

Abstract
An understanding of the folding states of α-helical membrane proteins in detergent systems is important for functional and structural studies of these proteins. Here, we present a rapid and simple method for identification of the folding topology and assembly of transmembrane helices using paramagnetic perturbation in nuclear magnetic resonance spectroscopy. By monitoring the perturbation of signals from glycine residues located at specific sites, the folding topology and the assembly of transmembrane helices of membrane proteins were easily identified without time-consuming backbone assignment. This method is validated with Mistic (membrane-integrating sequence for translation of integral membrane protein constructs) of known structure as a reference protein. The folding topologies of two bacterial histidine kinase membrane proteins (SCO3062 and YbdK) were investigated by this method in dodecyl phosphocholine (DPC) micelles. Combing with analytical ultracentrifugation, we identified that the transmembrane domain of YbdK is present as a parallel dimer in DPC micelle. In contrast, the interaction of transmembrane domain of SCO3062 is not maintained in DPC micelle due to disruption of native structure of the periplasmic domain by DPC micelle.
Keywords: folding topology, helical membrane protein, histidine kinase, NMR spectroscopy, paramagnetic relaxation enhancement
Introduction
Integral membrane proteins (IMPs) play critically important roles in many cellular functions, such as intercellular communication, energy transduction, ion regulation, and transport. Despite the pressing need for knowledge of the 3D structures of IMPs,1 the study of the structural biology of membrane proteins remains underdeveloped due to the difficulty of obtaining stable samples under suitable conditions.
Recent advances in the application of nuclear magnetic resonance (NMR) techniques have allowed remarkable progress in determining the solution structure of membrane proteins in various detergent systems.2 However, some detergents can alter the folding state of helical membrane proteins, resulting in destabilization or inactivation.3,4 Therefore, before initiating the 3D structural determination of helical membrane protein, the folding state of the protein and the assembly of its transmembrane helices should be well characterized in the presence of detergent. Knowledge of the assembly of the transmembrane helices is often important to understand a variety of biological systems, for example, the dimerization of transmembrane domains upon activation or the binding to a ligand molecule.5
However, until now there has been no simple method that can verify the folding topology and assembly of helical membrane proteins in detergent solution. For soluble proteins or β-barrel membrane proteins, simple 2D 1H-15N correlation NMR can provide a useful tool for identification of the folding state and for screening of suitable buffer conditions. However, NMR spectra of helical membrane proteins typically are not well-dispersed, leading to low spectral resolution. It is therefore very difficult to determine the folding state of helical membrane proteins using typical 2D NMR spectra.
From this perspective, bacterial histidine kinase (HK), an enzyme that plays an important role in signal transduction in bacteria and that typically contains two transmembrane helices, may provide a useful model for the development of a method to identify the folding state. Although HKs are important for bacterial metabolism and for antibiotic resistance in pathogenic bacteria,6 the signal transduction mechanisms via transmembrane helices are not clearly understood.
The recent development of paramagnetic relaxation enhancement (PRE) techniques using site-directed spin-labeling provides a useful tool for the detection of protein–protein interactions and the characterization of protein dynamics and for the structural determination of membrane proteins in solution NMR.7–11 By simple attachment of a probe molecule via a disulfide bond to a specific cysteine residue, this method provides information about long-range distances within the membrane protein. Thus, to obtain well-folded targets of HKs with proper helical assembly, screening with PRE method is applicable. However, costly and time-consuming backbone assignment is required for structural study using the general PRE method and NMR. Monitoring of specific glycine signals in heteronuclear single quantum coherence (HSQC) spectra combined with use of the PRE technique enabled us to identify the assembly of the transmembrane helices of HK without backbone assignment. In addition, we also identified the dimeric interaction of HK using a mixture of unlabeled and 15N-labeled proteins attached to the PRE probe. This approach allows an understanding of the folding topology of helical membrane proteins by solution NMR. We suggest that this method can be used as a fast screening method to select sample conditions for well-folded helical membrane proteins in a variety of solubilizing systems.
Results
Considering that 1H-15N correlation signals from the backbone amides of glycine residues appear in a distinct region in 2D HSQC or transverse relaxation-optimized spectroscopy (TROSY) spectra, the NMR signal from a specific glycine residue can be easily assigned by introducing a site-directed mutation. In many cases, glycine residues located in the loop region can be mutated without generating major changes in the overall structure. In additional, the signal intensities of glycine residues in this region are relatively high due to their flexibility. These advantages led us to design an experiment for the identification of the folding state via the detection of reductions in glycine signal intensity by paramagnetic perturbation. To identify the folding topology of transmembrane domain, we assume that two different models present in detergent micelles (Fig. 1). If the interaction between two transmembrane helices is strong enough in detergent micelles, two transmembrane helices would be located in the range 25 Å (the model A in Fig. 1). On the other hand, if the interaction is not stable in detergent micelles, each transmembrane helix may be separated and surrounded by different micelles, respectively (the model B in Fig. 1). Under the assumptions of these models, we designed PRE method with specific glycine residue as shown in Figure 1. In the case of model A, the signal intensity of glycine residue on HSQC spectrum should be reduced by paramagnetic perturbation, but it should not be affected in the case of model B, resulting that the folding state of two transmembrane helices can be identified as model A or B.
Figure 1.
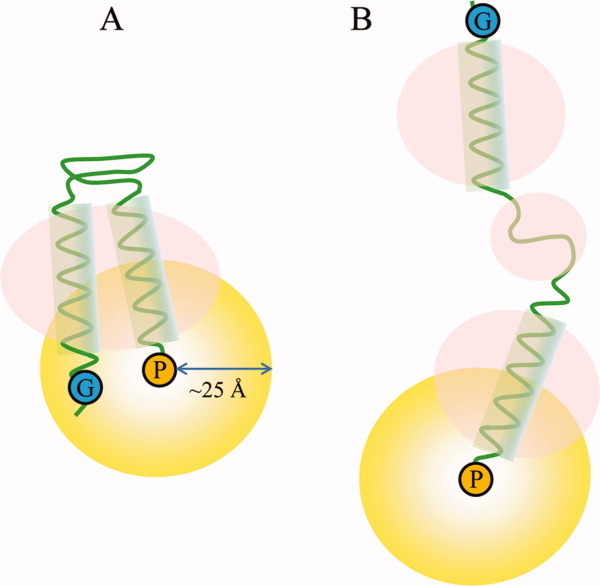
Schematic models representing the design of an experiment to identify the potential folding states of protein domains in the presence of detergent micelles (pink circles, detergent micelles; yellow circles, distance range detectable by PRE). (A) Detergents form a micelle on the transmembrane domain without affecting the folding state; (B) Detergents disrupt the overall folding states of extra-membrane and transmembrane domains. G (in blue circles) and P (in yellow circles) represent glycine residues and the paramagnetic probe, respectively.
As a reference experiment and to validate this method, we tested the topology of Mistic (membrane-integrating sequence for translation of IMP constructs).12 This sequence has four helices and its structure in a detergent micelle is already known. To introduce a unique cysteine at the loop between helices 3 and 4 for MTSL (1-oxyl-2,2,5,5-tetramethyl-Δ3-pyrroline-3-methyl)methanethiosulfonate) attachment, we constructed a double mutant of Mistic (Cys3Gly and Lys86Cys). To assign the glycine residues at positions 22, 47, and 107, we prepared the mutants Gly22Ala, Gly47Ala, and Gly107Ala. The 1H-15N correlation signals of the glycine residues were in good agreement with those obtained in a previous report (Supporting Information Fig. S1).12 The signal intensity of the cross peak of the Gly22 residue was decreased significantly by the paramagnetic perturbation, whereas the signals from the other two glycines (Gly47 and Gly107) were unaffected (Fig. 2). After the addition of DTT (dithiothreitol), the signal intensity of Gly22 was restored. This result indicates that the first loop (between helices 1 and 2) is close to the third loop (between helices 3 and 4), whereas the second loop (between helices 2 and 3) is far from the third loop [Fig. 2(E)]. The ratio of signal intensity (peak height) of Gly22 between paramagantic and diamagnetic species (Ipara/Idia) was <0.85 (0.19/0.26) that represent ∼14–23 Å away from the spin-label nitroxide on Cys86 residue according to the method by Battiste et al.11 In fact, the distance between the nitrogen of Gly22 backbone and the nitrogen of Lys86 side chain is ∼15 Å in Mistic protein (PDB: 1YGM). Therefore, the topology of the Mistic protein could be identified as shown in Figure 2(E); this topology corresponds to the expectation based on the structure of the Mistic protein.
Figure 2.
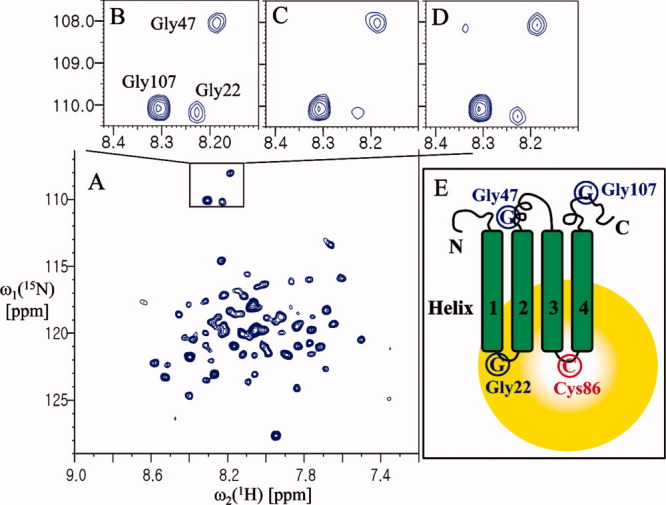
Analysis of the folding topology of Mistic. (A) The 2D 1H-15N TROSY spectrum of Mistic (Cys3Gly and Lys86Cys) at 37°C in 20 mM Bis-tris pH 6.0, ∼30 mM LDAO, and 2 mM DTT. (B) The 1H-15N correlation peaks of Gly22, Gly47, and Gly107 of the protein before spin-labeling. (C) The 1H-15N correlation peaks of the glycine residues of the spin-labeled protein. (D) The same sample as in (A), after incubation with 20 mM DTT for 1 h at 37°C. (E) Schematic representation of the folding topology of Mistic. The peak intensities of Gly22 were measured relative to a reference peak that was not affected by paramagnetic perturbation (at 8.26 and 123.05 ppm for the 1HN and 15N chemical shifts, respectively). The resulting values were: B, 0.33; C, 0.19; and D, 0.26.
To test this method on membrane proteins of unknown structure, we prepared transmembrane domains from two HKs: YbdK from Bacillus subtilis (YbdK-TM) and SCO3062 from Streptomyces coelicolor (SCO3062-TM; Supporting Information Fig. S2). The proteins were cloned into the Vitreoscilla hemoglobin (VHb) fusion vector,13 expressed in the BL21 (DE3) host strain at 18°C overnight, and purified in the presence of dodecyl phosphocholine (DPC) micelles. To attach the MTSL at a specific position, a cysteine residue was introduced at the C-terminus of each protein. Mutation of a residue in the C-terminal region did not affect the overall spectrum, indicating that the folding of the target proteins was unchanged. DPC micelles yielded NMR spectra of higher quality than those prepared using other detergents that we tested in this study for both proteins (data not shown).
YbdK is a 320-residue HK found in the Gram-positive bacterium Bacillus subtilis that has an intramembrane-sensing HK (IM-HK) domain architecture.14 Because IM-HK lacks an extracytoplasmic-sensing domain, it was proposed that YbdK senses its stimulus either directly inside or at the surface of the cytoplasmic membrane. Thus, the method by which IM-HK senses external stimuli in the transmembrane helices is of interest. The 1H-15N correlation peaks of Gly5 and Gly7 of YbdK-TM were assigned based on the YbdK-TM (Gly7Ala) mutant (Supporting Information Fig. S3). The signal intensities of the Gly5 and Gly7 residues were significantly decreased after paramagnetic spin-labeling at the C-terminal of the YbdK-TM (Ser73Cys) mutant [Fig. 3(C)]. The signal was recovered after the addition of DTT [Fig. 3(D)], confirming that the reduction in signal intensity was due to paramagnetic relaxation. This result indicates that the two transmembrane helices interact with each other. Therefore, if the target protein for analysis is monomer in deterget micelle, the folding topology of target protein can be modeled as Figure 3(E). However, this result can not distinguish oligomeric state of the protein. In general, bacterial HKs are known to act as dimers for autophosphorylation in the cytoplasmic dimerization and histidine phosphotransfer (DHp) domain.6,14,15 Thus, it is of interest to determine whether the intramembrane-sensing domain of YbdK-TM forms a dimer or not.
Figure 3.
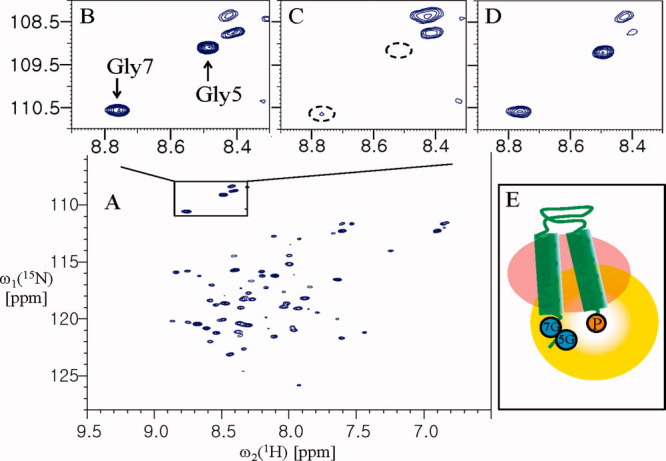
Analysis of the folding topology of YbdK-TM. (A) The 2D 1H-15N HSQC spectrum of YbdK-TM(Ser73Cys) at 40°C in 20 mM sodium acetate pH 4.8, ∼70 mM DPC, and 2 mM DTT. (B) The 1H-15N correlation peaks of Gly5 and Gly7 before spin-labeling. (C) The 1H-15N correlation peaks of the glycine residues of the spin-labeled protein. (D) The same sample as for (C) but after incubation with 20 mM DTT for 2 h at room temperature. (E) A possible model of the folding topology of YbdK-TM, if YbdK-TM is monomer in DPC micelles. The 5G and 7G (in the blue circles) and P (in the yellow circle) represent residues Gly5 and Gly7, and the paramagnetic probe at Cys73 position, respectively. The peak intensities of Gly7 were measured relative to a reference peak that was not affected by paramagnetic perturbation (at 8.11 and 112.74 ppm for the 1HN and 15N chemical shifts, respectively). The resulting values were as follows: B, 3.20; C, 0.32; and D, 2.54.
Although intermolecular interaction studies of proteins using PRE are generally used only after backbone assignment,8 here we designed a simple experiment to detect the interprotomer interaction of YbdK-TM using paramagnetic perturbation. We monitored the cross peaks of two glycines (Gly5 and Gly7) of 15N-labeled YbdK-TM in the presence of a threefold molar excess of spin-labeled YbdK-TM (Ser73Cys) with nitrogen at natural abundance. As shown in Figure 4(C), the cross peak from the Gly5 residue of 15N-labeled YbdK-TM disappeared completely on addition of spin-labeled YbdK-TM (Ser73Cys), and the signal intensity of the Gly7 residue decreased markedly. This result indicates that the two glycine residues at the N-terminus of YbdK-TM are located close to the C-terminus of YbdK-TM (Ser73Cys) and demonstrates the existence of an interaction between the two protomers because analytical ultarcentrifugation (AUC) analysis for YbdK-TM showed that the molecular weight of YbdK-TM in DPC micelle solution was estimated to 15.3 kDa, which correspond to a dimer size.16 Semiquantitatively, the intermolecular distance between the nitrogen of Gly5 of 15N-labeled YbdK-TM and the spin-label nitroxide on YbdK-TM (Ser73Cys) should be less than 14 Å because the cross peak from the Gly5 residue of 15N-labeled YbdK-TM completely quenched,11 whereas the Gly7 residue of 15N-labeled YbdK-TM is ∼14–23 Å away from the spin-label nitroxide on YbdK-TM (Ser73Cys) because the ratio of signal intensity of Gly7 between paramgantic and diamagnetic species is 0.42 (1.14/2.74). Threfore, the topology of YbdK-TM in DPC micelles can be modeled as shown in Figure 4(E). Although it is not likely that the extended protomers could reassemble in an antiparallel geometry due to the presence of the detergent, antiparallel arrangement of two extended protomers might give a similar PRE effect using our protocol (Supporting Information Fig. S4). We could exclude this possibility based on the additional glycine mutation near the C-terminus (Fig. 5). From the result of Figure 4(C), the possibility of model D (parallel, extended dimer) in Supporting Information Figure S4 was eliminated because the signal of Gly5 residue was completely quenched. From the result of Figure 5, the possibility of model E (antiparallel, extended dimer) in Supporting Information Figure S4 is also excluded because the signal intensity of Gly71 residue of 15N-labeled YbdK-TM (Ser71Gly) was reduced by spin-labeling on C-terminus of nonlabeled YbdK-TM (Ser73Cys), meaning that the C-terminus of two protomers are parallel arrangement as shown in Figure 5(E). The Gly71 residue of 15N-labeled YbdK-TM (Ser71Gly) is located within 25 Å range from the spin-label nitroxide on nonlabeled YbdK-TM (Ser73Cys) because the ratio of signal intensity of Gly71 between paramgantic and diamagnetic species is 0.47 (1.43/3.02). For analysis of SCO3062-TM (a putative HK), which has a periplasmic sensor domain and two transmembrane helices, a SCO3062-TM (Ser2Gly and Arg146Cys) mutant was prepared to introduce a cysteine residue at the C-terminus such that the Gly2 residue position could be assigned by comparing the NMR spectra of the mutant and wild-type (Supporting Information Fig. S5). Monitoring of the 1H-15N correlation peak of Gly2 showed that the signal intensity of Gly2 at the N-terminus was not affected by the introduction of the MTSL paramagnetic probe at the C-terminus [Fig. 6(C)]. This result indicates that the N-terminus of helix 1 is located far from the C-terminus of helix 2, beyond the 25 Å range that can be detected using this method. Therefore, the topology of SCO3062-TM in the presence of DPC micelles was assumed to be that shown in Figure 6(E). In the case of SCO3062-TM, it is likely that the presence of DPC micelles affects the global fold of the periplasmic domain. In fact, the 2D 1H-15N HSQC spectrum of the periplasmic domain of SCO3062 was well-dispersed and homogeneous in the absence of the DPC micelles, whereas in the presence of DPC micelles it appeared very similar to that of SCO3062-TM (Supporting Information Fig. S6). Therefore, we conclude that the assembly of the transmembrane helices of SCO3062 is disturbed by misfolding of the periplasmic domain when DPC micelles are present.
Figure 4.
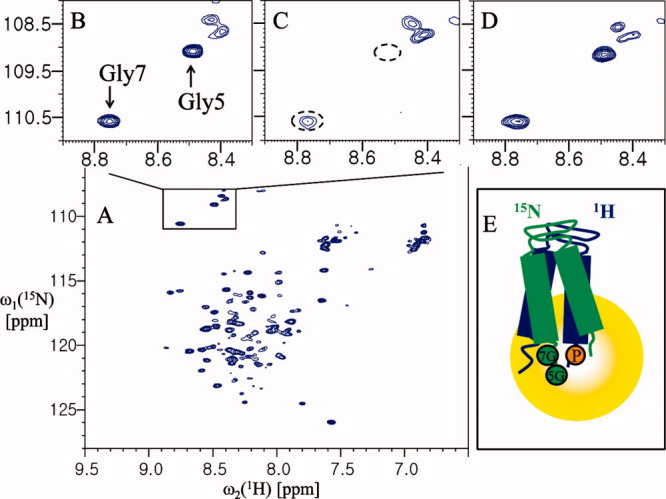
Analysis of the interprotomer interactions of YbdK-TM. (A) The 2D 1H-15N HSQC spectrum of 0.4 mM YbdK-TM at 40°C in 20 mM sodium acetate pH 4.8 and ∼70 mM DPC. (B) The 1H-15N correlation peaks of Gly5 and Gly7. (C) The 1H-15N correlation peaks of the glycine residues of the protein (0.4 mM) in the presence of 1.2 mM spin-labeled YbdK-TM (Ser73Cys) with nitrogen at natural abundance. (D) The same sample as for (C) but after incubation with 20 mM DTT for 2 h at 37°C. (E) Model of dimer topology of YbdK-TM in DPC micelle. The 5G and 7G (in the green circles), and P (in the yellow circle) represent residues Gly5 and Gly7, and the paramagnetic probe at Cys73 position, respectively. The peak intensities of Gly7 were measured relative to a reference peak that was not affected by paramagnetic perturbation (at 8.11 and 112.82 ppm for the 1HN and 15N chemical shifts, respectively). The resulting values were as follows: B, 3.74; C, 1.14; and D, 2.74.
Figure 5.
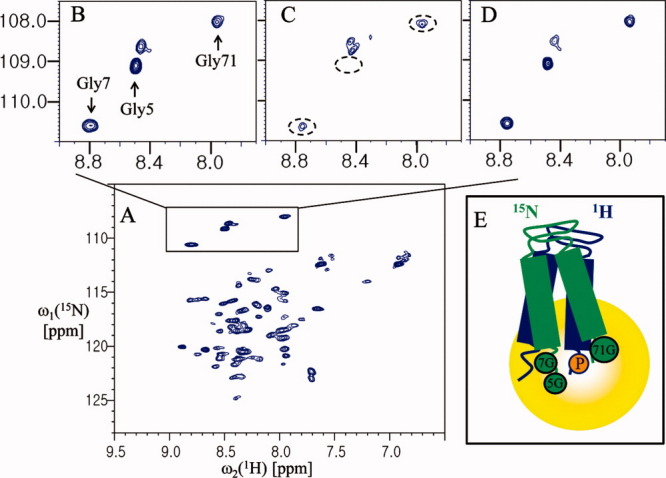
Analysis of the dimer topology of YbdK-TM. (A) The 2D 1H-15N HSQC spectrum of 0.4 mM YbdK-TM (S71G) at 40°C in 20 mM sodium acetate pH 4.8, and ∼70 mM DPC. (B) The 1H-15N correlation peaks of Gly5, Gly7, and Gly71. (C) The 1H-15N correlation peaks of the glycine residues of the protein (0.4 mM) in the presence of 1.2 mM spin-labeled YbdK-TM (Ser73Cys) with nitrogen at natural abundance. (D) The same sample as (C) but after incubation with 20 mM DTT for 2 h at 37°C. (E) A model of the parallel dimer of YbdK-TM. 5G, 7G and 71G (in green circles), and P (in the yellow circle) represent the Gly5, Gly7, and Gly71, and the paramagnetic probe at Cys73 position, respectively. The peak intensities of Gly71 were measured relative to a reference peak that was not affected by paramagnetic perturbation (at 8.10 and 113.0 ppm for the 1HN and 15N chemical shifts, respectively). The resulting values were as follows: B, 2.92; C, 1.43; and D, 3.02.
Figure 6.
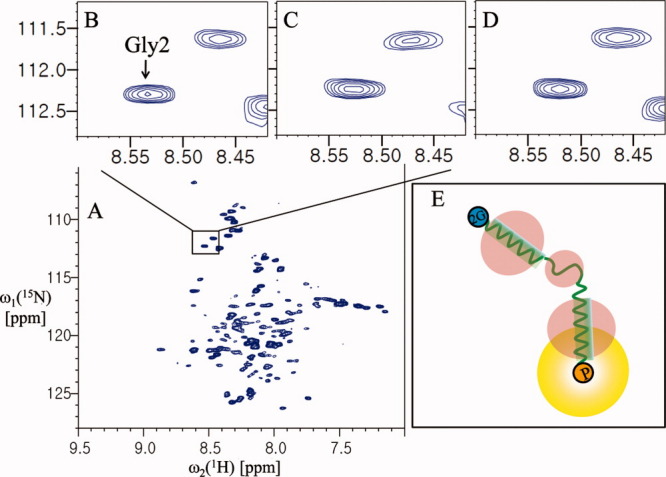
Analysis of the folding topology of SCO3062-TM. (A) The 2D 1H-15N TROSY spectrum of 0.4 mM SCO3062-TM in 20 mM sodium acetate pH 4.0, ∼70 mM DPC, and 2 mM DTT. (B) The 1H-15N correlation peaks of Gly2 before spin-labeling. (C) The 1H-15N correlation peaks of the glycine residue of the spin-labeled protein. (D) The same sample as for (C) but after incubation with 20 mM DTT for 2 h at room temperature. (E) A model of the folding topology of SCO3062-TM in DPC micelles. 2G (in the blue circles) and P (in the yellow circles) represent Gly2 and the paramagnetic probe at Cys146 position, respectively. The peak intensities of Gly2 were measured relative to a reference peak that was not affected by paramagnetic perturbation (at 8.35 and 111.40 ppm for the 1HN and 15N chemical shifts, respectively). The resulting values were as follows: B, 0.66; C, 0.66; and D, 0.64.
Discussion
In conclusion, we successfully identified the folding topology and the assembly of α-helical membrane proteins in detergent micelles by monitoring the NMR signals of glycine residues with paramagnetic perturbation but without backbone assignment. Using Mistic protein as a reference experiment, we could validate that the folding topology of four helices protein could be analyzed by this method in detergent micelles. In addition, we were able to analyze the folding topologies of the transmembrane helices of bacterial HKs rapidly. By monitoring the glycine signals (Gly5, Gly7, and Gly71) of YbdK-TM on HSQC spectra, we demonstrated the parallel dimer assembly of YbdK-TM combining with AUC. We excluded the possibility of extended dimer formation of YbdK-TM as model D and E in Supporting Information Figure S4 by the glycine mutation positioned at 71 of C-terminus. In contrast, no interaction between two transmembrane helices of SCO3062-TM was observed because the folding state of periplasmic domain of SCO3062 was disrupted by DPC micelle. Based on our results, 3D structural determination and functional studies of YbdK-TM in DPC micelles can be initiated, although SCO3062 requires further investigation due to its instability. In the case of two α-helical transmebrane proteins, if the target protein is present as monomer, the folding topology of the target protein directly can be identified as Figure 3(E) by this method. However, in cases of oligomeric proteins, this method is only useful for dimeric assembly and topology of the proteins combining with AUC. If protein is dimer like YbdK-TM, calculation of the intramolecular distance between two helices by PRE method is not simple because PRE affects both intramolecular and intermolecular distances. Although more accurate distance calculation for structure refinement by PRE method can be obtained from total correlation time of dimagnetic protein and paramagnetic rate enhancements (R2sp) of spin labeled protein,9,11,12 we suppose that this semiquantitative method is sufficient for fast screening of the folding topology and the dimeric assembly of α-helical membrane proteins in detergent micelles before structural determination. We believe that this method provides fast preliminary results to begin 3D structure determination and functional study of α-helical membrane protein in detergent micelle.
Materials and Methods
The Mistic gene (1-110) from B. subtilis was cloned into the pET-28a (Novagen) vector containing a hexahistidine tag at the N-terminus.12 The above vector was transformed into Escherichia coli strain BL21 (DE3). The cells were grown in M9 minimal medium enriched with 15NH4Cl as the sole nitrogen source (99% 15N; Cambridge Isotope Laboratories) at 18°C. Protein expression was induced with 0.2 mM isopropyl-β-d-thiogalactopyranoside (IPTG) at an OD600 of 1.0. The cells were grown overnight. The cells were harvested, resuspended in 20 mM Tris pH 7.9, 200 mM NaCl, 10% glycerol, 20 mM imidazole, 5 mM β-mercaptoethanol, and lysed using a cell disruptor (Cabinet Cell Disrupter, Constant Systems) at 5°C and 25 kpsi (pounds-per-square-inch). Inclusion bodies and other cell debris were pelleted at 10,000 g for 20 min. The membranes were pelleted by centrifugation at 200,000 g for 2 h. The membranes were solubilized in 20 mM Tris pH 7.9, 200 mM NaCl, 10% (v/v) glycerol, 5 mM b-mercaptoethanol, 20 mM n-lauryl-N,N-dimethylamine-N-oxide (LDAO), and 20 mM imidazole using a homogenizer and mild agitation for 60 min at 4°C. Following centrifugation at 200,000 g for 1 h at 4°C, the supernatant was incubated for 2 h under gentle stirring with 10 mL of Ni-NTA resin (Qiagen) equilibrated with the above buffer (but with the added inclusion of 3 mM LDAO). The resin with the bound protein was then packed into a gravity-flow column and washed with five bed volumes of the same buffer containing 30 mM imidazole. The Mistic protein was eluted by the addition of 500 mM imidazole. Final purification was performed by size exclusion chromatography on a prep-grade HiLoad 16/60 Superdex 75 column (GE Healthcare) in a buffer containing 20 mM Bis-tris pH 6.0 and 3 mM LDAO. The final samples were concentrated at 4°C using an Amicon concentrator (Millipore) with a 10 kDa molecular weight cut-off membrane. Gly22Ala, Gly47Ala, and Gly107Ala mutants were prepared using a QuikChange® Site-Directed Mutagenesis Kit (Stratagene) for the glycine assignments at positions 22, 47, and 107. Single cysteine mutants of Mistic (Cys3Gly and Lys86Cys) were prepared for the attachment of MTSL and used to study the topology of the protein. Following the removal of the histidine tag by thrombin digestion and size exclusion chromatography, TROSY spectra were recorded at 37°C in a solution containing 20 mM Bis-tris pH 6.0, ∼30 mM LDAO, and 0.3 mM Mistic. For the mutants that were used to assign the glycines, we recorded TROSY spectra under the same conditions but without cleavage of the histidine tag. All NMR experiments for the Mistic proteins were performed using a Bruker DRX900 spectrometer equipped with a cryogenic, triple resonance probe.
The YbdK-TM (Leu2-Ser65) construct was cloned into a VHb fusion vector (pPosKJ) with an additional sequence (GlySerHisMetGlySerGlySerLeu) at the N-terminus.16 This allowed thrombin cleavage to allow removal of the hemoglobin fusion protein. After transformation of the plasmid into Escherichia coli strain BL21(DE3), the protein was expressed and purified according to the protocol described previously.16 Briefly, the protein was expressed at 18°C by adding 0.4 mM IPTG at an OD600 of 0.6 and growing the cells overnight. Following the extraction of YbdK-TM from the membrane fraction of the cells using 14 mM DPC, the protein was purified using Ni-NTA resin containing 2 mM DPC. Further purification steps were conducted in the presence of 2 mM DPC. To assign Gly7 at the N-terminal and to introduce MTSL at the C-terminal, we prepared YbdK-TM (Gly7Ala) and YbdK-TM (Ser73Cys) mutants (the numbering used is from the additional sequence). HSQC spectra were recorded at 40°C in a solution containing 20 mM sodium acetate pH 4.8, ∼70 mM DPC, and 0.4 mM protein, using a Bruker 500 MHz spectrometer equipped with a triple resonance cryoprobe.
The gene encoding SCO3062-TM (Met1-Gln147) with a thrombin site at the N-terminus was cloned into the C-terminal region of the pPosKJ vector. The protein was expressed and purified as previously described.17 The SCO3062-TM (Arg2Gly) mutant that was constructed for glycine assignment was prepared using the Stratagene mutagenesis kit; the R146 residue at the C-terminal of the mutant was replaced with cysteine for investigation of the topology of SCO3062-TM. TROSY spectra were acquired at 40°C in a buffer containing 20 mM sodium acetate pH 4.0, ∼70 mM DPC, and 0.4 mM protein using the 900 MHz NMR spectrometer. To prepare the periplasmic domain of SCO3062, we transformed the recombinant pPosKJ expression vector SCO3062 (Glu28-124Arg) into Escherichia coli strain BL21(DE3). The cells were grown in M9 minimal medium containing 15NH4Cl at 37°C until the OD600 reached ∼0.6. The hemoglobin-fused periplasmic domain of SCO3062 was expressed by adding 0.5 mM IPTG at 18°C; the cells were harvested after overnight growth and lysed by sonication in 50 mM Tris pH 7.9, and 20% glycerol. Following centrifugation at 10,000 g for 20 min, the supernatant was loaded onto a HisTrap column (GE Healthcare) equilibrated with 50 mM Tris pH 7.5, 500 mM NaCl, and 10 mM imidazole. After washing of the column with five column volumes of the previous buffer in which the imidazole concentration had been increased to 20 mM, and the protein was eluted with the same buffer containing 200 mM imidazole. The hemoglobin fusion protein was cleaved by thrombin digestion and the periplasmic domain of SCO3062 was purified using a prep-grade HiLoad 16/60 Superdex 75 column (GE Healthcare) equilibrated with 20 mM HEPES pH 7.0, and 50 mM NaCl. HSQC spectra of the protein were recorded at 40°C on the 500 MHz spectrometer.
During purification for the proteins, 2 mM DPC for YbdK-TM and SCO3062-TM and 3 mM LDAO for Mistic, which are higher than CMC (critical micelle concentration) of DPC and LDAO, respectively, were required to stabilize the proteins during purification steps. For the Mistic protein, we observed homogeneous HSQC spectra at ∼30 mM LDAO with 0.3 mM Mistic.
From the previous reports,16,17 we optimized DPC concentration for YbdK-TM and SCO3062-TM and estimated that ∼80 and ∼71 DPC molecules bound on YbdK-TM and SCO3062-TM, respectively. The homogeneous HSQC spectra for the proteins were obtained at the ratio of concentration of protein:DPC = 1:∼100–300. Therefore, we used ∼70 mM DPC for both proteins (0.4 mM) in this report.
Paramagnetic spin-labeling with MTSL was achieved in accordance with established protocols with minor modifications.9 MTSL (1 mM, from a 200 mM MTSL stock in acetonitrile) was added to protein solutions containing 25 mM HEPES pH 7.0, 150 mM NaCl, and detergent (3 mM LDAO for Mistic, and 2 mM DPC for YbdK-TM and SCO3062-TM) and incubated at room temperature overnight. Excess MTSL was removed by passage through a PD-10 desalting column. After attachment of MTSL, no remarkable change of the chemical shifts of specific glycine residues was observed. To remove MTSL for diamagnetic protein, 20 mM DTT added and incubated at 37°C or room temperature until no signal recovery is observed any more.
Glossary
Abbreviations:
- AUC
analytical ultracentrifugation
- CMC
critical micelle concentration
- DHp
dimerization and histidine phosphotransfer
- DPC
dodecyl phosphocholine
- DTT
dithiothreitol
- HK
histidine kinase
- HSQC
heteronuclear single quantum coherence
- IMPs
Integral membrane proteins
- IM-HK
intramembrane-sensing histidine kinase
- IPTG
isopropyl-β-d-thiogalactopyranoside
- LDAO
n-lauryl-N,N-dimethylamine-N-oxide
- Mistic
membrane-integrating sequence for translation of integral membrane protein constructs
- MTSL
1-oxyl-2,2,5,5-tetramethyl-Δ3-pyrroline-3-methyl)methanethiosulfonate
- NMR
nuclear magnetic resonance
- PRE
paramagnetic relaxation enhancement
- TROSY
transverse relaxation optimized spectroscopy
- VHb
Vitreoscilla hemoglobin.
References
- 1.Drews J. Drug discovery: a historical perspective. Science. 2000;287:1960–1964. doi: 10.1126/science.287.5460.1960. [DOI] [PubMed] [Google Scholar]
- 2.Kim HJ, Howell SC, Van Horn WD, Jeon YH, Sanders CR. Recent advances in the application of solution NMR spectroscopy to multi-span integral membrane proteins. Prog Nucl Magn Reson Spectrosc. 2009;55:335–360. doi: 10.1016/j.pnmrs.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Palazzo G, Lopez F, Mallardi A. Effect of detergent concentration on the thermal stability of a membrane protethe case study of bacterial reaction center solubilized by N,N-dimethyldodecylamine-N-oxide. Biochim Biophys Acta. 2010;1804:137–146. doi: 10.1016/j.bbapap.2009.09.021. [DOI] [PubMed] [Google Scholar]
- 4.Lunds S, Orlowski S, Foresta B, Champeíl P, Maire M, Møller JV. Detergent structure and associated lipids determinants in the stabilization of solubilized Ca2+-ATPase from sarcoplasmic reticulum. J Biol Chem. 1989;264:4907–4915. [PubMed] [Google Scholar]
- 5.George SR, O'Dowd BF, Lee SP. G-Protein-coupled receptor oligomerization and its potential for drug discovery. Nat Rev Drug Discov. 2002;1:808–820. doi: 10.1038/nrd913. [DOI] [PubMed] [Google Scholar]
- 6.Khorchid A, Ikura M. Bacterial histidine kinase as signal sensor and transducer. Int J Biochem Cell Biol. 2006;38:307–312. doi: 10.1016/j.biocel.2005.08.018. [DOI] [PubMed] [Google Scholar]
- 7.Fanucci1 GE, Cafiso DS. Recent advances and applications of site-directed spin labeling. Curr Opin Struct Biol. 2006;16:644–653. doi: 10.1016/j.sbi.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 8.Rumpel S, Becker S, Zweckstetter M. High-resolution structure determination of the CylR2 homodimer using paramagnetic relaxation enhancement and structure-based prediction of molecular alignment. J Biomol NMR. 2008;40:1–13. doi: 10.1007/s10858-007-9204-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liang B, Bushweller JH, Tamm LK. Site-directed parallel spin-labeling and paramagnetic relaxation enhancement in structure determination of membrane proteins by solution NMR spectroscopy. J Am Chem Soc. 2006;128:4389–4397. doi: 10.1021/ja0574825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Page RC, Lee S, Moore JD, Opella SJ, Cross TA. Backbone structure of a small helical integral membrane protea unique structural characterization. Protein Sci. 2009;18:134–146. doi: 10.1002/pro.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Battiste JL, Wagner G. Utilization of site-directed spin labeling and high-resolution heteronuclear nuclear magnetic resonance for global fold determination of large proteins with limited nuclear overhauser effect data. Biochemistry. 2000;39:5355–5365. doi: 10.1021/bi000060h. [DOI] [PubMed] [Google Scholar]
- 12.Roosild TP, Greenwald J, Vega M, Castronovo S, Riek R, Choe S. NMR structure of mistic, a membrane-integrating protein for membrane protein expression. Science. 2005;307:1317–1321. doi: 10.1126/science.1106392. [DOI] [PubMed] [Google Scholar]
- 13.Kwon SY, Choi YJ, Kang TH, Lee KH, Cha SS, Kim GH, Lee HS, Kim KT, Kim KJ. Highly efficient protein expression and purification using bacterial hemoglobin fusion vector. Plasmid. 2005;53:274–282. doi: 10.1016/j.plasmid.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 14.Mascher T. Intramembrane-sensing histidine kinases: a new family of cell envelope stress sensors in Firmicutes bacteria. FEMS Microbiol Lett. 2006;264:133–144. doi: 10.1111/j.1574-6968.2006.00444.x. [DOI] [PubMed] [Google Scholar]
- 15.Sevvana M, Vijayan V, Zweckstetter M, Reinelt S, Madden DR, Herbst-Irmer R, Sheldrick GM, Bott M, Griesinger C, Becker S. A ligand-induced switch in the periplasmic domain of sensor histidine kinase CitA. J Mol Biol. 2008;377:512–523. doi: 10.1016/j.jmb.2008.01.024. [DOI] [PubMed] [Google Scholar]
- 16.Kim YP, Yeo KJ, Kim MH, Kim YC, Jeon YH. Structural characterization of the intra-membrane histidine kinase YbdK from Bacillus subtilis in DPC micelles. Biochem Biophys Res Commun. 2010;391:1506–1511. doi: 10.1016/j.bbrc.2009.12.106. [DOI] [PubMed] [Google Scholar]
- 17.Yeo KJ, Kwak SN, Kim HJ, Cheong C, Kim MH, Jeon YH. Expression and characterization of the integral membrane domain of bacterial histidine kinase SCO3062 for structural studies. Biochem Biophys Res Commun. 2008;376:409–413. doi: 10.1016/j.bbrc.2008.09.002. [DOI] [PubMed] [Google Scholar]