

Abstract
Lipopolysaccharide (LPS) biosynthesis represents an underexploited target pathway for novel antimicrobial development to combat the emergence of multidrug-resistant bacteria. A key player in LPS synthesis is the enzyme d-arabinose-5-phosphate isomerase (API), which catalyzes the reversible isomerization of d-ribulose-5-phosphate to d-arabinose-5-phosphate, a precursor of 3-deoxy-d-manno-octulosonate that is an essential residue of the LPS inner core. API is composed of two main domains: an N-terminal sugar isomerase domain (SIS) and a pair of cystathionine-β-synthase domains of unknown function. As the three-dimensional structure of an enzyme is a prerequisite for the rational development of novel inhibitors, we present here the crystal structure of the SIS domain of a catalytic mutant (K59A) of E. coli d-arabinose-5-phosphate isomerase at 2.6-Å resolution. Our structural analyses and comparisons made with other SIS domains highlight several potentially important active site residues. In particular, the crystal structure allowed us to identify a previously unpredicted His residue (H88) located at the mouth of the active site cavity as a possible catalytic residue. On the basis of such structural data, subsequently supported by biochemical and mutational experiments, we confirm the catalytic role of H88, which appears to be a generally conserved residue among two-domain isomerases.
Keywords: d-arabinose-5-phosphate isomerase, antimicrobial development, lipopolysaccharide biogenesis, 3-deoxy-d-manno-octulosonate synthesis, three-dimensional protein structure, Gram-negative bacteria
Introduction
Bacterial diseases still pose a severe threat to human health, mainly because of the emergence and spread of multidrug-resistant bacteria. For this reason, it is imperative that antimicrobial drug development keeps up with the evolution of antibiotic resistance mechanisms, to contain the diffusion of pathogenic bacteria and associated diseases worldwide. An effective strategy to design novel antimicrobials is to target and interfere with biochemical pathways that are essential for bacterial survival. One such pathway is the biogenesis of the complex glycolipid lipopolysaccharide (LPS), a fundamental component of the outer layer of the outer membrane (OM) of Gram-negative bacteria, and a well-established virulence factor that stimulates the mammalian innate immune response.1,2
LPS is responsible for the peculiar permeability properties of the OM, preventing the entry of toxic molecules including bile salts, detergents, and lipophilic antibiotics.3 Structurally, LPS can be divided into three elements: lipid A (the hydrophobic moiety that anchors LPS to the OM), the core oligosaccharide, and the O-antigen. The core oligosaccharide provides the link between the highly conserved membrane-embedded lipid A and the structurally diverse O-antigen polysaccharide chain; it can be further subdivided into the inner and outer cores.2 Whereas the structure of the outer core is somewhat variable, the inner core region is more conserved, with one structural element, the 3-deoxy-d-manno-octulosonate (Kdo) residue, being present in all inner cores analyzed in LPS molecules to date.4 LPS is essential in most Gram-negative bacteria, and the Kdo2-lipid A moiety represents the minimal structure indispensable for cell growth.2,5 Thus, blocking Kdo synthesis compromises cell viability and therefore represents an ideal target for novel antimicrobial development.
Kdo synthesis is initiated by d-arabinose-5-phosphate isomerase (API), an enzyme that catalyzes the reversible isomerization of d-ribulose-5-phosphate (Ru5P) to the Kdo precursor, d-arabinose-5-phosphate (A5P; Fig. 1). The reaction catalyzed by API (KdsD in E. coli) is an aldose-ketose isomerization in which a hydrogen atom is transferred between the C1 and C2 atoms of the substrate. The carbon-bound hydrogen can move by one of two mechanisms: a hydride shift or a proton transfer via an acid–base mechanism involving a cis-enediol intermediate.6 The hydride shift mechanism appears to operate in the xylose isomerase family of enzymes.7 In the rabbit phosphoglucose isomerase PGI, which catalyzes the interconversion of glucose 6-phosphate to fructose 6-phosphate, an active site glutamate (E357) is the base catalyst for isomerization and H388 is the acid catalyst for ring opening.8,9 On the contrary, the isomerization reaction catalyzed by the glucosamine 6-phosphate synthase from E. coli, which converts fructose 6-phosphate into glucosamine 6-phosphate, is likely to involve the formation of a Schiff base with K603, a ring opening step catalyzed by H504, and proton transfer from C1 to C2 catalyzed by E488.10 The proposed mechanism for the action of E. coli GmhA involves an enediol switch, with E65 and H180 acting as an acid and a base, respectively, to promote the conversion of sedoheptulose 7-phoshate into d-glycero-d-manno-heptopyranose-7-phosphate.11 Very recently, however, a modified mechanism has been proposed for Burkholderia pseudomallei GmhA: this enzyme contains an active site Zn2+ ion that orients the side chains of E68 and Q175, acting as the base and the acid, respectively, to promote the isomerization reaction.12 Interestingly, the isomerase activity of the K-antigen specific API KpsF, a paralogous enzyme to KdsD, has also been shown to be inhibited by bivalent metals, particularly Zn2+, Cd2+, and Hg2+.13
Figure 1.
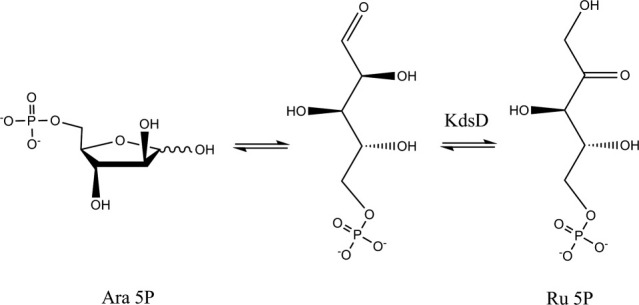
Kdo biosynthesis. Schematic representation of the first reaction catalyzed by KdsD, where d-arabinose-5-phosphate (Ara 5P) is converted into d-ribulose-5-phosphate (Ru 5P).
API is highly conserved among phylogenetically diverse bacterial species, therefore potential inhibitors would likely offer broad protection. In E. coli, API is encoded by two paralogous genes kdsD and gutQ, which may substitute for each other; however, inactivation of both genes is lethal, indicating that API indeed has an essential function.14–16 E. coli KdsD is a 328-residue enzyme, active as a homotetrameric protein. Each of the four subunits contains two distinct domains; an N-terminal sugar isomerase (SIS) domain (residues 1–200), followed by a pair of cystathionine-β-synthase domains (CBS; residues 213–325) of unknown function.
To design novel KdsD inhibitors, it is essential to gain a thorough structural understanding of the enzyme fold, the architecture of the active site, and the location of the key residues involved in catalysis. With regard to current knowledge on the three-dimensional structures of E. coli KdsD, only the crystal structure of the CBS domain has been determined as part of a large-scale structural genomics program (PDB code 3FNA), while the SIS domain remains unrevealed. The SIS domain is a common structural module in several proteins that share sugar isomerization or sugar binding activities but differ with regard to their overall structures and sequences (in some cases <20% sequence identity). On this basis, a model for the three-dimensional structure of the E. coli KdsD SIS domain was recently proposed, based on a homology-modeling approach, and potential catalytically important residues were identified.17
In this communication, we address such questions and report the crystallographic analysis of a catalytically inactive, His-tagged recombinant mutant (K59A) of E. coli KdsD (KdsDK59A), and describe the three-dimensional structure of its SIS domain at 2.6-Å resolution. We selected this mutant for our studies based on previous unsuccessful attempts to crystallize the structure of the wild-type form. On the basis of our structural results, in comparison with other known SIS domains, we highlight several residues that could be important for KdsD function, and highlight a key structural difference affecting the location of the C-terminal region relative to the active site. Such a key finding brought us to predict a catalytic role for a His residue (H88) not previously considered; such a role in KdsD catalysis was subsequently confirmed by our supporting functional investigations.
Results
X-ray diffraction data analysis
X-ray diffraction data were collected at 2.6-Å resolution (at the European Synchrotron Radiation Facility, Grenoble, France) and refined to Rfactor and Rfree values of 26.1% and 30.5%, respectively (Table I). Contrary to expectations, only a tetramer of the SIS domain was contained in the crystal asymmetric unit, suggesting that cleavage of the CBS domain had occurred during crystallization. To further investigate such findings, the purified recombinant protein was incubated at 20°C for 1 week and then analyzed by SDS-PAGE. Indeed, cleavage was confirmed; two bands were observed at molecular weights of ∼35 and 22 kDa, corresponding to the full-length and the cleaved protein, respectively. Western blotting with an antibody that recognizes the N-terminal His-tag indicated that the lower band did, in fact, correspond to the SIS domain (results not shown).
Table I.
Data Collection and Refinement Parameters of KdsDK59A SIS Domain Crystals
| Data collection statistics | |
| Space Group | P21 |
| Unit cell dimensions | |
| a, b, c (Å) | 55.9, 67.4, 82.2 |
| α, β, γ (°) | 90, 106.9, 90 |
| No. of unique reflections | 17031 (2512) |
| Average I/σ(I)a | 5.9 (2.4) |
| Completeness (%)a | 94.3 (95.5) |
| Redundancya | 2.7 (2.7) |
| Rmerge (%)a | 0.122 (0.355) |
| Refinement statistics | |
| Resolution range (Å) | 2.6–40.0 |
| R (%) | 26.1 |
| Rfree (%) | 30.5 |
| RMSD deviations | |
| Bond Lengths (Å) | 0.002 |
| Bond Angles (°) | 0.430 |
| No. of molecules/au | 4 |
| Total protein atoms (Bavg, Å2) | 5086 (36.0) |
| Total solvent atoms (Bavg, Å2) | 9 (26.1) |
| Ramachandran plot (%) | |
| Favored regions | 92.0 |
| Allowed regions | 7.9 |
Rmerge = ∑|I − (I)|/∑ I × 100, where I is the intensity of a reflection and (I) is the average intensity; Rfree was calculated from 5% of randomly selected data, for cross validation; R-factor = ∑|Fo − Fc|/∑|Fo| × 100.
The values in parentheses represent the highest resolution shell (2.60–2.74 Å). Procheck was used to define the favored and allowed regions of the Ramachandran Plot.18
Overall structure of the SIS domain
In agreement with other SIS domains of known structure, the E. coli KdsDK59A SIS domains are organized in a homotetramer (the four chains are labeled A through D), with each structurally identical subunit adopting the canonical Rossmann fold (a central four-stranded parallel β-sheet and seven α-helices) [Figs. 2(A,B) and 4]. Electron density is absent for the N-terminal His-tag and is present from residue 10/11 up to residue 182/183, depending on the chain. The tetramer subunits are organized so that helices α1, α3 (containing the mutation K59 → A), and α6 of chain-A (or -C) are oriented in an antiparallel manner to the corresponding helices in chain-B (or -D), in a head-to-toe manner. Subunits A and B (or C and D), interact mainly via hydrophobic interactions with an interface that extends over a surface area of 2109 Å2 (2095 Å2 for monomers C and D), as calculated using the PISA tool available from the European Bioinformatics Institute (http://www.ebi.ac.uk).
Figure 2.
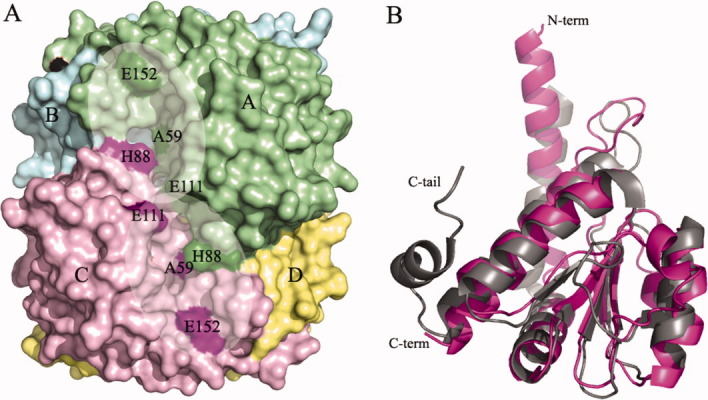
Molecular models and details of the KdsDK59A SIS domain crystal structure. A: Surface representation of the SIS domain tetramer present in the asymmetric unit, and the active site cavity formed by chains A (green) and C (pink), highlighting the two active sites (shading). The catalytic residues (A59, H88, E111, and E152) of the two active sites of chains A and C are highlighted in dark green or pink, respectively. B: Superimposition of the E. coli KdsDK59A mutant SIS domain monomer (purple) on MJ1247 from M. jannaschi (gray) achieved using the C-alpha match program (http://bioinfo3d.cs.tau.ac.il/c_alpha_match/). The C-tail of MJ1247 and the N- and C-termini of KdsDK59A are labeled. Figures were generated using Pymol Version 1.1r1 (http://www.pymol.org).
Figure 4.
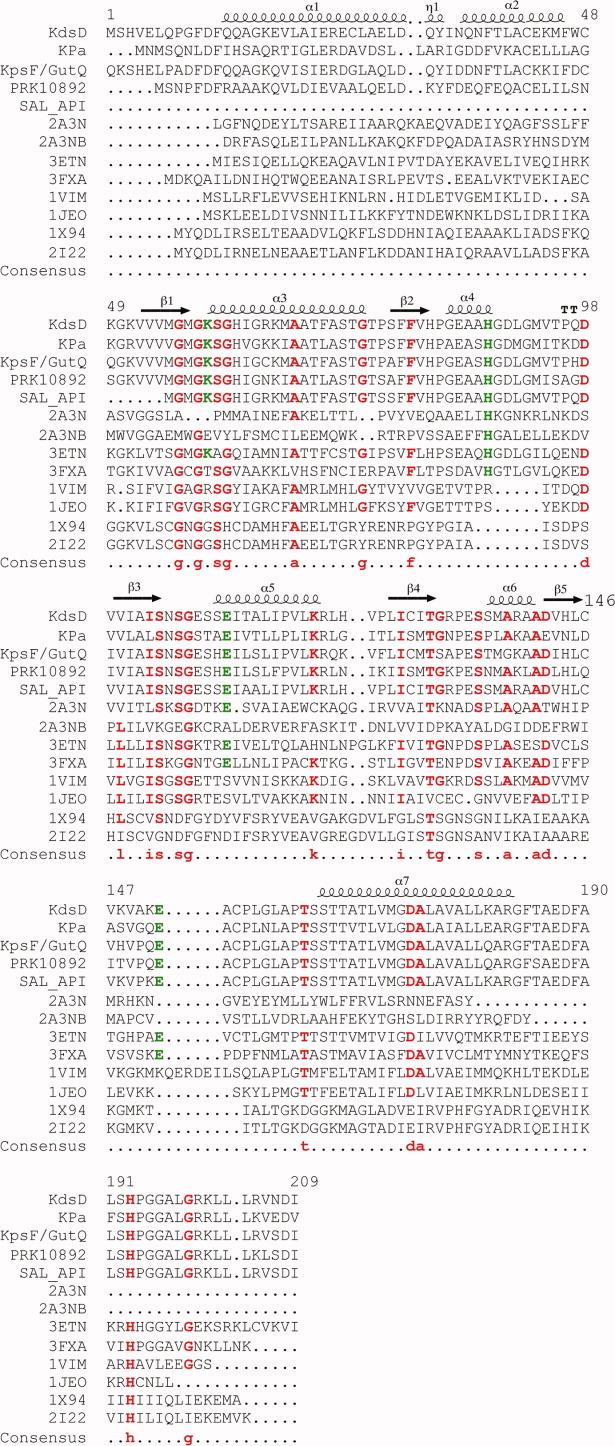
Multiple sequence alignments of SIS domains. Residues 1–209 of KdsD were aligned against single-domain (PDB entries 1VIM, 1JEO, 2I22, 3ETN, 3FXA, and 1X94) and the two-domain (2A3N) phosphosugar isomerase of known structure, and four, two-domain proteins (KdsD from Pseudomonas aeruginosa, KpsF/GutQ family protein from Pectobacterium carotovorum, d-arabinose-5-phosphate from Salmonella enterica, and a putative polysialic acid capsule expression protein from Vibrio parahaemolyticus), of unknown structure, alongside the secondary structure arrangement of KdsDK59A (residues 10–183). The sequences of the two diverse SIS domains of 2A3N are shown covering residues 1–171 (2A3N) and 172–343 (2A3NB). Consensus amino acids, including H88 and H193, and residues (K59, E111, and E152) known to be catalytically important in KdsDK59A are indicated in red and green font, respectively. Multiple sequence and secondary structure alignments were generated using the program MultAlin (http://multalin.toulouse.inra.fr/multalin/) and ENDscript1.1.
The atomic coordinates of the KdsDK59A SIS domain tetramer were used to search for homologs using the Profunc program available from http://www.ebi.ac.uk, which attempts to predict protein function using both sequence- and structure-based methods. There were 20 hits with six “certain” matches (E-value < 1.00E-06), with structural similarities >97.5%, despite low amino acid sequence identities that ranged from 19.1 to 38.1%, respectively (Table II). The C-alpha main chain atoms of subunit A of the SIS domain of KdsDK59A were superimposed with the SIS domain monomers shown in Table II using the C-alpha matching program (http://bioinfo3d.cs.tau.ac.il/c_alpha_match/). As expected, the structure of the KdsDK59A SIS domain is highly conserved (RMSD of the core domain ranging from 1.05 to 1.82 Å) despite low primary structure identity (<40%; Table II). As an example, in Figure 2(B), we show the structural comparison between the KdsDK59A SIS domain chain-A and the hypothetical MJ1247 protein from Methanococcus jannaschi (PDB code 1JEO).19
Table II.
Sequence and Monomer Structural Similarities Between Single- and Two-Domain Isomerases in Comparison with the SIS Domain of KdsDK59A
| PDB code | Sequence similarity (%) | Monomer RMSD (Å) |
|---|---|---|
| Single-domain isomerases | ||
| 1VIMa | 26.01 | 1.41 (152 res.) |
| 2I22b | 20.81 | 1.57 (128 res.) |
| 1JEOc | 21.97 | 1.43 (135 res.) |
| 1X94d | 20.23 | 1.53 (153 res.) |
| 3ETNe | 38.15 | 1.33 (163 res.) |
| 3FXAf | 31.21 | 1.05 (167 res) |
| Two-domain isomerases | ||
| 2A3N(A)g | 17.16 | 1.67 (123 res.) |
| 2A3N(B)h | 12.65 | 1.8 (117 res.) |
Hypothetical protein from Archeoglobus fulgidus.
Escherichia coli phosphoheptose isomerase.
Hypothetical MJ1247 protein from Methanococcus jannaschi.
Phosphoheptose isomerase from Vibrio cholerae.
Hypothetical phosphosugar isomerase from Bacteroides fragilis.
Sugar-phosphate isomerase from Listeria monocytogenes.
Residues 2–171 of glucosamine-fructose-6-phosphate aminotransferase from Salmonella typhimurium.
Residues 172–343 of glucosamine-fructose-6-phosphate aminotransferase from Salmonella typhimurium.
With regard to the tetrameric assembly, the homolog with the highest structural similarity to KdsDK59A (RMSD of 1.49 Å, covering 471/685 residues) is a hypothetical phosphosugar isomerase from Bacteroides fragilis (PDB code 3ETN; unpublished), with a proposed role in capsule formation.
Active sites and subunit interfaces
A complete analysis of protein cavities and pockets revealed the presence of two deep surface crevices, located at opposite sides of the tetramer [Fig. 2(A)]. The two crevices are almost identical and involve residues belonging to chains A/C, or the equivalent residues from chains B/D [Fig. 3(A)]. Each crevice has an internal twofold symmetry and provides, therefore, two identical adjacent active sites located in subunit A and C or B and D, respectively [Figs. 2(A) and 3(A)]. Regarding the A/C crevice, the active site from subunit A is lined by residues M57, G58, A59, S60, S104, N105, S106, S109, S110, E111, E152, G157, P160, S163, and T164 from chain A, and H82, G84, and H88 from chain C [Figs. 3(A) and 4]. The active site in subunit C is lined by the same residues with chain A and C reversed. The interface between the two symmetric active sites is marked by residues M57, H82, G84, and E111 from both A and C subunits. On the basis of the KdsDK59A structure, we confirm that the residues proposed to be catalytically important by previous homology modeling and mutagenesis studies (K59, mutated to alanine in our construct, E111 and E152)17 are located in the active site. An additional His residue, H193 (whose mutation to alanine yields an enzyme that is inactive both in vivo and in vitro), however, is not visible in the KdsDK59A structure,17 although it was confirmed to be present via in-gel digestion analysis by mass spectrometry (data not shown). The relevance of H193 was further evaluated in this work by the construction of a truncated KdsD construct comprising residues 1–183 of the SIS domain (KdsD 1–183; see below).
Figure 3.

A: Formation of the active site cavity: the two active sites are indicated by shading and catalytic residues present at the interface between monomers B (cyan) and D (yellow) are shown as sticks. For simplicity, monomers A and C are not shown. B: The active site interface between KdsDK59A monomers B (cyan) and D (yellow), with monomer A (green) superimposed on MJ1247 (gray), illustrating the positions of the catalytic residues in sticks. The position of the C-tail His residue (H176) in MJ1247 is shown in relation to H88 in KdsDK59A monomer D. Monomer C is not shown for simplicity. Figures were generated using Pymol Version 1.1r1 (http://www.pymol.org).
In the reported homology model, it was also suggested that a serine-/threonine-rich region could contribute to binding of the phosphate moiety of the substrate, as seen for the E. coli phosphoheptose isomerase (GmhA), crystallized in complex with its substrate, sedoheptulose-7-phosphate (PDB code 2I22).11,17 In agreement with this proposal, the four KdsDK59A chains contribute 11 serine/threonine residues to the active site cavity, involving S60, T70, S109, S163, T164, and T167 (Fig. 4).
To gain insight into the possible roles of the above identified residues, KdsDK59A was superimposed on GmhA (PDB code 2I22).11 Subunits A and B superimpose well with subunits A and D of GmhA, respectively; however, the remaining subunits do not superimpose, likely due to a conformational change that occurs on substrate binding at the active sites created by GmhA chains B and C. Several residues from GmhA have been confirmed to play established roles in substrate binding: S55, T120, D169, and Q172, which correspond to KdsDK59A residues S60, N105, T161, and T164, respectively, suggesting that these residues could indeed adopt a similar role in KdsD. The structure comparison highlighted an additional residue that could also be functionally important T70, which corresponds to GmhA residue E65, shown to be fundamental for this enzyme both in vitro and in vivo.11
With reference to the positions of the catalytically relevant residues of KdsDK59A in the active site of subunit A, H82 and E111 from both subunits (A and C) fall in a distant part of the cavity, furthest from the solvent, whereas A59 is located near the mouth of the cavity, opposite to H88 in subunit C [Fig. 3(A)]. Residue E152 is the most external residue, located in shallow cavity on the surface of the protein [Figs. 2(A) and 3(A)]. As previously mentioned, H193, located in the linker region between the SIS domain and the first CBS domain and shown to be essential for full isomerase activity, is located in the C-terminal segment of the crystallized SIS domain but not defined by electron density. The location of H193 in a flexible region of the crystallized protein does not allow us to reach firm conclusions on the role played by this residue, which may or may not become part of the active site, considering also that the corresponding residue (H176) in the sequence of the hypothetical protein MJ1247 from M. jannaschi has been shown to adopt a catalytic role.19
Concerning KdsDK59A residue H88, there appears to be no sequence-equivalent residue in GmhA or MJ1247 (Fig. 4). However, despite belonging to topologically diverse regions and to different subunits within the tetrameric assembly, the catalytic C-terminal histidine of MJ1247 (H176)19 matches the same three-dimensional location of H88 in KdsDK59A. H88 is housed in KdsDK59A helix α4, located between β-strands 2 and 3, whereas MJ1247 H176 is in the C-terminal region (C-tail) of the protein [Figs. 2(B) and 3(B)]. If we focus on the active site cavity of KdsDK59A subunit A, the position of H88 from chain C matches the position of MJ1247 H176 from chain B. Thus, for H176 to adopt the same spatial position as H88, the C-tail of MJ1247 subunit B folds in the active site of subunit A, whereas in KdsDK59A, this space is occupied by helix α4 from subunit C [Fig. 3(B)]. Similarly, MJ1247 H176 from chain D matches the position of KdsDK59A H88 from chain A in the active site of KdsDK59A subunit C [Fig. 3(B)]. Analogous rules apply for the active sites in subunits B and D of KdsDK59A. In light of the catalytic role of H176 in MJ1247, one could postulate that H88 plays a similar role in substrate binding/catalysis.
With regard to the superimposition of KdsDK59A with the hypothetical protein from Archeoglobus fulgidus (1VIM), subunits A and B superimpose well, however, C and D do not. Focusing on active site A in KdsDK59A, as for MJ1247, the C-tail His (H179) from subunit B is located in the same three-dimensional position as H88 in subunit C. Similarly, if we look at active site B, H179 from subunit A in 1VIM matches the position of H88 from subunit D. Contrary to the observations made for active sites C and D in MJ1247, these two His residues do not superimpose, a likely consequence of the conformational changes that are evident in these two subunits of 1VIM in comparison with KdsDK59A. The C-tail contribution to the formation of each of the four MJ1247 active sites and the two active sites in 1VIM appears to be a common structural feature of some single-domain sugar isomerases but not for the majority of sugar isomerases that contain additional domains, like KdsD (Fig. 4). GmhA and the phosphoheptose isomerase from Vibrio cholerae (1X94) are single-domain isomerase exceptions as they contain sequence-conserved C-terminal His residues, however structurally, they are not located in close proximity to the active site.
Interestingly, a BLAST primary sequence search (http://www.ncbi.nlm.nih.gov/) for homologs shows that H88 is highly conserved in all two-domain sugar isomerases (Fig. 4). The putative glucosamine-fructose-6-phosphate aminotransferase from S. typhimurium (PDB code 2A3N) contains two domains, although both are SIS domains with diverse sequences, nevertheless considered as a two-domain isomerase in our analyses. The first SIS domain contains a His residue at position 82, equivalent to H88 in subunit A, whereas H247, from the second SIS domain corresponds to H88 in the B subunit (Fig. 4). Interestingly, from our multiple alignments, we observe that the presence of a H88 equivalent is exclusively coupled to the presence of an additional five-residue segment that immediately follows the His residue; isomerases that lack a H88 display a gap in this region (Fig. 4). This loop region is also present in two single-domain exceptions: the phosphosugar isomerase from B. fragilis (PDB code 3ETN) that contains both His residues at positions 79, corresponding to KdsD 88 and at positions 186 and 187 in the C-tail, and the sugar-phosphate isomerase from Listeria monocytogenes (PDB code 3FXA), which hosts the conserved His residue equivalent to the position of H88, in addition to a C-terminal His at position 188, which is also located in the active site (Fig. 4). Together, our findings strongly infer a possible catalytic role for KdsD H88, which possibly extends to the majority of two-domain isomerases and some single-domain cases.
Functional analyses
The catalytic relevance of H88, indicated by our crystallographic analysis and further suggested by sequence comparisons with other sugar isomerases, was investigated by constructing the enzyme site-specific mutant KdsDH88A and by assessing its ability to catalyze the isomerization of A5P to R5P in vitro (see Materials and Methods). Indeed, such functional experiments confirmed the implications brought about by the structural analyses. The effect of the H88 → A mutation affected the isomerization of A5P into R5P, with the specific activity of KdsDH88A being reduced to 9.5% of the wild-type protein activity (Table III).
Table III.
Complementation Analysis and Specific Activity of Wild Type and Mutant Recombinant His-tag KdsD Proteins
| Growth conditiona |
||||||
|---|---|---|---|---|---|---|
| Complementing plasmid | Codon change | + Ara | + Glu | Protein | Specific activityb (mU/mg) | Residual activityc |
| pRSETb | — | + | —d | — | ||
| pRSETb/kdsD | — | + | + | KdsD | 29.55 ± 0.89 | 100 |
| pRSETb/kdsDH88A | CAT → GCT | + | + | KdsDH88A | 2.81 ± 0.19 | 9.5 |
| pRSETb/kdsDK59A | AAA → GCA | + | —d | KdsDK59A | 0.72 ± 0.06e | 2.4 |
| pRSETb/kdsD1–183 | — | + | —d | KdsD1–183 | nd | nd |
nd: not determined.
LD-ampicillin agar plates supplemented with Ara (+ Ara) and Glu (+ Glu) for induction and repression, respectively, of BB-8 chromosomal kdsD gene.
Specific activity was determined as reported in Materials and Methods. Each measurement is the mean of at least five independent determinations. Standard deviations never exceeded 10%.
Percentage (%)-specific activity in comparison with the wild-type protein.
Efficiency of plating ≤10−4.
Data from Sommaruga et al.17
To further explore the role of the H88A mutated residue, in vivo complementation studies were performed. The mutant plasmid was introduced into the BB-8 conditional mutant strain, where kdsD expression is induced by arabinose and the gutQ paralogous copy is inactivated (see Material and Methods section).16 Growth of the BB-8 strain transformed with the mutagenized plasmids was assessed under permissive (with arabinose) and nonpermissive conditions (with glucose; Table III). Albeit low, the residual activity displayed by the KdsDH88A mutant was sufficient to support cell growth when the protein was overexpressed in vivo from a plasmid in the conditionally defective API mutant BB-8 harboring pRSETb/kdsDH88A. The importance of H193 for KdsD activity was further supported by analysis of the KdsD construct carrying the 1–183 residues of the SIS domain. Indeed, the pRSETb/kdsD1–183 mutant plasmid is unable to complement the BB-8 strain under nonpermissive conditions (Table III).
Discussion
There are numerous biochemical pathways that remain to be exploited in the field of antimicrobial development, to design new inhibitors that can help combat the ever-increasing incidence of bacterial multidrug resistance. LPS biogenesis is an ideal target as it is essential for OM synthesis, in the absence of which Gram-negative bacteria cannot survive.2 As previously mentioned, LPS contains a highly conserved inner core component, Kdo.4 The first reaction of the Kdo synthesis is catalyzed by the API enzyme (KdsD in E. coli; Fig. 1). This reaction involves the reversible isomerization of d-ribulose-5-phosphate to d-arabinose-5-phosphate, although the exact catalytic mechanism of KdsD remains to be determined.
Following unsuccessful crystallization trials of wild-type recombinant KdsD, a catalytically inactive mutant K59A, expressed in recombinant form with an N-terminal His-tag, was crystallized and X-ray diffraction data were collected, permitting us to solve its SIS domain three-dimensional structure. In agreement with other API members, KdsDK59A is tetrameric, comprising four structurally identical subunits that adopt a Rossmann fold [Fig. 2(A,B)]. The four active sites are formed at the interfaces between two subunits (A/C or B/D), resulting in the formation of two extended surface grooves encompassing residues from subunits A and C and from subunits B and D, respectively [Fig. 2(A)]. The grooves contain two symmetrically adjacent active sites, one for each component subunit. The structure of KdsD confirms that K59 (here mutated to A59), E111, and E152, predicted to be catalytically relevant by previous homology modeling studies, are located in the active sites17 [Fig. 2(A)]. On the contrary, residue H193, highly conserved in both single- and two-domain isomerases (Fig. 4), also predicted by homology modeling and confirmed to be functionally relevant, is not defined, together with a few C-terminal residues, in the KdsDK59A structure. Although any firm conclusion on the involvement of H193 in the active site structure will require further crystal structure analyses, other roles for this residue can be suggested. In fact, H193 may play a key structural role, in agreement with dynamic light scattering (DLS) findings carried out on a SIS domain construct truncated at G183, which show that the protein is monomeric in solution (data not shown). Such findings suggest a role for H193 in KdsD oligomerization, in keeping with functional studies that suggest its involvement in catalysis.17
Furthermore, the active site cavities are particularly rich in serine and threonine residues, consistent with the hypothesis that such residues are involved in substrate binding, as reported for GmhA.11 Comparisons of the superimposed subunits of KdsDK59A and GmhA also allowed us to identify the residues S60, K66, T70, N105, T161, and T164 as possibly important functional residues, given that equivalent residues in GmhA are critical. The importance of such residues, however, remains to be confirmed in functional studies of the relevant mutants. Most importantly, from the KdsDK59A structure, we identified H88 as a new residue involved in catalysis.
H88 could not be predicted by previous homology modeling studies for two main reasons: (i) H88 is donated to the active site by the polypeptide chain adjacent to the subunit which houses the active site (i.e., H88 from chain C is part of the active site of subunit A and viceversa; the same applies to subunits B and D). Therefore, the structure of the full quaternary assembly would have been required to predict the strategic location of H88. (ii) The homology model was based on a single-domain isomerase, where the C-terminal tail of the protein is part of the active site. As previously mentioned, H193 at the C-terminus of the SIS domain was confirmed to be important for KdsD function. This residue is highly conserved in both single- and two-domain enzymes and, based on our structural comparisons, with regard to space and geometry, an active site location for H193 would be acceptable. Nevertheless, complementation and DLS studies of a truncated SIS domain construct indicate that the residues following G183 are necessary for bacterial growth and tetramer formation, respectively, stressing a structural role for H193 in proper active site formation in the oligomeric species.
The possibility of H88 being functionally relevant in other isomerases was raised following a sequence BLAST search that showed that this residue is highly conserved in enzymes that typically contain an additional domain to the SIS domain but not in those composed of the SIS domain only (Fig. 4). To determine the importance of H88 for the isomerization of A5P into R5P, in vitro functional assays were carried out confirming that H88 is, in fact, crucial for KdsD activity (Table III). Although the purified mutant protein showed only 9.5% of residual activity compared with the wild-type protein, its overexpression in the in vivo complementation assay was sufficient to rescue bacterial growth due to the increased amount of protein in the cell (Table III). Interestingly, in the Pseudomonas aeruginosa KdsD homolog, the corresponding histidine residue (H85) mutated to Ala gives rise to a protein inactive in vitro, as its specific activity is below the detection limit of the assay, and in vivo as, even when overexpressed, it does not complement the conditionally defective API mutant (unpublished).
KdsD is an ideal target for the design of novel antimicrobial drugs as it exerts an essential function in LPS biogenesis, a vital component of the OM of Gram-negative bacteria, and deletion mutants are lethal.14–16 Furthermore, it pertains to the wide API family of enzymes that is highly conserved in phylogenetically diverse bacteria, therefore KdsD inhibitors could offer cross-species protection. In this context, the crystal structure of KdsDK59A provides a detailed experimental picture of the catalytic site, which is dependent on the quaternary assembly of the protein, likely being specific for two-domain isomerases. Moreover, the data here reported allow targeting in silico docking studies for the design/discovery of new antimicrobial drug leads to a specific region of KdsD.
Materials and Methods
Cloning, expression, and purification of KdsD proteins
Plasmids pRSETb/kdsD and pRSETb/kdsDK59A, which express KdsD (accession number NP_417664.1) and KsdDK59A, respectively, with an His6-tag at the N-terminal end have been previously described.17 pRSETb/kdsD was used as template to generate the H88A substitution using the QuickChange site-directed mutagenesis kit (Stratagene), obtaining pRSETb/kdsDH88A. The KdsD protein carrying the 1–183 residues of the SIS domain was constructed by amplification of the kdsD gene from pRSETb/kdsD template using oligonucleotides AP122Bam 5′-CGA GAT GGA TCC GTC GCA CGT AGA GTT ACA AC-3′ and EcSISrevNco 5′-CAT CAT GCC ATG GTT AGC CGC GTG CTT TTA ACA GCG-3′. The amplified fragment was digested with BamH1 and NcoI restriction enzymes and cloned into the pRSETb vector using standard cloning procedures, generating pRSETb/kdsD1–183.
Wild-type and mutant proteins were then expressed, purified by Ni-chelate affinity chromatography and dialyzed into 20 mM Tris pH 8.0 as previously described.17
Complementation assays
E. coli BB-8 is a conditional mutant in which gutQ is substituted by a chloramphenicol resistance (cat) cassette, expression of kdsD is under control of the arabinose-inducible araBp promoter, and thus cannot grow in the absence of this sugar.16 For the complementation analysis, BB-8 transformed by pRSETb/kdsD, pRSETb/kdsDK59A, and pRSETb/kdsDH88A plasmids was grown at 37°C until stationary phase in LD supplemented with 0.2% arabinose (Ara) and 100 μg/mL ampicillin (Amp). The cultures were then serially diluted, replicated on LD–Amp agar plates with 0.2% Ara or 0.2% glucose (Glu), and incubated overnight at 37°C.
Crystallization
Following unsuccessful crystallization attempts of wild-type KdsD, crystallization trials of full length, E. coli KdsDK59A were set up in 96-well sitting drop plates (Greiner) using the Oryx 8.0 crystallization robot (Douglas Instruments), at a protein concentration of 13.5 mg/mL. Small (∼50 μm) crystals, difficult to reproduce, grew after 2 weeks at 20°C in a 0.3 μL crystallization drop containing 70% protein and 30% well solution (20% PEG 8K and 0.1 M HEPES, pH 7.5). Crystals were flash frozen in liquid nitrogen in the crystallization solution supplemented with 24% glycerol as the cryoprotectant.
Data collection and processing
Diffraction data were collected at a resolution of 2.6 Å at the European Synchrotron Radiation Facility (Grenoble, France; beam line ID23-1). Data were processed using imosflm and SCALA and assigned to the monoclinic P21 space group.20,21 Four SIS domain chains were eventually shown to be present per asymmetric unit (see below), with a solvent content estimated to be 33.1% (Table I).
Molecular replacement, model building, and refinement
The three-dimensional structure of the SIS domain of KdsDK59A was solved by molecular replacement using the structure of a putative phosphosugar isomerase from B. fragilis (PDB code 3ETN) as a search model (42.6% sequence identity). During molecular replacement, it became apparent that only the SIS domain was present as a tetramer in the asymmetric unit, suggesting that full-length KdsDK59A had undergone proteolysis during crystallization, resulting in the removal of the CBS domain. This was subsequently confirmed by SDS-PAGE (not shown). The structure was refined using REFMAC 5.422 and fitted to the generated electron density maps using Coot.23 All data were refined to satisfactory final Rfactor, Rfree factors and geometric parameters (Table I). The atomic coordinates and structure factors for the SIS domain of E. coli KdsDK59A have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org) under PDB code 2XHZ.24
Quality of the structural data
Electron density corresponding to residues 11–182 of the KdsDK59A SIS domain was visible in chains A–D. A region of scarce electron density is seen, which corresponds to residues 86–90 in chains A–D, corresponding to α-helix number 4, located in a flexible region between β-strands 2 and 3. The geometric parameters were checked using Procheck, assessing that all residues fell in the Ramachandran plot allowed or favorably allowed regions.18
Determination of KdsD activity
KdsD activity was assayed continuously using A5P (Sigma–Aldrich) as a substrate, as previously described.17 The conversion of A5P into Ru5P was monitored by following the increase in absorbance at 280 nm. At this wavelength, the molar absorption coefficient for Ru5P is 58.6 M−1 cm−1. One Unit of enzyme is defined as the amount of enzyme that produces 1 μmol of product per minute under standard assay conditions. Protein content was determined using the Coomassie Plus Protein Assay Reagent (Pierce) and bovine serum albumin as the standard reference protein.
Acknowledgments
LJG is a recipient of Assegno di Ricerca (2009) from the University of Milano. The authors are grateful to Prof. R. Grandori and Dr. M. Samalikova (Università di Milano-Bicocca) for the mass spectrometry studies.
Glossary
Abbreviations:
- A5P
d-arabinose-5-phosphate
- API
d-arabinose-5-phosphate isomerase
- CBS
cystathionine-β-synthase domain
- GmhA
phosphoheptose isomerase
- Kdo
3-deoxy-d-manno-octulosonate
- KdsD
E. colid-arabinose-5-phosphate isomerase
- KdsDK59A
His-tagged recombinant K59A mutant of KdsD
- LPS
lipopolysaccharide
- OM
outer membrane
- PDB
Protein Data Bank
- Ru5P
ribulose-5-phosphate
- SIS
sugar isomerase domain.
References
- 1.Raetz CR, Guan Z, Ingram BO, Six DA, Song F, Wang X, Zhao J. Discovery of new biosynthetic pathways: the lipid A story. J Lipid Res. 2009;50:S103–S108. doi: 10.1194/jlr.R800060-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Raetz CR, Whitfield C. Lipopolysaccharide endotoxins. Annu Rev Biochem. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nikaido H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol Mol Biol Rev. 2003;67:593–656. doi: 10.1128/MMBR.67.4.593-656.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Holst O. The structures of core regions from enterobacterial lipopolysaccharides—an update. FEMS Microbiol Lett. 2007;271:3–11. doi: 10.1111/j.1574-6968.2007.00708.x. [DOI] [PubMed] [Google Scholar]
- 5.Schnaitman CA, Klena JD. Genetics of lipopolysaccharide biosynthesis in enteric bacteria. Microbiol Rev. 1993;57:655–682. doi: 10.1128/mr.57.3.655-682.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rose IA. Mechanism of the aldose-ketose isomerase reactions. Adv Enzymol Relat Areas Mol Biol. 1975;43:491–517. doi: 10.1002/9780470122884.ch6. [DOI] [PubMed] [Google Scholar]
- 7.Asboth B, Naray-Szabo G. Mechanism of action of d-xylose isomerase. Curr Protein Pept Sci. 2000;1:237–254. doi: 10.2174/1389203003381333. [DOI] [PubMed] [Google Scholar]
- 8.Jeffery CJ, Hardre R, Salmon L. Crystal structure of rabbit phosphoglucose isomerase complexed with 5-phospho-d-arabinonate identifies the role of Glu357 in catalysis. Biochemistry. 2001;40:1560–1566. doi: 10.1021/bi0018483. [DOI] [PubMed] [Google Scholar]
- 9.Lee JH, Chang KZ, Patel V, Jeffery CJ. Crystal structure of rabbit phosphoglucose isomerase complexed with its substrate d-fructose 6-phosphate. Biochemistry. 2001;40:7799–7805. doi: 10.1021/bi002916o. [DOI] [PubMed] [Google Scholar]
- 10.Teplyakov A, Obmolova G, Badet-Denisot MA, Badet B, Polikarpov I. Involvement of the C terminus in intramolecular nitrogen channeling in glucosamine 6-phosphate synthase: evidence from a 1.6 A crystal structure of the isomerase domain. Structure. 1998;6:1047–1055. doi: 10.1016/s0969-2126(98)00105-1. [DOI] [PubMed] [Google Scholar]
- 11.Taylor PL, Blakely KM, de Leon GP, Walker JR, McArthur F, Evdokimova E, Zhang K, Valvano MA, Wright GD, Junop MS. Structure and function of sedoheptulose-7-phosphate isomerase, a critical enzyme for lipopolysaccharide biosynthesis and a target for antibiotic adjuvants. J Biol Chem. 2008;283:2835–2845. doi: 10.1074/jbc.M706163200. [DOI] [PubMed] [Google Scholar]
- 12.Harmer NJ. The structure of sedoheptulose-7-phosphate isomerase from Burkholderia pseudomallei reveals a zinc binding site at the heart of the active site. J Mol Biol. 2010;400:379–392. doi: 10.1016/j.jmb.2010.04.058. [DOI] [PubMed] [Google Scholar]
- 13.Meredith TC, Woodard RW. Characterization of Escherichia colid-arabinose-5-phosphate isomerase encoded by kpsF: implications for group 2 capsule biosynthesis. Biochem J. 2006;395:427–432. doi: 10.1042/BJ20051828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meredith TC, Woodard RW. Escherichia coli YrbH is a d-arabinose-5-phosphate isomerase. J Biol Chem. 2003;278:32771–32777. doi: 10.1074/jbc.M303661200. [DOI] [PubMed] [Google Scholar]
- 15.Meredith TC, Woodard RW. Identification of GutQ from Escherichia coli as a d-arabinose-5-phosphate isomerase. J Bacteriol. 2005;187:6936–6942. doi: 10.1128/JB.187.20.6936-6942.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sperandeo P, Pozzi C, Deho G, Polissi A. Non-essential KDO biosynthesis and new essential cell envelope biogenesis genes in the Escherichia coli yrbG-yhbG locus. Res Microbiol. 2006;157:547–558. doi: 10.1016/j.resmic.2005.11.014. [DOI] [PubMed] [Google Scholar]
- 17.Sommaruga S, Gioia LD, Tortora P, Polissi A. Structure prediction and functional analysis of KdsD, an enzyme involved in lipopolysaccharide biosynthesis. Biochem Biophys Res Commun. 2009;388:222–227. doi: 10.1016/j.bbrc.2009.07.154. [DOI] [PubMed] [Google Scholar]
- 18.Lovell SC, Davis IW, Arendall WB, 3rd, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins. 2003;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 19.Martinez-Cruz LA, Dreyer MK, Boisvert DC, Yokota H, Martinez-Chantar ML, Kim R, Kim SH. Crystal structure of MJ1247 protein from M. jannaschi at 2.0 A resolution infers a molecular function of 3-hexulose-6-phosphate isomerase. Structure. 2002;10:195–204. doi: 10.1016/s0969-2126(02)00701-3. [DOI] [PubMed] [Google Scholar]
- 20.Evans PR. Proceedings of the CCP4 study weekend on data collection and processing. UK: CLRC Daresbury Laboratory; 1993. [Google Scholar]
- 21.Leslie AG. The integration of macromolecular diffraction data. Acta Crystallogr Sect D. 2006;62:48–57. doi: 10.1107/S0907444905039107. [DOI] [PubMed] [Google Scholar]
- 22.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr Sect D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 23.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr Sect D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 24.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]