions presented in Figure 5 clearly contains two parts, one of which aligns almost perfectly with the isotopic distribution of the same peptide derived from nonoxidized GCase. This allowed us to carry out deconvolution of the bimodal isotopic distributions of the peptic fragments derived from GCase-ox by subtracting from them the normalized isotopic distributions of the same peptides derived from intact GCase. This procedure produces a nearly symmetrical second component of the isotopic distribution of each peptide. In each of the three cases in question, it is close to the profile of the exchange reaction end point, and therefore, it represents an alternative (non-native) protein conformation that is highly unprotected within the corresponding protein segment.
Figure 5.

Isotopic distributions of peptide ions representing peptic fragments (185–209, 232–240, 220–227, and 447–456) of GCase (blue) and GCase-ox (red) following 1 and 100 min of HDX in solution. The gray traces represent isotopic distributions within the same peptide for acid denatured GCase, which allowed for complete exchange (e.g., the maximum level of amide exchange for the peptide) and was analyzed under the same conditions as native GCase. The deconvoluted isotopic distribution attributed to disordered GCase-ox is shown as thin black lines in the top two panels. In the lower right panel, peptides with an oxidized M450 are indicated with a star.
In each of these cases, the ratio of the two components of the peptide ion isotopic distribution can be used to estimate the fraction of the protein molecules that sampled the “disordered” conformation at least once during the exchange in solution. The term “disordered” refers not to the entire protein, but to the segment of the polypeptide chain represented by the peptic fragment. The percentage of GCase-ox molecules that have sampled the conformation(s), which are highly disordered in the (185–209) region, is 25% for the 1-min time interval and 30% for the 100-min time interval. The percentage of GCase-ox molecules that have sampled conformation(s), which are highly disordered in segments (232–240) and (384–396), is negligible at the 1-min time interval; and within the 100-min time period, this percentage becomes 25%. As the protein molecules in the GCase-ox sample have on average 1.5 modifications per molecule and unmodified species are virtually absent, the slow-exchanging fraction of GCase-ox cannot be a signature of remaining intact (nonoxidized) protein molecules in the sample.
Figure 6 shows all GCase segments whose protection changes as a result of protein oxidation mapped to its amino acid sequence and the quaternary structure. Differences in protection are identified by subtracting the %HDX measured for GCase from GCase-ox to generate ΔHDX. Therefore, a positive ΔHDX indicates an increased HDX for GCase-ox. In Figure 6(A), the second pair of bars below the sequence are colored according to their level of ΔHDX, which highlights the seven segments that displayed moderate (250–258, 282–296, and 412–417) and more pronounced (133–142, 185–209, 232–240, and 384–396) increases in HDX. Most importantly, none of the segments whose deuterium uptake is accelerated in the oxidized protein actually contains the covalently modified residues. Although the protein segments whose protection is affected by GCase oxidation are dispersed throughout the sequence [Fig. 6(A)], almost all of them appear to form well-defined clusters in the three-dimensional structure of the protein [Fig. 6(B–E)]. The only segment (185–209) which is not a part of this cluster (highlighted in Fig. 6) is in fact located on the opposite side of the catalytic cavity of GCase.
Figure 6.
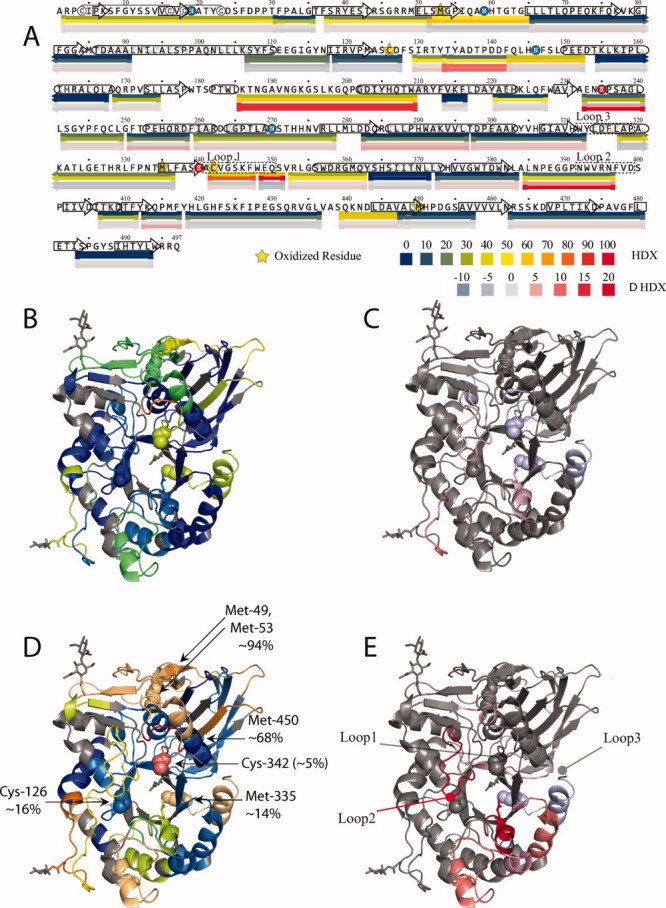
Local backbone amide protection deduced from HDX-MS measurements mapped to the protein sequence (A) and the structure (B–E) are colored based on HDX (upper color legend) or ΔHDX (difference between oxidized and intact forms of GCase, lower color legend). The four bars in each row represent the following (from top to bottom): HDX 1 min (mapped to Structure B); HDX 100 min (mapped to Structure D); ΔHDX 1 min (mapped to Structure C); and ΔHDX 100 min (mapped to Structure E).
Discussion
Intact GCase displays a very uneven distribution of backbone protection across the protein sequence at physiological pH, and the observed pattern is consistent with both the crystal structure12,19 and earlier HDX-MS measurements carried out at neutral or mildly basic pH.7 A remarkable feature of the observed pattern of the protein backbone protection is a noticeable flexibility of several loops surrounding the entrance into the protein catalytic site, particularly Loop 1 (using the terminology of Ref. 2020), which is immediately adjacent to one of the two catalytic glutamic acid residues (Fig. 3). At the same time, the protein segment containing the other catalytic glutamic acid (E235) appears to be extremely stable and does not show any detectable uptake of deuterium even following 2 hours of exchange (Fig. 3).
Conformational flexibility and structural fluctuations have been known to play an important role in enzyme activity21 and were focal points of extensive investigation in the past several years. The emerging consensus on protein dynamics being an essential element of enzyme catalysis22 advanced the understanding of enzymatic phenomena by exploiting the relationship between folding landscapes and catalysis,23 a logical extension of the concepts of folding funnels24,25 and folding–binding landscapes.26 For example, one of the emerging concepts stipulates an intrinsic ability of a protein under native conditions to sample different conformations that meet functional requirements, a typical example being the ability of several enzymes to sample open and closed forms in the absence of the substrate, succeeded by the stabilization of one form (usually closed) upon the substrate binding.27 Furthermore, utilization of the folding landscape concept allows the enzyme catalysis to be viewed using the “catalytic networks” paradigm, where multiple conformations may be sampled by the protein both serially and in parallel in the mechanism, invoking the notion of a very “rugged” multidimensional free-energy surface with multiple minima and transition states.28
These views of protein conformational dynamics as a defining element of its enzymatic activity are fully consistent with the backbone flexibility pattern of GCase at physiological pH of the lysosome as observed in this work. The very uneven distribution of the backbone protection within the catalytic site (Fig. 3) highlights the fine balance between the structure and the dynamics that is needed for the proper functioning of the enzyme. Various structural elements within the catalytic site provide both a rigid binding template for the substrate (e.g., the protein segment that forms a short helix incorporating catalytic residue E235) and flexible segments that facilitate substrate entry into the catalytic cavity with proper alignment (e.g., Loop 1 covering the entrance to the cavity and immediately bordering the E340 catalytic residue). The presence of both rigid and moderately flexible elements in the active site had previously been observed in HDX-MS studies of other enzymes, such as thermolysin,29 and was suggested to reflect a carefully tuned balance required for enzymatic function, indicating the prevalence of stochastic elements in the function of thermolysin rather than supporting a deterministic mechanism. More generally, the fine balance between the well-defined structure and large-scale dynamic motions was shown to be a critical element in protein binding events that are not necessarily related to the catalysis, such as assembly of protein oligomers.30,31
In the case of GCase, the fine balance between conformational stability and dynamics is affected by protein oxidation. Indeed, the most stable segment within the catalytic site (represented by peptic fragment 232–240) becomes noticeably destabilized, showing a slight increase in deuterium uptake following 1-min exposure to D2O. Acceleration of the exchange kinetics becomes much more apparent at longer exchange times; in fact, the evolution of the isotopic distribution of this peptide provides evidence for the existence of an alternative, non-native conformation in solution at lysosomal pH (Fig. 5), where this segment lacks any protection. This alternative conformation is likely to be enzymatically inactive GCase. Indeed, structural disorder is frequently considered to be incompatible with efficient catalysis, and examples of highly flexible, molten globule-like proteins possessing catalytic activity32,33 are extremely rare. However, the rate of sampling of this disordered conformation by GCase-ox at lysosomal pH is not very high, as suggested by the relatively low abundance of ionic species representing the fully unprotected peptide even after 100 min of exchange (black traces in Fig. 5).
Peptide fragment 384–396 is another segment located close to the catalytic site that exhibits similar bimodal exchange as that observed for fragment 232–240. This structural element (Loop 2) displays a surprisingly high degree of backbone protection within the intact GCase, but becomes noticeably destabilized on protein oxidation (Fig. 6). The evolution of the isotopic distribution of this peptide derived from GCase-ox also provides evidence for the existence of an alternative, non-native conformation in solution at lysosomal pH (red trace in Fig. 5). As the abundance of the ionic species (384–396) representing this conformation following 100 min of exchange is the same as for peptide 232–240 as discussed above, it is very likely that these two unfolding events represent a single conformation that displays a high degree of disorder at the catalytic site of GCase-ox. As the HDX-MS measurements in this work were carried out under equilibrium conditions, the relatively low sampling rate of this alternative conformation by GCase-ox is likely to reflect its low Boltzmann weight. Therefore, the noticeable decrease of biological activity of GCase-ox is unlikely to be a direct result of structural changes within the active site which, in turn, adversely affect catalytic activity of the protein. Rather, inactivation of the enzyme on oxidation is likely to involve indirect mechanisms, as discussed below.
The loss of biological activity can be caused by the decreased stability and/or bioavailability of the enzyme, which are triggered by changes in higher order structure and conformational dynamics in protein domains that are remote from the catalytic sites. Careful examination of the changes in backbone protection outside of the catalytic site of GCase triggered by the protein oxidation reveals several noncontiguous peptides that represent protein segments localized in a single protein domain and located in close proximity to each other in the three-dimensional structure of GCase. Among these peptides, peptide (185–209) appears to be most affected by the oxidation, as its isotopic distribution reveals the presence of an alternative, highly unprotected conformation even after 1 min of exchange (Fig. 5), with other peptides in this region being affected to a less significant extent (Fig. 6). Disulfide-linked aggregates were observed by comparing reduced and nonreduced sodium dodecyl sulfate polyacrylamide gel electrophoresis (data not shown); however, the changes observed during the HDX time course experiment are not explained by covalent disulfide aggregates because of the comparable HDX levels between GCase and GCase-ox after only 1 min of exchange and the low pH of the experiment that inhibits further disulfide scrambling. It seems plausible that even transient loss of structure in this region may either lead to increased aggregation propensity or interfere with enzyme trafficking. Both of these two phenomena (aggregation and disruption of trafficking) could negatively affect enzyme's biological activity without interfering directly with the protein–substrate interaction.
The presence of the alternative, non-native conformations of GCase-ox in aqueous solution at lysosomal pH becomes evident on examination of the protein ion charge state distributions of GCase and GCase-ox in nano-ESI-MS acquired at pH 4.5 (Fig. 1). The extent of multiple charging of protein ions usually reflects their compactness in solution.14 While the three most abundant peaks of both GCase and GCase-ox ions correspond to charge states +14 to +16 (+15 being the most abundant), the distribution of GCase-ox ions trails toward higher charge states, revealing the presence of an alternative conformation, whose compactness is lower than that of the native state. Conformations whose surface areas are close to each other often give rise to overlapping charge state distributions, and a distinction can only be made using chemometrics tools.34,35 However, in the case of GCase, it is quite clear that a relatively structured conformation (represented by charge states +16 to +19) is present alongside the natively folded protein in solution, and its degree of compactness is very close to that of the native conformation. Another non-native conformation of GCase has a significantly higher degree of structural disorder and is represented by charge states +23 to +30. Even this conformation, however, appears to retain a significant amount of higher order structure as the average charge state is significantly lower than that observed in acid-unfolded protein (gray trace in Fig. 1).
The abundance of protein ions at different charge states can be used (with discretion) to estimate fractional concentrations of various conformers in solution.36 It is clear that of the two non-native states of GCase-ox, the more compact state has a higher Boltzmann weight. Therefore, we assign this conformation as likely having a compromised (unfolded) segment 185–209. The second significantly less compact state of GCase-ox has a noticeably lower Boltzmann weight (based on the intensity of ionic signal for charge states +23 to +30, m/z 2100 to 2800) and is likely related to the loss of structure at and around the catalytic site of the protein. The relatively high degree of structural disorder of this conformation suggests that it may also be unfolded around segment 185–209. Lastly, the nano-ESI-MS revealed higher propensity of GCase-ox to dimerization. Intriguingly, the GCase-ox dimers not only appear to be more abundant when compared with intact GCase dimers but also have larger surface area (as suggested by the higher charge density of GCase-ox dimers). Taken together, these observations indicate that the partial loss of structure within GCase-ox catalyzes formation of protein dimers (and, by extension, higher oligomers).
One intriguing feature of the observed changes of GCase backbone flexibility on protein oxidation is a lack of clear correlation between the location of residues affected by oxidation and the location of peptides whose protection is diminished as a result of oxidation. This is in stark contrast to the behavior of a chemically stressed (oxidized) monoclonal antibody, where the segments incorporating oxidized residues suffer the most significant loss of structure, as revealed by the recently reported HDX-MS measurements.37 There are five residues (three methionine and two free cysteine residues) in the GCase sequence that have been detected as oxidized; however, the extent of their oxidation is residue specific. The residue that displays the highest degree of oxidation (M53, ca. 94% oxidized) is located within the protein segment that is localized on the protein surface and also displays the highest degree of flexibility in the intact protein (Fig. 3). Both factors are likely to facilitate the oxidation process. Oxidation of GCase does not lead to any detectable changes in the kinetics of deuterium uptake within this segment. Unlike this methionine residue, M450 (ca. 68% oxidized) is located within a stable structural element, but is exposed to the solvent. Backbone protection of this structural element (or any other element in its immediate vicinity) is also not affected by protein oxidation (Figs. 5 and 6).
Three other residues prone to oxidation are located in relatively close proximity to the structural elements whose levels of amide protection are sensitive to GCase oxidation (C126, 16%; C342, 5%; and M335, 14%). It is possible that the subtle changes in local structure triggered by these PTMs lead to increased conformational dynamics and decreased stability in some neighboring elements of the secondary structure, as revealed by accelerated kinetics of local uptake of deuterium in HDX experiments (vide supra). However, it is rather surprising that the structural element that suffers the most as a result of GCase oxidation (232–240) is the most remote segment from the sites of oxidation (ranging from 11 to 42 Å based on the crystal structure). Although the detailed molecular mechanism giving rise to this phenomenon remains unknown, it is possible to invoke a “relay” scheme, where subtle conformational changes induced by oxidation events are propagated through the entire protein structure causing only slight changes in the tertiary structure without affecting the stable elements of the secondary structure in the protein interior. However, when this “wave” of conformational changes (e.g., slight repositioning of helices with respect to each other) reaches the exterior segments of the protein, it may result in local unfolding due to the fewer ternary contacts that provide local stabilization via hydrogen bonding or hydrophobic packing. It appears likely that the helix-loop segment represented by peptic fragment 185–209 can be susceptible to this relay-type transduction of local conformational changes triggered by oxidation at remote sites by virtue of being a terminal point in this chain.
In any event, a significant decrease of local conformational stability of this segment (185–209) of GCase-ox, in particular, the sampling of non-native conformations in which this entire region is unstructured, exposes several hydrophobic residues to solvent (Fig. 6). The exposure of these residues (previously sequestered from solvent in the interhelical interfaces) is almost certain to trigger aggregation. Apart from the increased aggregation propensity, significant changes in the protein conformation are very likely to reduce bioavailability of GCase. Therefore, even though the catalytic activity of GCase may not be significantly affected by the protein oxidation directly (via either direct covalent modifications or loss of the higher order structure within the catalytic cavity), the impaired bioavailability and increased aggregation propensity are certain to make a significant negative impact on the biological activity of this protein, explaining the observed 40–60% drop in GCase in vitro activity.
Conclusions
MS has already proven itself as an extremely useful tool for characterizing covalent structure of biopharmaceutical products, including identification and localization of both enzymatic and nonenzymatic PTMs. In the past several years, MS also emerged as a potent technique for the analysis of higher order structure and dynamics of biopharmaceuticals.38 This work further demonstrates the great utility of MS for both tasks, when mapping of oxidation sites within a large (60 kDa) protein drug is followed by the detailed analysis of conformational consequences of these nonenzymatic PTMs.
Oxidation of methionine and free cysteine residues is one of the most common nonenzymatic PTMs that affect the stability of biopharmaceutical products. Although some oxidation events are relatively harmless and do not affect protein behavior to any appreciable extent both in vivo and during storage, others may exert significant impact on protein behavior in a variety of ways (e.g., reduction of the protein drug efficacy triggered by structural changes in the active site or its vicinity and reduction of bioavailability due to increased aggregation propensity, and an elevated immunogenic response caused by the presence of non-native conformations).
The intact form of the recombinant GCase is remarkably stable, but the forced oxidation of GCase reduces its biological activity (the ability to catalyze hydrolysis of glucocerebroside in the acidic environment of lysosomes) by 40–60%, although no residues located in the catalytic cavity of the enzyme are modified. Application of HDX-MS to probe conformational plasticity of both intact and oxidized forms of GCase at physiologically relevant (lysosomal) pH reveals diminished backbone protection of some key structural elements within the catalytic site. However, the observed changes occur on a time scale that is significantly longer than catalysis, effectively ruling out loss of structure in the catalytic site as a factor contributing to the reduction of the biological activity. At the same time, oxidation leads to destabilization of conformation on a much larger scale in protein segments remote to both the catalytic cavity and the sites of oxidation. This is likely to increase the aggregation propensity of GCase and also interfere with its trafficking. Therefore, it appears that oxidation exerts its negative impact on the biological activity of GCase indirectly by diminishing the overall conformational stability of the protein without necessarily interfering with the enzyme's ability to interact with the substrate.
Materials and Methods
Gene-activated GCase (Shire HGT) was produced in an immortalized human cell line (HT-1080) utilizing targeted recombination with a promoter that activates the endogenous GCase39 in the presence of the mannosidase I inhibitor kifunensine, which results in a molecule with predominantly high-mannose-type glycans.12 Frozen purified drug product was used for baseline HDX experiments and frozen purified stress-oxidized drug substance was used for HDX analyses of GCase-ox. Hydrogen peroxide (30%, w/v) was purchased from Fisher Scientific (Pittsburgh, PA), and deuterium oxide (D2O) was purchased from Cambridge Isotope Laboratories (Andover, MA). All other chemicals were of analytical grade or higher.
GCase oxidation was carried out by adding hydrogen peroxide at a final concentration of 0.1% (v/v) to purified velaglucerase alfa drug substance, from various lots manufactured by the commercial process (Shire HGT), and held at 25°C for 3 h. The resulting GCase-ox was buffer exchanged, diluted, and frozen below −65°C. Quantitation of the extent of covalent modification in GCase-ox for each site of oxidation was carried out by comparing the signal abundance in extracted ion chromatograms for the monoisotopic peaks. Unmodified and oxidized peptides containing the modification sites were then compared in the LC-MS chromatogram.
GCase activity for β-glucocerebroside was measured using a synthetic surrogate substrate, p-nitrophenyl-β-d-glucopyranoside. The assay is performed at pH 5.0, an optimal pH for the surrogate substrate with GCase, for 1 h at 37°C with a reported value expressed as units per milligram of protein. Activity is determined by the release of p-nitrophenol from p-nitrophenyl-β-glucopyranoside with 1 U corresponding to the conversion of 1 μmol of substrate per unit time (1 min) at 37°C. After completion of the enzymatic reaction, the pH of the samples is raised by the addition of glycine carbonate solution (pH 10.7), and the resulting p-nitrophenolate is measured by colorimetric detection (i.e., 405 nm).
HDX-MS experiments were carried out using a previously described protocol.40,41 Briefly, measurements of local backbone dynamics by HDX-MS were initiated by diluting the protein stock solutions (65 μM GCase in 50 mM sodium citrate, 0.01% polysorbate-20, pH 6.0) to a final concentration of 3 μM GCase in the exchange buffer (50 mM sodium citrate D2O, pH 4.5, uncorrected for the isotope effect) equilibrated to 25°C. The final H:D ratio was 1:10. At the indicated exchange time, the solution was placed in an ice bath for 30 s and followed by adding and rapidly mixing an equal volume of prechilled quench solution [2.0M guanidine HCl, 4.0% formic acid, 200 mM tris(2-carboxyethyl)phosphine HCl], yielding pH 2.4. All subsequent steps were performed in an ice bath to ensure a low level of H/D back exchange. The quenched sample was digested online using an immobilized pepsin column with a flow rate of 0.09 mL min−1 and 0.1% formic acid as mobile phase (pH 2.5). The resulting peptides were collected and desalted with an inline nanotrap cartridge and resolved with a C18 column (3 μm, 2.1 mm × 50 mm; BioSuite™) using a rapid gradient from 5 to 65% acetonitrile containing 0.1% formic acid at a flow rate of 0.3 mL min−1. The total time for digestion and desalting was 4 min, and all peptides were detected within 15 min of quenching the exchange reaction. Replicate measurements were made to ensure the precision of mass measurements; the standard deviation for all measurements was less than 0.25 Da with a typical average value of 0.1 Da. MS measurements were carried out with a hybrid quadrupole/time-of-flight MS (QStar-XL, MDS Sciex/Applied Biosystems, Toronto, Canada) equipped with a standard Turbospray™ source.
Proteolytic peptides were unequivocally identified with accurate mass analysis or from MS/MS data. Glycopeptides were recognized by their characteristic neutral average mass loss of 162.14 Da and readily identified by MS after accounting for the mass of a prominent well-characterized high-mannose glycan (Mavg = 1865.67 Da). The extent of deuterium incorporation was calculated by monitoring the average molecular mass increase for each identified peptide. The theoretical maximum deuterium incorporation value was calculated for each peptide based on the dilution factor and number of exchangeable amides, with the assumption that isotopic labeling on all side chains and the N-terminal amine is lost by back exchange during sample workup.15
Protein ion charge-state distributions were collected by static nano-ESI-MS for intact GCase and GCase-ox at lysosomal pH. Samples were buffer exchanged into 50 mM ammonium acetate pH 4.5 by repeated concentration and dilution using Amicon centrifugal concentrators with a 10 kDa molecular weight cutoff (Millipore, Billerica, MA) and a fixed angle rotor operated at 5000g and 4°C. Spectra for 6 μM solutions of GCase and GCase-ox were collected with the QStar-XL using a nanospray source with an ionization potential (IP) of 950, declustering potential (DP) of 120, and focusing potential (FP) of 220. Denatured samples were formed by diluting into 50% acetonitrile with 5% acetic acid to a final protein concentration of 3 μM, and spectra were collected with the QStar-XL using the Turbospray™ source (IP, 4500; DP, 75; and FP, 265). Deconvolution was performed with the Bayesian protein reconstruct tool of BioAnalyst v1.1.5 (MDS Sciex/Applied Biosystems, Toronto, Canada).
Acknowledgments
The authors are very grateful to Mark DeRose and Dr. Gaozhong Zhu for providing the GCase samples and for their helpful discussions.
References
- 1.Kacher Y, Brumshtein B, Boldin-Adamsky S, Toker L, Shainskaya A, Silman I, Sussman JL, Futerman AH. Acid beta-glucosidase: insights from structural analysis and relevance to Gaucher disease therapy. Biol Chem. 2008;389:1361–1369. doi: 10.1515/BC.2008.163. [DOI] [PubMed] [Google Scholar]
- 2.Butters TD. Gaucher disease. Curr Opin Chem Biol. 2007;11:412–418. doi: 10.1016/j.cbpa.2007.05.035. [DOI] [PubMed] [Google Scholar]
- 3.Grabowski GA. Phenotype, diagnosis, and treatment of Gaucher's disease. Lancet. 2008;372:1263–1271. doi: 10.1016/S0140-6736(08)61522-6. [DOI] [PubMed] [Google Scholar]
- 4.Charrow J. Enzyme replacement therapy for Gaucher disease. Expert Opin Biol Ther. 2009;9:121–131. doi: 10.1517/14712590802573395. [DOI] [PubMed] [Google Scholar]
- 5.Beck M. New therapeutic options for lysosomal storage disorders: enzyme replacement, small molecules and gene therapy. Hum Genet. 2007;121:1–22. doi: 10.1007/s00439-006-0280-4. [DOI] [PubMed] [Google Scholar]
- 6.Beck M. Therapy for lysosomal storage disorders. IUBMB Life. 2010;62:33–40. doi: 10.1002/iub.284. [DOI] [PubMed] [Google Scholar]
- 7.Kornhaber GJ, Tropak MB, Maegawa GH, Tuske SJ, Coales SJ, Mahuran DJ, Hamuro Y. Isofagomine induced stabilization of glucocerebrosidase. ChemBioChem. 2008;9:2643–2649. doi: 10.1002/cbic.200800249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hruska KS, LaMarca ME, Scott CR, Sidransky E. Gaucher disease: mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA) Hum Mutat. 2008;29:567–583. doi: 10.1002/humu.20676. [DOI] [PubMed] [Google Scholar]
- 9.Grace ME, Newman KM, Scheinker V, Berg-Fussman A, Grabowski GA. Analysis of human acid beta-glucosidase by site-directed mutagenesis and heterologous expression. J Biol Chem. 1994;269:2283–2291. [PubMed] [Google Scholar]
- 10.Goker-Alpan O, Giasson BI, Eblan MJ, Nguyen J, Hurtig HI, Lee VMY, Trojanowski JQ, Sidransky E. Glucocerebrosidase mutations are an important risk factor for Lewy body disorders. Neurology. 2006;67:908–910. doi: 10.1212/01.wnl.0000230215.41296.18. [DOI] [PubMed] [Google Scholar]
- 11.Mata IF, Samii A, Schneer SH, Roberts JW, Griffith A, Leis BC, Schellenberg GD, Sidransky E, Bird TD, Leverenz JB, Tsuang D, Zabetian CP. Glucocerebrosidase gene mutations: a risk factor for Lewy body disorders. Arch Neurol. 2008;65:379–382. doi: 10.1001/archneurol.2007.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brumshtein B, Salinas P, Peterson B, Chan V, Silman I, Sussman JL, Savickas PJ, Robinson GS, Futerman AH. Characterization of gene-activated human acid-beta-glucosidase: crystal structure, glycan composition, and internalization into macrophages. Glycobiology. 2010;20:24–32. doi: 10.1093/glycob/cwp138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stadtman ER. Protein oxidation and aging. Free Radic Res. 2006;40:1250–1258. doi: 10.1080/10715760600918142. [DOI] [PubMed] [Google Scholar]
- 14.Kaltashov IA, Abzalimov RR. Do ionic charges in ESI MS provide useful information on macromolecular structure? J Am Soc Mass Spectrom. 2008;19:1239–1246. doi: 10.1016/j.jasms.2008.05.018. [DOI] [PubMed] [Google Scholar]
- 15.Bai Y, Milne JS, Mayne L, Englander SW. Primary structure effects on peptide group hydrogen exchange. Proteins. 1993;17:75–86. doi: 10.1002/prot.340170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krishna MMG, Hoang L, Lin Y, Englander SW. Hydrogen exchange methods to study protein folding. Methods. 2004;34:51–64. doi: 10.1016/j.ymeth.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 17.Davies MJ. The oxidative environment and protein damage. Biochim Biophys Acta. 2005;1703:93–109. doi: 10.1016/j.bbapap.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 18.Xiao H, Hoerner JK, Eyles SJ, Dobo A, Voigtman E, Mel'cuk AI, Kaltashov IA. Mapping protein energy landscapes with amide hydrogen exchange and mass spectrometry. I. A generalized model for a two-state protein and comparison with experiment. Protein Sci. 2005;14:543–557. doi: 10.1110/ps.041001705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dvir H, Harel M, McCarthy AA, Toker L, Silman I, Futerman AH, Sussman JL. X-ray structure of human acid-beta-glucosidase, the defective enzyme in Gaucher disease. EMBO Rep. 2003;4:704–709. doi: 10.1038/sj.embor.embor873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Premkumar L, Sawkar AR, Boldin-Adamsky S, Toker L, Silman I, Kelly JW, Futerman AH, Sussman JL. X-ray structure of human acid-beta-glucosidase covalently bound to conduritol-B-epoxide. Implications for Gaucher disease. J Biol Chem. 2005;280:23815–23819. doi: 10.1074/jbc.M502799200. [DOI] [PubMed] [Google Scholar]
- 21.Yon JM, Perahia D, Ghélis C. Conformational dynamics and enzyme activity. Biochimie. 1998;80:33–42. doi: 10.1016/s0300-9084(98)80054-0. [DOI] [PubMed] [Google Scholar]
- 22.Nagel ZD, Klinman JP. A 21(st) century revisionist's view at a turning point in enzymology. Nat Chem Biol. 2009;5:543–550. doi: 10.1038/nchembio.204. [DOI] [PubMed] [Google Scholar]
- 23.Roca M, Messer B, Hilvert D, Warshel A. On the relationship between folding and chemical landscapes in enzyme catalysis. Proc Natl Acad Sci USA. 2008;105:13877–13882. doi: 10.1073/pnas.0803405105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dill KA, Chan HS. From Levinthal to pathways to funnels. Nat Struct Biol. 1997;4:10–19. doi: 10.1038/nsb0197-10. [DOI] [PubMed] [Google Scholar]
- 25.Onuchic JN, Luthey-Schulten Z, Wolynes PG. Theory of protein folding: the energy landscape perspective. Annu Rev Phys Chem. 1997;48:545–600. doi: 10.1146/annurev.physchem.48.1.545. [DOI] [PubMed] [Google Scholar]
- 26.Tsai C-J, Kumar S, Ma B, Nussinov R. Folding funnels, binding funnels, and protein function. Protein Sci. 1999;8:1181–1190. doi: 10.1110/ps.8.6.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bahar I, Chennubhotla C, Tobi D. Intrinsic dynamics of enzymes in the unbound state and relation to allosteric regulation. Curr Opin Struct Biol. 2007;17:633–640. doi: 10.1016/j.sbi.2007.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Benkovic SJ, Hammes GG, Hammes-Schiffer S. Free-energy landscape of enzyme catalysis. Biochemistry. 2008;47:3317–3321. doi: 10.1021/bi800049z. [DOI] [PubMed] [Google Scholar]
- 29.Liu Y-H, Konermann L. Conformational dynamics of free and catalytically active thermolysin are indistinguishable by hydrogen/deuterium exchange mass spectrometry. Biochemistry. 2008;47:6342–6351. doi: 10.1021/bi800463q. [DOI] [PubMed] [Google Scholar]
- 30.Griffith WP, Kaltashov IA. Highly asymmetric interactions between globin chains during hemoglobin assembly revealed by electrospray ionization mass spectrometry. Biochemistry. 2003;42:10024–10033. doi: 10.1021/bi034035y. [DOI] [PubMed] [Google Scholar]
- 31.Griffith WP, Kaltashov IA. Protein conformational heterogeneity as a binding catalyst: ESI-MS study of hemoglobin H formation. Biochemistry. 2007;46:2020–2026. doi: 10.1021/bi062032q. [DOI] [PubMed] [Google Scholar]
- 32.Vamvaca K, Vögeli B, Kast P, Pervushin K, Hilvert D. An enzymatic molten globule: efficient coupling of folding and catalysis. Proc Natl Acad Sci USA. 2004;101:12860–12864. doi: 10.1073/pnas.0404109101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pervushin K, Vamvaca K, Vogeli B, Hilvert D. Structure and dynamics of a molten globular enzyme. Nat Struct Mol Biol. 2007;14:1202–1206. doi: 10.1038/nsmb1325. [DOI] [PubMed] [Google Scholar]
- 34.Mohimen A, Dobo A, Hoerner JK, Kaltashov IA. A chemometric approach to detection and characterization of multiple protein conformers in solution using electrospray ionization mass spectrometry. Anal Chem. 2003;75:4139–4147. doi: 10.1021/ac034095+. [DOI] [PubMed] [Google Scholar]
- 35.Frimpong AK, Abzalimov RR, Eyles SJ, Kaltashov IA. Gas-phase interference-free analysis of protein ion charge-state distributions: detection of small-scale conformational transitions accompanying pepsin inactivation. Anal Chem. 2007;79:4154–4161. doi: 10.1021/ac0704098. [DOI] [PubMed] [Google Scholar]
- 36.Kuprowski MC, Konermann L. Signal response of coexisting protein conformers in electrospray mass spectrometry. Anal Chem. 2007;79:2499–2506. doi: 10.1021/ac0620056. [DOI] [PubMed] [Google Scholar]
- 37.Burkitt W, Domann P, O'Connor G. Conformational changes in oxidatively stressed monoclonal antibodies studied by hydrogen exchange mass spectrometry. Protein Sci. 2010;19:826–835. doi: 10.1002/pro.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leverence R, Mason AB, Kaltashov IA. Noncanonical interactions between serum transferrin and transferrin receptor evaluated with electrospray ionization mass spectrometry. Proc Natl Acad Sci USA. 2010;107:8123–8128. doi: 10.1073/pnas.0914898107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zimran A, Loveday K, Fratazzi C, Elstein D. A pharmacokinetic analysis of a novel enzyme replacement therapy with gene-activated human glucocerebrosidase (GA-GCB) in patients with type 1 Gaucher disease. Blood Cells Mol Dis. 2007;39:115–118. doi: 10.1016/j.bcmd.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 40.Bobst CE, Abzalimov RR, Houde D, Kloczewiak M, Mhatre R, Berkowitz SA, Kaltashov IA. Detection and characterization of altered conformations of protein pharmaceuticals using complementary mass spectrometry-based approaches. Anal Chem. 2008;80:7473–7481. doi: 10.1021/ac801214x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bobst CE, Zhang M, Kaltashov IA. Existence of a noncanonical state of iron-bound transferrin at endosomal pH revealed by hydrogen exchange and mass spectrometry. J Mol Biol. 2009;388:954–967. doi: 10.1016/j.jmb.2009.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]