

Abstract
The bacterial enzyme 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidase (MTAN) plays a central role in three essential metabolic pathways in bacteria: methionine salvage, purine salvage, and polyamine biosynthesis. Recently, its role in the pathway that leads to the production of autoinducer II, an important component in quorum-sensing, has garnered much interest. Because of this variety of roles, MTAN is an attractive target for developing new classes of inhibitors that influence bacterial virulence and biofilm formation. To gain insight toward the development of new classes of MTAN inhibitors, the interactions between the Helicobacter pylori-encoded MTAN and its substrates and substrate analogs were probed using X-ray crystallography. The structures of MTAN, an MTAN-Formycin A complex, and an adenine bound form were solved by molecular replacement and refined to 1.7, 1.8, and 1.6 Å, respectively. The ribose-binding site in the MTAN and MTAN-adenine cocrystal structures contain a tris[hydroxymethyl]aminomethane molecule that stabilizes the closed form of the enzyme and displaces a nucleophilic water molecule necessary for catalysis. This research gives insight to the interactions between MTAN and bound ligands that promote closing of the enzyme active site and highlights the potential for designing new classes of MTAN inhibitors using a link/grow or ligand assembly development strategy based on the described H. pylori MTAN crystal structures.
Keywords: nucleosidase, X-ray crystallography, quorum-sensing, activated methyl cycle
Introduction
The enzyme 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidase (MTAN) is known to catalyze the N-glycosyl hydrolysis of three different adenosyl derivatives, placing it at a central hub in bacterial metabolism.1,2 The substrates for MTAN, 5′-methylthioadenosine (MTA), 5′-deoxyadenosine, and AdoHcy, are produced from S-adenosylmethionine (AdoMet) through the synthesis of polyamines, AdoMet radical reactions, and by AdoMet-dependent methylation reactions, respectively. The products from the MTAN enzymatic reaction feed directly into the purine and methionine salvage pathways. Because adenine and methionine, as well as AdoMet, are energetically expensive for bacteria to synthesize de novo, by leveraging these salvage pathways, bacteria avoid the costly production of these important metabolites. The polyamines, such as spermine and spermidine, produced from AdoMet are important for interacting with nucleic acids and regulating DNA replication.3–7 Because AdoMet lies at a metabolic nexis of so many important pathways, the importance of appropriately degrading AdoHcy, MTA, and 5′-deoxyadenosine cannot be overstated. Inappropriate accumulation of these MTAN substrates promotes feedback inhibition of AdoMet-dependent methyltransferases, proteins of the polyamine biosynthetic pathway, and the AdoMet-radical dependent biotin synthase, respectively.2–5
In addition to the direct metabolic effects, every common biological polymer (nucleic acids, proteins, and polysaccharides) is modified by AdoMet-dependent methyltransferases.7 The effects of these methylation reactions are manifold, so it follows that the level of AdoMet-dependent methyl transfer reactions and the concentration of downstream products from these reactions reflect the metabolic health of any bacteria and that disruption of this pathway would be detrimental to bacterial cells.7,8 Indeed, many bacteria exhibit a profound response to downstream metabolites of AdoHcy degradation. Some of these diffusible signaling molecules allow species specific and interspecies bacterial communication, or quorum-sensing, between neighboring bacteria.9
There are now many known classes of compounds that act as quorum-sensing signaling molecules.9 Autoinducer-2 (AI-2) molecules are furanosyl compounds synthesized from the carbohydrate moiety of AdoHcy (Fig. 1). Two enzymes are required for the synthesis of AI-2 from AdoHcy. MTAN catalyzes the irreversible hydrolysis of the N-glycosidic bond in AdoHcy to produce adenine and S-ribosylhomocysteine.1 In the second step, the S-ribosylhomocysteine Lyase (LuxS) enzyme then catalyzes the transformation of S-ribosylhomocysteine to homocysteine and 4,5-dihydroxy-2,3-pentanedione, which is the precursor to both types of AI-2 compounds.10 4,5-Dihydroxy-2,3-pentanedione spontaneously cyclizes and reacts with water forming one of two possible diastereomers, (2S,4S)-2-methyl-2,3,3,4-tetrahydroxytetrahydrofuran (S-THMF) or (2R,4S)-2-methyl-2,3,3,4-tetrahydroxytetrahydrofuran (R-THMF), depending on direction of nucleophilic attack on carbon 2. Additionally, S-THMF can react with borate to produce a furanosyl boron diester (S-THMF-borate). Both R-THMF and S-THMF-borate are diffusible AI-2 molecules that can promote quorum-sensing in bacterial populations. Although S-THMF-borate has been shown to affect only Vibrio species, R-THMF can affect the metabolism and gene expression of many different bacterial species.11–14 The downstream gene expression and metabolic effects mediated by AI-2 molecules are still being characterized, but those identified to date are significant. Some of these include antibiotic production, sporulation, expression of virulence factors such as type III secretion systems, flagellar biosynthesis, and biofilm formation.15–19
Figure 1.

Metabolic pathway encompassing MTAN. This series of reactions illustrates the degradation of AdoHcy after AdoMet is used as a general methyl-donor. The enzyme that catalyzes each step is boxed. The chemical steps following 4,5-dihydroxy-2,3-pentanedione production that lead to the production of the THMF compounds and S-THMF-borate occur spontaneously without the need for any enzymes.
In various Helicobacter pylori, the presence or absence of AI-2 produces mixed and sometimes contradictory results that appear to be strain specific. In H. pylori strain G27, the production of AI-2 directly affects flagellar morphogenesis and thereby regulates bacterial motility.15 The H. pylori SSI strain, when lacking a functional luxS gene, exhibits a decrease in viability in mice, whereas the X47 strain of H. pylori that also lacked a functional luxS gene showed no effect.20 Although no studies have yet examined the consequence of inhibiting MTAN activity in H. pylori in vivo, inhibiting MTAN in E. coli and V. cholerae strains decreases the production of AI-2 in these bacteria and produces defects in biofilm formation.16
Structural and kinetic studies of MTAN from multiple species have led to a clear understanding of the roles of active site residues necessary for catalysis and the details of the enzymatic mechanism [Fig. 2(A)].21–25 Using the numbering of the H. pylori MTAN (HpMTAN) enzyme for clarity, catalysis is initiated by proton transfer from D198 to the N7 atom of the substrate. The resulting delocalized positive charge on the adenine moiety leads to stretching, and ultimately breaking, of the N-glycosidic bond of the substrate. This produces an oxocarbenium ion that is attacked by a nucleophilic water molecule coordinated by the C3′-hydroxyl of the substrate, and the side chains of R194 and E13, with E13 acting as a general base to activate the nucleophilic water. A multiple sequence alignment of MTAN sequences from 50 bacterial species representing diverse genera clearly exhibits the importance of these catalytic residues [Fig. 2(B)]. Interestingly, this sequence alignment highlights other conserved residues that may possess important functions not yet described.
Figure 2.
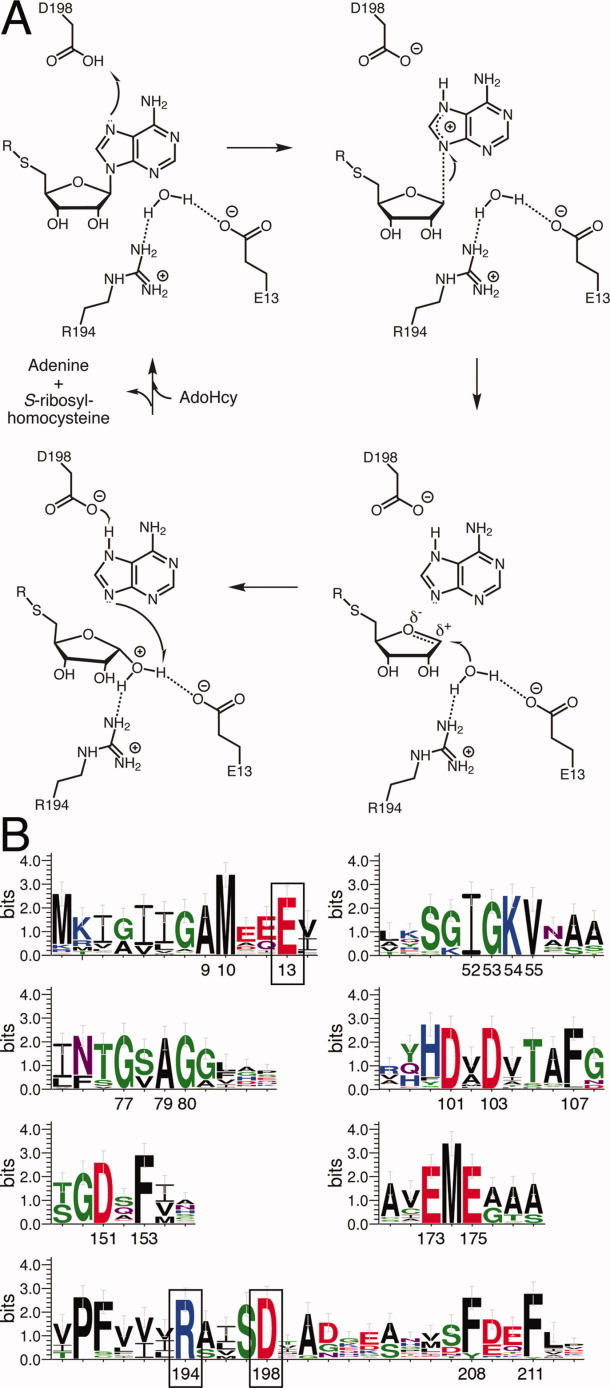
Proposed MTAN Enzymatic Mechanism. (A) In HpMTAN, the side chain of D198 acts as a general-acid by donating a proton to N7 of the adenine moiety of the substrate (upper left). The reaction proceeds through the formation of early and late transition states shown in the following two steps. In the final step, a water molecule coordinated by the side chains of E13 and R194 attacks the oxocarbenium intermediate. (B) Sequence Requirements of MTAN. The WebLogo was made using a sequence alignment of MTAN sequences from 50 different bacterial species. The regions containing conserved residues are shown. The numbering is based on the numbering of HpMTAN. As a reference, the residues shown in panel A to be important for the enzyme mechanism are boxed. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Although not essential for bacterial viability, MTAN has been the focus of intense efforts for the design of inhibitors and characterization of MTAN inhibitory effects on quorum-sensing.16,26–29 These inhibitors prevent biofilm formation in both E. coli O157:H7 and V. cholerae N16961.16 This inhibitory activity correlates with a measurable decrease in AI-2 production in V. harveyi under the same conditions. In the same study, bacteria cultured in the presence of a powerful MTAN inhibitor exhibit quorom-sensing defects after transfer of those bacteria to culture media lacking that inhibitor, thereby indicating that the inhibitory effects of MTAN inhibitors are persistent through multiple generations. A recent study examining mutants of Neisseria meningitidis lacking functional MTAN or LuxS enzymes showed similar growth defects. However, it was proposed that those growth defects were not a consequence of preventing quorum-sensing, but more likely arose from metabolic defects in the activated methyl cycle.30
Most of the current MTAN inhibitors are adenosine analogs that are known to inhibit human methylthioadenosine phosphorylase (MTAP), a homolog of the bacterial MTANs, and exhibit exceptional Kd values.27 However, the design work has neglected to specifically target the bacterial proteins over the human homolog. The Kd values for a complex of human MTAP with the extremely tight binding inhibitors developed to date are generally on the same order of magnitude as the Kd values for a complex of MTAN with the same inhibitors. In fact, when comparing the Kd values of 9-deazaadenine Immucillin-A derivatives for human MTAP versus HpMTAN, the inhibitors all bind the human protein more strongly than they bind HpMTAN.27 The two regions of the MTAN active site that differ from the MTAP active site include: the ribose binding pocket that both coordinates ribose and activates the nucleophile that attacks the oxocarbenium intermediate during catalysis, and the 5′-alkylthio binding region that interacts with the homocysteine moiety of AdoHcy.31 Here, we report our initial structural studies on the H. pylori-encoded MTAN that provide the basis for developing new classes of inhibitors that can leverage the differences between the HpMTAN enzyme and human MTAP.
Results
Description and relation to other MTAN structures
Structures of bacterial and plant MTAN and its homologous MTAP enzymes have been solved in multiple forms that thoroughly detail the interactions between the enzymes and the substrates, products, transition state analogs, and other inhibitors.21–23,26,31–36 Briefly, every MTAN structure solved to date, including the HpMTAN structure presented here, shows the enzyme as an obligatory homodimer. The dimer interface is formed primarily by a cluster of four helices, two from each monomer, plus a single loop composed of roughly 20 residues that interacts directly with residues forming the active site in the opposing molecule. Although this shared active site could promote cross talk between the two active sites of the dimer, no allosteric effects have been described in any of the previously published studies with these enzymes.
Each monomer is shown to possess a mixed α+β fold with a central β-sheet encased in a surrounding shell of six α-helices. The monomers of all MTAN structures have been observed in either of two forms depending on the ligand-bound state of the enzyme (Fig. 3). When a ribose-containing compound occupies the ribose binding region of the active site, a closed form of the enzyme is observed. In this form, the C-terminal 25 residues of MTAN form a kinked helix (helix α6) where the short arm of the helix forms a cap over the active site to sequester the substrate and products from the aqueous environment. If the ribose binding region of the active site is empty, the N-terminus of the helix α6 disengages the active site and the kink relaxes into random coil, resulting in an open form of the enzyme. This opening of the active site is presumed to promote product release and allows enzyme cycling.
Figure 3.
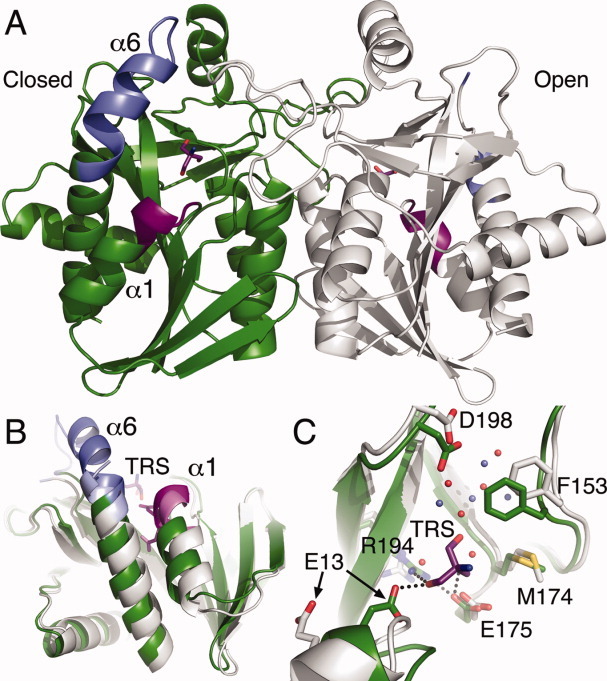
Structure of HpMTAN. (A) The dimeric structure of HpMTAN lacking substrates or products exhibits both the open and closed forms of the enzyme. The open form (HpMTAN-HOH in the text) is shown with a gray backbone ribbon, and the closed form (HpMTAN-TRS in the text) is shown in green. The blue (helix α6) and violet (helix α1) regions highlight the protein backbone regions that are have altered structure when the enzyme switches between the two forms. The tris molecule in the closed active site and an ethylene glycol molecule in the open active site are shown. Violet bonds represent carbon, red represent oxygen, and blue represent nitrogen. (B) Superposition of the HOH and TRS molecules. The colors for each molecule are the same as that shown in panel A. The superposition of these two molecules clearly exhibits the changes in the positioning and relative orientation of helices α1 and α6. The position of the tris molecule (TRS) relative to these helices is also shown. (C) Active site structure changes in response to tris binding. The coloring of TRS is the same as described in Fig 3A. Additionally, bonds from carbon atoms in HpMTAN-HOH are in white, whereas bonds from carbon atoms from HpMTAN-TRS are green. Yellow represents bonds from sulfur atoms. The water molecules from HpMTAN-HOH are shown as red spheres, whereas those from the HpMTAN-TRS molecule are light blue spheres. Dashed lines represent newly formed hydrogen bonds resulting from tris binding and active site closing. Figures three, five, six, and seven were made using PyMOL.37
For the HpMTAN structures, the MTAN protein crystallized in a P3121 space group with unit cell parameters a = b = 81.3 Å, c = 135.5 Å, α = β = 90°, γ = 120° with two molecules per asymmetric unit (Table I). In this case, each of the two molecules represents a different form of the enzyme [Fig. 3(A)]. The active site in molecule A represents the closed form of the enzyme (HpMTAN-TRS), which contains five bound water molecules and one molecule of tris[hydroxymethyl]aminomethane (tris) in the active site. The active site in molecule B represents the open form of the enzyme and contains bound water molecules (HpMTAN-HOH) and a single molecule of ethylene glycol.
Table I.
X-ray Diffraction and Refinement Statistics
| MTAN | MTAN-FMA | MTAN-ADE-TRS | |
|---|---|---|---|
| Resolution range (Å) (highest shell) | 50.00–1.70 (1.74–1.70) | 50.00–1.80 (1.85–1.80) | 50.00–1.60 (1.66–1.60) |
| Space group | P3121 | P3121 | P3221 |
| a, b (Å) | 81.3 | 81.7 | 81.4 |
| c (Å) | 135.5 | 134.5 | 67.6 |
| Total reflections (unique reflections) | 813,325 (57,817) | 236,192 (47,622) | 377,229 (33,924) |
| Completeness (%) | 100.0 (100.0) | 97.3 (96.3) | 98.7 (97.7) |
| Redundancy | 14.1 | 5.0 | 11.1 |
| Average I/σ(I) | 9.3 | 10.0 | 11.5 |
| Rsym (%) (highest shell) | 7.1 (36.7) | 6.1 (36.1) | 5.0 (38.7) |
| Atoms/A.S.U. | 3,872 | 3,871 | 1,983 |
| Rwork (%) | 17.00 | 17.06 | 14.14 |
| Rfree (%) | 19.53 | 20.74 | 17.07 |
| Average B-factor protein (Å2) | 19.47 | 18.02 | 21.29 |
| Average B-factor ligand (Å2) | 25.12 | 14.19 | 16.95 |
| Average B-factor solvent (Å2) | 28.60 | 29.91 | 33.31 |
| Ligand occupancy (%) | 92 | 100 | 100 |
| r.m.s.d. bonds (Å) | 0.006 | 0.006 | 0.005 |
| r.m.s.d. angles (°) | 1.005 | 1.017 | 0.945 |
| Ramachandran favored (%) | 97.1 | 96.5 | 96.6 |
| Ramachandran disallowed (%) | 0.0 | 0.0 | 0.0 |
| Coordinate Error (Å) | 0.18 | 0.19 | 0.17 |
Rsym = Σ|Ii − 〈Ii〉|/Σ|Ii|
Rwork = Σ||Fobs| − |Fcalc||/Σ|Fobs|
Rfree is the same as Rwork, but calculate using 5.0% of randomly chosen reflections.
Structural changes upon active site closing
When comparing the overall structures of HpMTAN-HOH and HpMTAN-TRS, the majority of the Cα atoms have few observed differences. However, the r.m.s. displacement is an unexpectedly high value of 1.25 Å. This value reflects the fact that 27% of the observed Cα atoms differ in position by at least 1.3 Å, where the most significant differences between the open and closed forms in the HpMTAN crystal structure occur in regions near the enzyme active site and reflect significant conformational changes observed in previously published structures of bacterial MTANs. The largest structural difference encompasses residues 198–224. Specifically, residues 200–208 of HpMTAN-HOH are disordered in the open form, but in the closed form of the enzyme, these residues form a loop that connects strand β11 to helix α6. Within this loop, residues 200–203 form a type II β-turn, whereas residues 204–208 extend helix α6 by five residues. The positional differences in the residues flanking the 200–208 region exemplify the magnitude of the conformational change within helix α6 [Fig. 3(B)]. Specifically, the position of the N199 Cα atom differs by 2.9 Å as a result of active site closing, whereas the position of the Cα atom of D209 changes by 5.5 Å.
The other significant conformational change occurs in the first helix that encompasses residues 10–21 [Fig. 3(C)]. Within this secondary structural element, a single-turn 310-helix, formed by residues 11–14, immediately precedes α-helix α1 in the open form of the enzyme. In transitioning to the closed form of the enzyme, however, the hydrogen bond between the backbone carbonyl of R11 and the backbone amide of I14 breaks, and a new bond is formed between the backbone carbonyl of R11 and the backbone amide of T15, thereby adding an additional turn to the N-terminus of helix α1. The primary consequence of this structural change is the movement of residue E13. The Cδ atom of E13 is shifted 6.7 Å toward the enzyme active site, which results in the formation of a through-water hydrogen bonded network between the side chains of E13 and R194. The water molecule in this newly formed network is the nucleophilic water molecule used to attack the oxocarbenium intermediate during catalysis. Other consequences of the structural changes near E13 will be discussed later.
As HpMTAN readily binds tris in the ribose binding pocket, enzymatic assays were designed to quantify the inhibition of HpMTAN by tris. Steady state kinetic experiments were performed by monitoring the loss of signal at 274 nm as a result of N-glycosidic cleavage of MTA. For MTA hydrolysis reactions lacking tris, the KM and kcat values were determined to be 44.9 μM ± 12.95 and 4.92 sec−1 ± 0.61, respectively. These values were then applied to the results from the reactions performed using different concentrations of tris to determine the Ki value. These data show that tris is a weak inhibitor of the HpMTAN with a Ki of 25.24 mM (Fig. 4).
Figure 4.
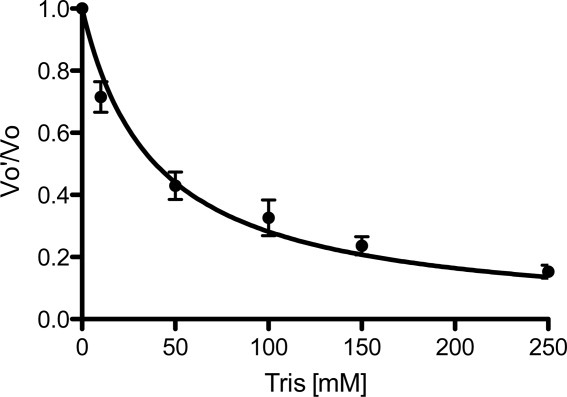
Tris is a weak inhibitor of HpMTAN. Results from steady state kinetic experiments using varying concentrations of tris are shown as compared to reactions performed lacking tris. VO′, initial velocity of reactions containing tris; VO, initial velocity of the uninhibited reaction.
Complex structure of HpMTAN with Formycin A
To confirm that the structural differences observed in helix α1 of HpMTAN-TRS are not structural artifacts of binding tris, we determined the crystal structure of a complex with the known nonhydrolysable transition state analog, Formycin A (FMA). Cocrystals produced using a complex of HpMTAN and FMA were isomorphous with the HpMTAN structure. In contrast to the HpMTAN structure, both molecules in the asymmetric unit of the HpMTAN-FMA cocrystal structure are in the closed conformation with one molecule of FMA and the nucleophilic water in both active sites of the homodimer [Fig. 5(A)]. This is consistent with the cocrystal structure of the S. aureus MTAN-FMA complex (SaMTAN-FMA) where the lone MTAN molecule in the asymmetric unit was observed in the closed form.34 In the SaMTAN-FMA case, the dyad axis relating the two molecules of the homodimer is coaxial with a crystallographic twofold axis of symmetry.
Figure 5.
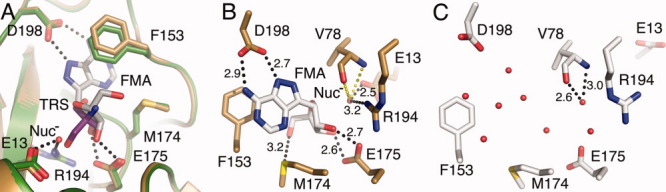
Structural comparison of the HpMTAN-FMA complex with HpMTAN. (A) The HpMTAN-TRS structure accurately represents the closed form of the enzyme. Components of the HpMTAN-TRS are colored as they are in Figure 3. Bonds to and from carbon in the FMA protein are gold, whereas the carbons in the FMA molecule are colored white. The nucleophilic water (Nuc−) and a tris hydroxyl group occupy nearly the same region of the active site. (B) and (C) Differences in ordered water molecules in the HpMTAN-FMA and HpMTAN-HOH structures. Note that the orientation of the structure in panel A is rotated 180° around the Y-axis when compared to panels B and C. (B) The FMA molecule and the nucleophilic water molecule form an extensive hydrogen bonding network. Black-dashed lines represent hydrogen bonds. The yellow dashed lines indicate potential hydrogen bonds between the nucleophilic water and backbone atoms of V78. However, distances between the backbone amide and carbonyl to the nucleophilic water are 3.6 and 3.7 Å, respectively. Additionally, the geometry of these two potential interactions is not commonly seen in hydrogen bonds. (C) The ordered water molecules (red spheres) within the HpMTAN-HOH active site fill the empty adenine binding and ribose binding pockets. The hydrogen bonds between the backbone atoms of V78 and the only water positioned near the nucleophilic water binding site indicate a possible docking site for the nucleophilic water molecule. When the substrate binds and E13 and R194 swing into the active site as observed in the HpMTAN-FMA stucture in Fig. 3B, those residues likely encounter this water in this docking site and reposition it. For all hydrogen bond distances shown in both (B) and (C), the units are angstroms.
Although the two molecules of the HpMTAN-FMA structure are not related by crystallographic symmetry, the r.m.s. displacement of the Cα positions of the two HpMTAN-FMA molecules is only 0.17 Å. Additionally, as both protein molecules in HpMTAN-FMA are in the closed conformation, they very closely resemble the HpMTAN-TRS structure. The r.m.s. displacement values comparing HpMTAN-TRS to molecules A and B of HpMTAN-FMA are 0.22 Å and 0.21 Å, respectively, thereby indicating that the closed form observed in the HpMTAN-TRS molecule accurately represents the substrate bound form of HpMTAN [Fig. 5(B)].
As expected for the FMA-bound structure, the nucleophilic water molecule forms a hydrogen bonded network with residues E13 and R194, as well as the O3′ oxygen of FMA with hydrogen bonding distances of 2.5, 3.2, and 2.9 Å, respectively. When compared to the HpMTAN-FMA structure, one of the active site water molecules in the HpMTAN-HOH structure is bound near to the nucleophilic water molecule observed in the HpMTAN-FMA structure. Similar to that seen for the EcMTAN-GOL and EcMTAN-ADE structures, this water molecule forms hydrogen bonds of length 3.0 and 2.6 Å with the backbone amide and carbonyl moieties of V78, respectively [Fig. 5(C)]. As this water is only 2.1 Å from the position of the nucleophilic water observed in the HpMTAN-FMA structure, it may represent a temporary water-docking site to ensure that a water molecule remains in the active site to promote the second step of the enzymatic reaction following the conformational change to the closed form.21
Comparison of HpMTAN-TRS and an HpMTAN-adenine-tris ternary complex
To gain additional specific information that describes interaction space between HpMTAN and various ligands, we incubated HpMTAN with AdoHcy and used this sample to cocrystallize the complex of HpMTAN with its reaction products, adenine and S-ribosylhomocysteine. Crystallization experiments produced crystals with the same morphology as the previous two HpMTAN structures. However, diffraction experiments indicated a change in both space group and unit cell parameters. Specifically, the hand of the threefold screw changed and the length of this axis was cut in half, indicating only one MTAN molecule per asymmetric unit. Comparison of the HpMTAN structures shows that crystal packing is identical for all three (Supporting Information Figure S1). Therefore, the change in space group and unit cell size stems from the fact that the two molecules of the homodimer now have identical structure and can be related to each other by crystallographic symmetry. In this structure, the two molecules of the homodimer are related by a crystallographic twofold axis of symmetry, similar to that seen in the native EcMTAN and FMA-bound SaMTAN structures.21,34
It was anticipated that the resulting difference maps would show HpMTAN in the closed form with both S-ribosylhomocysteine and adenine in the active site. Indeed, the difference maps calculated following molecular replacement showed MTAN in the closed form and that it possessed two discrete regions of strong density within the enzyme active site. We identified one region of density as the adenine product from hydrolysis of AdoHcy, whereas the second region of density was identified as a tris molecule rather than the expected S-ribosylhomocysteine. Although this was not completely unexpected considering the 20-fold higher concentration of tris versus S-ribosylhomocysteine, it was anticipated that the affinity of the enzyme active site for S-ribosylhomocysteine would promote complexation even in the presence of tris. The ternary complex of HpMTAN, adenine, and tris will be referred to as the HpMTAN-ADE-TRS form.
The refined HpMTAN-ADE-TRS model is very similar in backbone structure to the HpMTAN-FMA and HpMTAN-TRS models. When comparing the HpMTAN-ADE-TRS and one of the HpMTAN-FMA molecules, an R.M.S. displacement of 0.24 Å was calculated when using all Cα atoms. The three residues, E13, E175, and R194, responsible for creating the hydrogen bonded network with the nucleophilic water and ribose in the HpMTAN-FMA structure, or binding tris in the HpMTAN-TRS and HpMTAN-ADE-TRS structures, are in identical positions in all three structures. The presence of a purine in the active site of the HpMTAN-ADE-TRS structure, however, promotes slight differences in the purine binding pocket of the active site when compared to the HpMTAN-TRS structure. Both D198, the general acid responsible for initiating the reaction, and F153, which interacts with adenine through π–π interactions, shift slightly from their positions in the HpMTAN-TRS structure to accommodate the aromatic system. This reflects similar features observed in the EcMTAN complex with adenine and methylthioribose indicating that the HpMTAN-ADE-TRS structure accurately represents the structure of the product-bound form of HpMTAN.
Discussion
Here, we report crystal structures of HpMTAN that represent three different stages of the catalytic reaction pathway. The HpMTAN structure depicts the protein in two different forms. The HpMTAN-HOH form shows the enzyme in the open conformation with an active site containing structured water and a single ethylene glycol molecule. The second form, HpMTAN-TRS, shows the enzyme in a closed form where tris is bound in the ribose binding pocket of the enzyme active site. The presence of two forms in the same crystal contrasts with what has been seen for other bacterial MTAN crystal structures, where both proteins forming the MTAN homodimer possess the same form. In all prior bacterial structures, one of two situations occurs. In one situation, one molecule of MTAN is present in the asymmetric unit and that molecule is related by crystallographic symmetry to the second molecule of the dimer. In the second situation, there is a dimer in the asymmetric unit where both molecules are in either the closed or the open form. The HpMTAN structure described here shows unambiguously that the two molecules of the MTAN homodimer can take different conformations and that each molecule of the MTAN dimer can act independently of the other molecule.
Comparison of the open and closed forms highlights conformational changes between HpMTAN-HOH and HpMTAN-TRS that promote substrate binding within the active site and subsequent N-glycosidic bond hydrolysis. The two most significant changes occur in helix α1 and helix α6, both of which form essential components of the enzyme active site. The conformational change in helix α1 is different from that seen in other structurally characterized MTANs. In the previously published bacterial MTAN structures where the enzyme is in the closed form, the first turn of helix α1 does not possess a hydrogen bonded network consistent with an α-helix.16,21,22,26,34 It is known from the structures of both the open and closed forms of EcMTAN that upon substrate binding, the third and fourth backbone hydrogen bonds within helix α1 are broken.22,26 This produces a significant kink between the first and second turns of helix α1 that results in the positioning of E12 (E13 in HpMTAN) within the active site and forms a hydrogen bond between E12 and the nucleophilic water in the active site.
The conformational change that occurs in helix α1 of HpMTAN differs slightly in that the first turn of helix 1 transitions from a 310-helix to an α-helix upon substrate binding and closing of the active site. The most likely reason for this difference between HpMTAN and other MTANs appears to stem from structural restraints imposed by a proline residue at position 16 of HpMTAN. This proline is present in a very small subset of known MTAN protein sequences that, in addition to H. pylori, include MTANs encoded by other closely related Campylobacterales as well as Francisella tularensis. In all of the previous MTAN structures, this proline is either lacking or replaced by a leucine or isoleucine residue. As the proline is in the second of three turns in helix α1, the hydrogen bonding between the first and second turns is not typical. The backbone carbonyl of E12, which would typically be hydrogen bonded with the amide proton at position 16, does not have a hydrogen bonded partner, whereas the backbone carbonyls of R11 and E13 are hydrogen bonding with backbone amides of I14 and I17, respectively. Therefore, P16 is central to the transition from a 310 conformation in the first turn of the helix to the α helical conformation in the last two turns.
The other important conformational change during active site closing involves residues 198–208. These residues form the loop immediately preceding helix α6 and the first five residues of that helix. This conformational change fixes the 200–208 region of HpMTAN, which is disordered in the HpMTAN-HOH molecule, producing two essential structural effects that directly affect catalysis. First, the position of the general acid, D198, required to initiate the enzymatic reaction becomes fixed in position to form hydrogen bonding interactions with N7 and N6 of the substrate's adenine moiety and can now donate a proton to N7. Second, the N-terminus of helix α6 forms a cap over the substrate binding site that sequesters the active site from the environment. These structural details highlight the changes required for substrate binding and enzymatic activity that have been for the most part previously described.22,26 However, the interactions between MTAN and the substrates required to promote the conformational change have not.
Ligand requirements for promoting the MTAN conformational change
The HpMTAN-FMA crystal structure, in combination with the information from the HpMTAN structure, highlights interactions between residues of the active site and ligands that stabilize the closed form of the enzyme. To better understand those interactions, a careful analysis and comparison of the open and closed forms from all molecules of the HpMTAN and HpMTAN-FMA active sites was performed. It is apparent from this comparison that two different criteria must be met to promote the transition from the open to the closed form. First, the bound substrate must be capable of bridging the conserved E175 and R194 residues. This can be assumed to occur first, primarily because the position of E175 does not differ in any of the determined structures and only the terminal guanidinium group in the R194 side chain changes position upon substrate binding. This forms the hydrogen bonded network between these two residues and the bound ligand.
The second requirement for promoting and stabilizing the conformational change relates to the uncoiling of the 310-helix formed by residues 11–14 and the ordering of the residues 198–208 near the N-terminus of helix α6. These changes appear to result from repulsion between the bound substrate and the side chain of A9. Following substrate binding, the loop containing A9, located between strand β1 and helix α1, is pushed away from the active site and drags M10 away from helix α6 (Fig. 6). This repositioning of residue M10 relaxes the 310-helix at the beginning of helix 1 and forces E13 into the space vacated by the atoms forming the peptide bond between A9 and M10, resulting in the formation of two new hydrogen bonds with the side chain of E13. One of these is formed to the amide proton of V78, whereas the other is to the ligand bound within the active site. The movement of residues M10 and E13 also allows helix α6 to move closer to the active site, which leads to ordering of the 198–208 region of HpMTAN.
Figure 6.
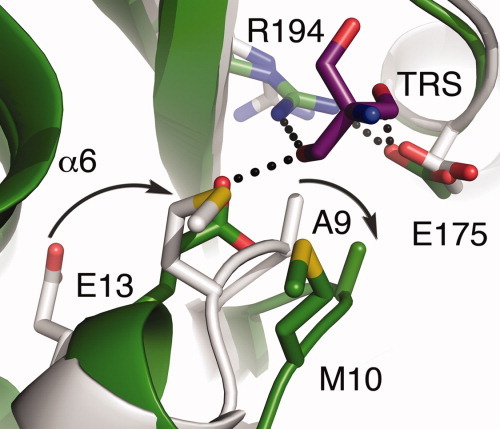
Intermolecular repulsive forces initiate the conformational change. The active sites of the HpMTAN-HOH and HpMTAN-TRS are shown in gray and green, respectively. Carbon atoms match the coloring of the respective backbone. The carbons of the tris molecule are purple. In all molecules, sulfur is yellow, nitrogen is blue, and oxygen is red. The curved arrows represent the relative motion of residues A9 and E13 as a consequence of tris binding. The black dots represent hydrogen bonds that help stabilize the closed form.
The nature of the bonding interaction between E13 and the bound ligand depends on the identity of the bound ligand within the enzyme active site. In the HpMTAN-FMA structure, E13 and R194 interact through the nucleophilic water, which is positioned and likely activated by the side chains of E13 and E175 as shown in Figure 7(A) and observed in previous structures containing FMA or other transition state analogs.16,21,22,26,34,36 In the HpMTAN-TRS form, however, the O3 atom of tris interacts directly with E13 and R194, resulting in the displacement of the catalytic water molecule. Indeed, the closest water molecule to O3 of the tris molecule is 5.9 Å away.
Figure 7.

The MTAN active site can accommodate a variety of hydrogen bonding patterns. Each of the three enzymes in the closed conformation clearly exhibit the orientation of the bound compounds in Fo–Fc omit maps contoured at 3σ. (A) Fo–Fc omit map of the FMA bound structure reflects characteristics seen in previous MTAN-FMA complexes. Both FMA and the nucleophilic water molecule (Nuc−) were omitted during map calculation. (B) The HpMTAN-TRS structure in the same orientation as (A) exhibits the Fo-Fc omit map where the tris molecule and four ordered water molecules within the adenine binding site were omitted from the map calculation. Note that the orientation of tris is such that the amine moiety is clearly pointed upward roughly along the Y-axis. (C) and (D) show the active site of the HpMTAN-ADE-TRS structure in two different orientations. Panel (D) is in the same orientation as (A) and (B) and shows the clear density for the three hydroxymethyl moieties. The orientation of tris in the active site is different from that observed in panel (B). Panel (C) is rotated roughly 90 degrees along the Y-axis to more clearly show the density for the amine moiety of tris. The Fo − Fc omit map was calculated while omitting both the tris and adenine molecules. (E) Stereo diagram of the HpMTAN-ADE-TRS active site showing the hydrogen bonding network with conserved residues of the active site. Dashed lines indicate the hydrogen bonds and distances between donors and acceptors are shown in angstroms.
In contrast to the ability of tris to promote closing of the enzyme active site, the previously determined EcMTAN crystal structure shows that glycerol, isopropanol, and 3,6,9,12,15,18,21,24-octaoxahexacosan-1-ol are all insufficient to promote this conformational change.21 In the EcMTAN structure, the glycerol molecule interacts with E174 in a manner analogous to how tris interacts with E175 in the HpMTAN structure. However, glycerol lacks the bulk of tris and cannot interact with E12, E174, and R193 of EcMTAN simultaneously and so cannot promote stable hydrogen bonding interactions between residues E174 and R193 of EcMTAN. In the HpMTAN-TRS structure, the tris molecule readily interacts with all three residues, forming the necessary hydrogen bonding interactions to stabilize the closed form of the enzyme. This suggests that any molecule that can bridge E13, E175, and R194 in HpMTAN, or the analogous residues of any MTAN, would also suffice to stabilize the closed form.
Of further interest is the orientation of the bound tris molecule. The density representing tris in the difference maps in both structures is of sufficiently high quality that it is possible to differentiate the three hydroxymethyl groups from the single amine moiety. Upon close inspection of the difference maps for these two structures, it is clear that tris binds differently [Fig. 7(B,D)]. Although it is not clear why tris binds in different orientations, the interactions promoting binding of tris in the HpMTAN-ADE-TRS structure are straightforward to rationalize. In this structure, steric hindrance is likely the predominant factor in defining tris orientation. The three comparatively bulky hydroxymethyl substituents point away from adenine, which directs the less bulky amine moiety toward adenine. Additionally, this amine also forms a hydrogen bond with the backbone carbonyl of V78 placing it approximately in the same position that would be occupied by the positively charged C1 atom of the oxocarbenium intermediate formed during the enzymatic reaction.
In contrast, the HpMTAN-TRS structure exhibits a tris molecule where the amine moiety and one of the hydroxymethyl groups swap positions [Fig. 7(B,D)]. The atoms, O1 and N of tris, do not appear to be forming hydrogen bonds with any components of the enzyme active site, but O1 does form a 3.0 Å hydrogen bond to an ordered water molecule in the adenine binding pocket. Although this is 0.2 Å shorter than the length of the hydrogen bond formed between O1 and the side chain of M10 in the HpMTAN-ADE-TRS structure, this seems unlikely to compensate for the loss of the short hydrogen bond between the amine of tris and the carbonyl of V78 in the HpMTAN-ADE-TRS structure. Although more studies would be required to understand the orientation preference of ligands within the HpMTAN active site, a present benefit of the variation in tris binding is that it offers additional information regarding the breadth of interaction space for the enzyme active site and various small molecule ligands. Indeed, as the tris molecule bound within HpMTAN-ADE-TRS active site forms six specific hydrogen bonding interactions within the active site versus the three hydrogen bonds formed to FMA, this information can be used to design scaffolds for building libraries of new classes of inhibitors that maximized the number of specific interactions between the inhibitor and the residues forming the ribose binding site [Fig. 7(E)]. Additionally, the tris scaffold could be amended to include chemical moieties to attempt to differentiate binding between bacterial MTANs and human MTAP.
The primary differences between the active sites of these two enzymes are found in the nucleophile binding site and the 5′-alkylthiol binding pocket.31 In bacterial MTANs, the 5′-alkylthiol binding pocket must be able to accommodate the homocysteine moiety of the AdoHcy. However, human MTAP uses only MTA as a substrate, so the 5′-alkylthiol binding pocket is shorter and has less volume when compared to that of the bacterial MTANs. Additionally, the MTAP nucleophile binding site must accommodate inorganic phosphate versus a water molecule for the MTANs. The MTAP site is formed by one arginine residue, one histidine residue, three threonine residues, and two backbone amides. Together, these form a pocket that has a slightly positive electrostatic potential. In contrast, the MTAN nucleophile binding pocket is formed by two glutamate residues, a backbone carbonyl, and an arginine residue, which together imparts a negative electrostatic potential. Although the affinity of tris alone has a Kd of ∼45 mM, it may be possible to assemble a tris-based inhibitor with a positively charged moiety and moderate bulk to interact strongly with the nucleophile binding site of the bacterial MTANs but promote charge repulsion with an empty MTAP nucleophile binding site. If the MTAP nucleophile binding site is filled with inorganic phosphate, the positively charged inhibitor could interact with the negatively charged phosphate, but any added bulk would likely hinder proper interactions between the inhibitor and regions of the protein forming the adenine and ribose binding sites and thereby prevent closure of the active site and weaken an inhibitor–MTAP complex. Addition of an adenine moiety or analog would further strengthen binding of such an inhibitor to MTAN. Recently, Edwards et al. have shown that human purine nucleoside phosphorylase, a homolog of both MTAN and MTAP, forms a complex with a tris-based Immucillin that has a Kd of 620 pM.38 Using this as a lead compound, structure-based design should lead to a chemical derivative that would preferentially bind bacterial MTANs over human MTAP and the human purine nucleoside phosphorylase.
For researchers attempting to develop therapies for bacterial infections, inhibiting quorum-sensing is an intriguing target that may limit the development of drug resistant bacterial strains, as the drugs would not kill the bacteria directly. Instead, by preventing the production of virulence factors, the host immune system may then be capable of clearing the infection rather than relying on the killing of the bacteria by the administered drug.15,16 The structural information presented here can be used in conjunction with grow/link and fragment-based design methodologies to create compounds that promote closing of the enzyme active site and stabilize this form to prevent enzyme turnover while also displacing the nucleophilic water molecule necessary for catalysis. The differences observed in the HpMTAN-ADE-TRS and HpMTAN structures are particularly important in this respect. Leveraging the information from the HpMTAN-ADE-TRS structure will allow researchers to shift from the use of adenosine-analogs as inhibitors toward compounds that will increase specificity toward MTAN by promoting differential binding between it and human MTAP. This bodes well for the development of potential treatments for H. pylori and other bacterial pathogens such as pathogenic E. coli, Neisseria meningitidis, Klebsiella pneumoniae, Salmonella typhimurium, S. aureus, Streptococcus pneumoniae, and V. cholerae, where MTAN plays an important role in bacterial metabolism and especially those where quorum-sensing plays an important role in pathogenesis.
Materials and Methods
Cloning of the pfs gene and expression of MTAN protein
The pfs gene encoding MTAN was PCR amplified from Helicobactor pylori strain J99 (ATCC) using primers 5′-CACCATGGGGCAAAAAATTGGCATTTT AGGGGC-3′ and 5′-CCGGATCCCTAAAGCTCATCC ACCATGCTTT-3′, digested with NcoI and BamHI (recognition sequences are underlined) and ligated into a derivative of pET32 (EMD Biosciences). The resulting plasmid was sequenced (U of Michigan Sequencing Core Facility) and then used to transform T7 Express competent cells (New England BioLabs) harboring the pRARE2 plasmid (EMD Biosciences). The transformants were cultured in LB broth containing 100 μg/mL ampicillin and 32 μg/mL chloramphenicol and incubated at 37°C until reaching an O.D.600 nm of 0.6–0.7. The incubator temperature was then decreased to 20°C, and MTAN expression was induced by adding isopropyl thiogalactoside to the culture at a final concentration of 1 mM. The induced culture was incubated for 16 h. Bacteria were harvested by centifugation and resuspended in metal chelation binding buffer (20 mM sodium phosphate pH 7.5, 0.5 M sodium chloride, 25 mM imidazole, 5 mM β-mercaptoethanol) and stored at −70°C until needed for protein purification.
MTAN purification
All subsequent purification steps were performed at 4°C. Thawed cells were lysed using lysozyme and sonication followed by treatment with DNase I. The lysate was clarified using centrifugation at 11,000 g and 4°C for 20 min. The supernatant was filtered using a 0.2 μM syringe filter and loaded onto a 5 mL HisTrap™ FF column (GE Healthcare) pre-equilibrated with metal chelation binding buffer. Following protein loading, the column was washed with 10 column volumes of metal chelation binding buffer and the remaining bound protein was eluted with a linear imidazole concentration gradient from 25 to 250 mM over 20 column volumes. Measuring absorbance at 280 nm allowed monitoring of protein elution. The eluted MTAN protein was pooled and recombinant rhinovirus 3C protease was added to cleave the affinity tag as the sample was dialyzed for ∼16 h against metal chelation binding buffer. The cleaved protein was again loaded onto a 5 mL HisTrap™ FF column to remove the affinity tag and the recombinant protease. The flow-through fractions containing MTAN were pooled and precipitated by adding solid ammonium sulfate to a final concentration of 2.4 M. The sample was centrifuged at 11,000 g and 4°C for 20 min. The resulting pellet was dissolved with size exclusion buffer that contained 20 mM sodium phosphate pH 7.5, 0.3 M sodium chloride, and 5 mM β-mercaptoethanol. MTAN was then separated from any remaining impurities using a HiLoad™ Superdex 200 size exclusion column that had been equilibrated with the size exclusion buffer. Fractions containing purified MTAN were pooled and dialyzed against crystallization buffer containing 20 mM Tris pH 8.5, 0.2 mM TCEP, and 1 mM EDTA. The MTAN concentration was determined by measuring the absorbance at 280 nm and using 3105 cm−1 M−1 as the extinction coefficient. MTAN was then concentrated using ultrafiltration to a final concentration of 16 mg/mL.
Enzyme kinetic experiments
The enzymatic assays to measure the HpMTAN kinetic parameters were performed at 37°C in a 100 mM HEPES and 50 mM KCl solution (pH 7.5). All reactions monitored the reduction of MTA concentration by measuring absorbance at 274 nm (Δɛ = 1.6 mM−1 cm−1) using a Biotek Synergy H4 plate reader (Winooski, VT, USA). Concentrations of MTA were varied from 10 to 125 μM. The inhibition assays were performed under the same environmental conditions using 25 μM MTA as substrate and varying tris concentrations between 0 and 250 mM. All reactions used an HpMTAN concentration of 25 nM.
Crystallization, structure determination, and refinement
Crystals of MTAN were produced using the hanging drop vapor diffusion method. Mixing 2 μL of MTAN protein solution with 2 μL of a well solution containing 0.1 M Tris pH 8.5 and 16% w/v polyethyleneglycol 8000 produced rod shaped crystals after 1 day of incubation at 18°C. For the cocrystal structures, crystallization experiments were the same as for the MTAN form with the exception of adding either FMA (Berry and Associates, Dexter, MI) or AdoHcy to a final concentration of 5 mM. Crystals were cryoprotected by adding ethylene glycol to the crystallization drop to a final concentration of 20% v/v and flash cooled to 95 K in a liquid nitrogen stream. X-ray diffraction data were collected at the LS-CAT ID-D beamline at the Advanced Photon Source (Argonne National Labs, Argonne, IL), using X-rays at 11.5 keV. Data were integrated and scaled using HKL2000. Diffraction data are shown in Table I.
The HpMTAN and FMA-bound crystals grew in the P3121 space group with unit cell parameters of a = b = 81.5 Å ± 0.2, c = 135.5 Å ± 0.5, α = β = 90°, γ = 120° with two molecules of MTAN in the asymmetric unit. Molecular replacement used the HpMTAN-FMA complex diffraction data and the SaMTAN-FMA crystal structure (accession 3BL6) as the search model in Phaser.39 As the HpMTAN and HpMTAN-FMA complex structures are isomorphous, the partially refined MTAN-FMA model was used as the basis to begin refinement of the HpMTAN structure.
Although they appeared identical to the HpMTAN and FMA cocrystals, the HpMTAN-ADE-TRS crystals grew in a P3221 space group with unit cell parameters a = b = 81.4 Å, c = 67.6 Å, α = β = 90°, γ = 120° and one molecule per asymmetric unit. A monomer from the HpMTAN-FMA structure was used as the molecular replacement search model for solving the HpMTAN-ADE-TRS complex. All three HpMTAN structures were initially refined using CNS 1.240 and manually refined in Coot.41 Five percent of each data set was set aside for cross-validation. To remove model bias, the initial molecular dynamics step included simulated annealing with a starting temperature of 5000 K for all three of the HpMTAN structures. Waters were added using CNS 1.2. The final refinement steps were performed using PHENIX 1.6.42
Accession numbers
The coordinates and structure factors for the HpMTAN, HpMTAN-FMA, and HpMTAN-ADE-TRS structures have been deposited in the Protein Data Bank with accession numbers 3NM4, 3NM5, and 3NM6, respectively. Coordinates used for molecular replacement were from PDB accession 3BL6.
Acknowledgments
The authors thank Daniel H. Lajiness for assistance with the manuscript.
Glossary
Abbreviations:
- AdoHcy
S-adenosylhomocysteine
- AdoMet
S-adenosylmethionine
- AI-2
autoinducer 2
- EcMTAN
E. coli MTAN
- FMA
formycin A
- HpMTAN
H. pylori MTAN
- LuxS
S-ribosylhomocysteine Lyase
- MTA
5′-methylthioadenosine
- MTAN
5′-methylthioadenosine/S-adenosylhomocysteine nucleosidase
- MTAP
5′-methylthioadenosine phosphorylase
- SaMTAN
S. aureus MTAN
- R-THMF
(2R,4S)-2-methyl-2,3,3,4-tetrahydroxytetrahydrofuran
- S-THMF
(2S,4S)-2-methyl-2,3,3,4-tetrahydroxytetrahydrofuran.
References
- 1.Duerre JA. A hydrolytic nucleosidase acting on S-adenosylhomocysteine and on S-methylthioadenosine. J Biol Chem. 1962;237:3737–3741. [Google Scholar]
- 2.Choi-Rhee E, Cronan JE. A nucleosidase required for in vivo function of the S-adenosyl-L-methionine radical enzyme, biotin synthase. Chem Biol. 2005;12:589–593. doi: 10.1016/j.chembiol.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 3.Borchardt RT, Creveling CR, Ueland PM. Biological methylation and drug design: experimental and clinical role of S-adenosylmethionine. Experimental biology and medicine. Clifton, N.J.: Humana Press; 1986. [Google Scholar]
- 4.Pajula RL, Raina A. Methylthioadenosine, a potent inhibitor of spermine synthase from bovine brain. FEBS Lett. 1979;99:343–345. doi: 10.1016/0014-5793(79)80988-6. [DOI] [PubMed] [Google Scholar]
- 5.Raina A, Tuomi K, Pajula RL. Inhibition of the synthesis of polyamines and macromolecules by 5′-methylthioadenosine and 5′-alkylthiotubercidins in BHK21 cells. Biochem J. 1982;204:697–703. doi: 10.1042/bj2040697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Riscoe MK, Ferro AJ, Fitchen JH. Methionine recycling as a target for antiprotozoal drug development. Parasitol Today. 1989;5:330–333. doi: 10.1016/0169-4758(89)90128-2. [DOI] [PubMed] [Google Scholar]
- 7.Sufrin JR, Meshnick SR, Spiess AJ, Garofalo-Hannan J, Pan XQ, Bacchi CJ. Methionine recycling pathways and antimalarial drug design. Antimicrob Agents Chemother. 1995;39:2511–2515. doi: 10.1128/aac.39.11.2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walsh CT. Posttranslational modifications of proteins: expanding nature's inventory. Greenwood Village, CO: Roberts and Co.; 2006. [Google Scholar]
- 9.Waters CM, Bassler BL. Quorum sensing: cell-to-cell communication in bacteria. Annu Rev Cell Dev Biol. 2005;21:319–346. doi: 10.1146/annurev.cellbio.21.012704.131001. [DOI] [PubMed] [Google Scholar]
- 10.Zhao G, Wan W, Mansouri S, Alfaro JF, Bassler BL, Cornell KA, Zhou ZS. Chemical synthesis of S-ribosyl-L-homocysteine and activity assay as a LuxS substrate. Bioorg Med Chem Lett. 2003;13:3897–3900. doi: 10.1016/j.bmcl.2003.09.015. [DOI] [PubMed] [Google Scholar]
- 11.Chen X, Schauder S, Potier N, Van Dorsselaer A, Pelczer I, Bassler BL, Hughson FM. Structural identification of a bacterial quorum-sensing signal containing boron. Nature. 2002;415:545–549. doi: 10.1038/415545a. [DOI] [PubMed] [Google Scholar]
- 12.Miller ST, Xavier KB, Campagna SR, Taga ME, Semmelhack MF, Bassler BL, Hughson FM. Salmonella typhimurium recognizes a chemically distinct form of the bacterial quorum-sensing signal AI-2. Mol Cell. 2004;15:677–687. doi: 10.1016/j.molcel.2004.07.020. [DOI] [PubMed] [Google Scholar]
- 13.Sztajer H, Lemme A, Vilchez R, Schulz S, Geffers R, Yip CY, Levesque CM, Cvitkovitch DG, Wagner-Dobler I. Autoinducer-2-regulated genes in Streptococcus mutans UA159 and global metabolic effect of the luxS mutation. J Bacteriol. 2008;190:401–415. doi: 10.1128/JB.01086-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rickard AH, Palmer RJ, Jr, Blehert DS, Campagna SR, Semmelhack MF, Egland PG, Bassler BL, Kolenbrander PE. Autoinducer 2: a concentration-dependent signal for mutualistic bacterial biofilm growth. Mol Microbiol. 2006;60:1446–1456. doi: 10.1111/j.1365-2958.2006.05202.x. [DOI] [PubMed] [Google Scholar]
- 15.Rader BA, Campagna SR, Semmelhack MF, Bassler BL, Guillemin K. The quorum-sensing molecule autoinducer 2 regulates motility and flagellar morphogenesis in Helicobacter pylori. J Bacteriol. 2007;189:6109–6117. doi: 10.1128/JB.00246-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gutierrez JA, Crowder T, Rinaldo-Matthis A, Ho MC, Almo SC, Schramm VL. Transition state analogs of 5′-methylthioadenosine nucleosidase disrupt quorum sensing. Nat Chem Biol. 2009;5:251–257. doi: 10.1038/nchembio.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sperandio V, Mellies JL, Nguyen W, Shin S, Kaper JB. Quorum sensing controls expression of the type III secretion gene transcription and protein secretion in enterohemorrhagic and enteropathogenic Escherichia coli. Proc Natl Acad Sci USA. 1999;96:15196–15201. doi: 10.1073/pnas.96.26.15196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sperandio V, Torres AG, Giron JA, Kaper JB. Quorum sensing is a global regulatory mechanism in enterohemorrhagic Escherichia coli O157:H7. J Bacteriol. 2001;183:5187–5197. doi: 10.1128/JB.183.17.5187-5197.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li M, Villaruz AE, Vadyvaloo V, Sturdevant DE, Otto M. AI-2-dependent gene regulation in Staphylococcus epidermidis. BMC Microbiol. 2008;8:4. doi: 10.1186/1471-2180-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee WK, Ogura K, Loh JT, Cover TL, Berg DE. Quantitative effect of luxS gene inactivation on the fitness of Helicobacter pylori. Appl Environ Microbiol. 2006;72:6615–6622. doi: 10.1128/AEM.01291-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee JE, Smith GD, Horvatin C, Huang DJ, Cornell KA, Riscoe MK, Howell PL. Structural snapshots of MTA/AdoHcy nucleosidase along the reaction coordinate provide insights into enzyme and nucleoside flexibility during catalysis. J Mol Biol. 2005;352:559–574. doi: 10.1016/j.jmb.2005.07.027. [DOI] [PubMed] [Google Scholar]
- 22.Lee JE, Cornell KA, Riscoe MK, Howell PL. Structure of Escherichia coli 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidase inhibitor complexes provide insight into the conformational changes required for substrate binding and catalysis. J Biol Chem. 2003;278:8761–8770. doi: 10.1074/jbc.M210836200. [DOI] [PubMed] [Google Scholar]
- 23.Lee JE, Cornell KA, Riscoe MK, Howell PL. Structure of E. coli 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidase reveals similarity to the purine nucleoside phosphorylases. Structure. 2001;9:941–953. doi: 10.1016/s0969-2126(01)00656-6. [DOI] [PubMed] [Google Scholar]
- 24.Allart B, Gatel M, Guillerm D, Guillerm G. The catalytic mechanism of adenosylhomocysteine/methylthioadenosine nucleosidase from Escherichia coli—chemical evidence for a transition state with a substantial oxocarbenium character. Eur J Biochem. 1998;256:155–162. doi: 10.1046/j.1432-1327.1998.2560155.x. [DOI] [PubMed] [Google Scholar]
- 25.Cornell KA, Swarts WE, Barry RD, Riscoe MK. Characterization of recombinant Eschericha coli 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidase: analysis of enzymatic activity and substrate specificity. Biochem Biophys Res Commun. 1996;228:724–732. doi: 10.1006/bbrc.1996.1723. [DOI] [PubMed] [Google Scholar]
- 26.Lee JE, Singh V, Evans GB, Tyler PC, Furneaux RH, Cornell KA, Riscoe MK, Schramm VL, Howell PL. Structural rationale for the affinity of pico- and femtomolar transition state analogues of Escherichia coli 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidase. J Biol Chem. 2005;280:18274–18282. doi: 10.1074/jbc.M414471200. [DOI] [PubMed] [Google Scholar]
- 27.Gutierrez JA, Luo M, Singh V, Li L, Brown RL, Norris GE, Evans GB, Furneaux RH, Tyler PC, Painter GF, Lenz DH, Schramm VL. Picomolar inhibitors as transition-state probes of 5′-methylthioadenosine nucleosidases. ACS Chem Biol. 2007;2:725–734. doi: 10.1021/cb700166z. [DOI] [PubMed] [Google Scholar]
- 28.Evans GB, Furneaux RH, Greatrex B, Murkin AS, Schramm VL, Tyler PC. Azetidine based transition state analogue inhibitors of N-ribosyl hydrolases and phosphorylases. J Med Chem. 2008;51:948–956. doi: 10.1021/jm701265n. [DOI] [PubMed] [Google Scholar]
- 29.Singh V, Schramm VL. Transition-state analysis of S. pneumoniae 5′-methylthioadenosine nucleosidase. J Am Chem Soc. 2007;129:2783–2795. doi: 10.1021/ja065082r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heurlier K, Vendeville A, Halliday N, Green A, Winzer K, Tang CM, Hardie KR. Growth deficiencies of Neisseria meningitidis pfs and luxS mutants are not due to inactivation of quorum sensing. J Bacteriol. 2009;191:1293–1302. doi: 10.1128/JB.01170-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee JE, Settembre EC, Cornell KA, Riscoe MK, Sufrin JR, Ealick SE, Howell PL. Structural comparison of MTA phosphorylase and MTA/AdoHcy nucleosidase explains substrate preferences and identifies regions exploitable for inhibitor design. Biochemistry. 2004;43:5159–5169. doi: 10.1021/bi035492h. [DOI] [PubMed] [Google Scholar]
- 32.Appleby TC, Erion MD, Ealick SE. The structure of human 5′-deoxy-5′-methylthioadenosine phosphorylase at 1.7 A resolution provides insights into substrate binding and catalysis. Structure. 1999;7:629–641. doi: 10.1016/s0969-2126(99)80084-7. [DOI] [PubMed] [Google Scholar]
- 33.Singh V, Shi W, Almo SC, Evans GB, Furneaux RH, Tyler PC, Painter GF, Lenz DH, Mee S, Zheng R, Schramm VL. Structure and inhibition of a quorum sensing target from Streptococcus pneumoniae. Biochemistry. 2006;45:12929–12941. doi: 10.1021/bi061184i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Siu KK, Lee JE, Smith GD, Horvatin-Mrakovcic C, Howell PL. Structure of Staphylococcus aureus 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidase. Acta Cryst. 2008;F64:343–350. doi: 10.1107/S1744309108009275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Edwards AA, Tipton JD, Brenowitz MD, Emmett MR, Marshall AG, Evans GB, Tyler PC, Schramm VL. Conformational states of human purine nucleoside phosphorylase at rest, at work, and with transition state analogues. Biochemistry. 2010;49:2058–2067. doi: 10.1021/bi902041j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Siu KK, Lee JE, Sufrin JR, Moffatt BA, McMillan M, Cornell KA, Isom C, Howell PL. Molecular determinants of substrate specificity in plant 5′-methylthioadenosine nucleosidases. J Mol Biol. 2008;378:112–128. doi: 10.1016/j.jmb.2008.01.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.DeLano WL. The PyMOL molecular graphics system. Palo Alto, CA: DeLano Scientific LLC; 2002. [Google Scholar]
- 38.Edwards AA, Mason JM, Clinch K, Tyler PC, Evans GB, Schramm VL. Altered enthalpy-entropy compensation in picomolar transition state analogues of human purine nucleoside phosphorylase. Biochemistry. 2009;48:5226–5238. doi: 10.1021/bi9005896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brunger AT. Version 1.2 of the Crystallography and NMR system. Nat Protoc. 2007;2:2728–2733. doi: 10.1038/nprot.2007.406. [DOI] [PubMed] [Google Scholar]
- 41.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Cryst. 2010;D66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Adams PD, Gopal K, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Pai RK, Read RJ, Romo TD, Sacchettini JC, Sauter NK, Storoni LC, Terwilliger TC. Recent developments in the PHENIX software for automated crystallographic structure determination. J Synchrotron Radiat. 2004;11:53–55. doi: 10.1107/s0909049503024130. [DOI] [PubMed] [Google Scholar]