

Abstract
Background
The rapidly increasing availability of whole-genome sequences has enabled the study of whole-genome evolution. Evolutionary mechanisms based on genome rearrangements have attracted much attention and given rise to many models; somewhat independently, the mechanisms of gene duplication and loss have seen much work. However, the two are not independent and thus require a unified treatment, which remains missing to date. Moreover, existing rearrangement models do not fit the dichotomy between most prokaryotic genomes (one circular chromosome) and most eukaryotic genomes (multiple linear chromosomes).
Results
To handle rearrangements, gene duplications and losses, we propose a new evolutionary model and the corresponding method for estimating true evolutionary distance. Our model, inspired from the DCJ model, is simple and the first to respect the prokaryotic/eukaryotic structural dichotomy. Experimental results on a wide variety of genome structures demonstrate the very high accuracy and robustness of our distance estimator.
Conclusion
We give the first robust, statistically based, estimate of genomic pairwise distances based on rearrangements, duplications and losses, under a model that respects the structural dichotomy between prokaryotic and eukaryotic genomes. Accurate and robust estimates in true evolutionary distances should translate into much better phylogenetic reconstructions as well as more accurate genomic alignments, while our new model of genome rearrangements provides another refinement in simplicity and verisimilitude.
Background
Introduction
Interest in the evolution of genome structure has been growing steadily in the last 10 years, sustained in part by the ever increasing number of sequenced genomes. In particular much work has been done on rearrangements (see, e.g., [1]), using the convention that each chromosome of the genome is represented by an ordered list of identifiers, each identifier referring to a syntenic block or, more commonly, to a member of a gene family. (In the following, we shall use the word "gene" in a broad sense to denote elements of such orderings and refer to such orderings as "gene orders".) Variations in the placement of homologous genes, as well as variations in gene content and multiplicity, among organisms can then be analyzed. Such data is of great interest to evolutionary biologists, but also to comparative genomicists and to any researcher interested in understanding evolutionary changes in pathogens, crop plants, and, more generally, the biome.
The most fundamental task in the analysis of such data is to estimate the amount of evolutionary change between two genomes--that is, to compute a pairwise evolutionary distance. The true distance, that is, the number of actual evolutionary events (rearrangements, duplications, and losses) that took place during the course of evolution, is what we want to obtain, but is not, of course, something that we can compute. Researchers have thus used a two-stage process, in which a well defined measure is first computed (such as an edit distance, that is, the smallest number of evolutionary events needed to transform one genome into the other), then a statistical model of evolution is used to infer an estimate of the true distance by deriving the effect of a given number of changes in the model on the computed measure and (algebraically or numerically) inverting the derivation to produce a maximum-likelihood estimate of the true distance under the model. This second step is usually called a distance correction and has long been used for sequence (DNA) data (see, e.g., [2]) as well as, more recently, for gene-order data, for which see [3-7].
Evolutionary events that affect the gene order of genomes include various rearrangements, which affect only the order, and gene duplications and losses, which affect both the gene content and, indirectly, the order. (Gene insertion, corresponding to lateral gene transfer or neofunctionalization, can be viewed as a special case of duplication.) Rearrangements themselves include inversion, transposition, block exchange, circularization and linearization, all of which act on a single chromosome, and translocation, fusion, and fission, which act on two chromosomes. All of these operations are subsumed in the double-cut-and-join (DCJ) [8,9], which has formed the basis for much of the algorithmic research on rearrangements over the last few years, including a statistically based method to estimate the true evolutionary distance between two genomes [7]. DCJ makes two cuts, which can be in the same chromosome or in two different chromosomes, producing four cut ends, then rejoins the four cut ends in any of the three possible ways. The DCJ model, however, is unrealistic in two major respects. First, if the two cuts are in the same chromosome, one of the two nontrivial rejoinings causes a fission, creating a new circular chromosome. However, circular chromosomes do not normally arise in organisms with linear chromosomes, and prokaryotic genomes normally consist of a single circular chromosome. Nor can this form of rejoining be forbidden as, without it, DCJ simply reduces to inversion. Secondly, DCJ is a model of rearrangements: it does not take into account evolutionary events that alter the gene content, such as duplications and losses.
Of these two problems, the first has not been seriously addressed: the model we present here is, to the best of our knowledge, the first model that naturally preserves the dichotomy between prokaryotic and eukaryotic genomes. While gene (or segment) duplications and losses have long been studied by geneticists and molecular biologist, their integration with rearrangements in a unified model has seen relatively little work to date. El-Mabrouk [10] gave an exact algorithm to compute edit distances for inversions and losses and also a heuristic to approximate edit distances for inversions, losses, and nonduplicating insertions (all of her results assume that genes cannot be duplicated). More recently, Yancopoulos and Friedberg [11] gave an algorithm to compute edit distances under deletions, insertions, duplications, and DCJ operations, under the constraint that each deletion can only remove a single gene. These and other approaches targeted the edit distance, not the true evolutionary distance. Swenson et al. [12] gave an algorithm to approximate the true evolutionary distance under deletions, insertions, duplications, and inversions for unichromosomal genomes and showed good results under simulations and for small-scale phylogenetic reconstruction. Rearrangements, duplications and losses have also been addressed in the framework of ancestral reconstruction (see, e.g., [13]). All of these approaches have focused on parsimony criteria and have used pre-assigned weights for the various operations.
In this paper, we propose a new evolutionary model which respects the dichotomy between prokaryotic and eukaryotic genomes and which takes gene duplications and losses into account. Using this new evolutionary model, we develop a statistically based method to estimate the true evolutionary distance in terms of the actual number of rearrangements, gene duplications, and gene losses. Finally, we provide extensive experimental results on a wide variety of genome structures to illustrate the robustness and high accuracy of our estimator.
Genomes as gene-order data
We denote the tail of a gene g by gt and its head by gh. We write +g to indicate an orientation from tail to head (gt → gh), -g otherwise (gh → gt). Two consecutive genes a and b can be connected by one adjacency of one of the following four types: {at, bt}, {ah, bt}, {at, bh}, and {ah, bh}. If gene c lies at one end of a linear chromosome, then we also have a singleton set, {ct} or {ch}, called a telomere. A genome can then be represented as a multiset of genes together with a multiset of adjacencies and telomeres. For example, the toy genome in Figure 1, composed of one linear chromosome, (+a, +b, -c, +a, +b, -d, +a), and one circular one, (+e, -f), can be represented by the multiset of genes {a, a, a, b, b, c, d, e, f} and the multiset of adjacencies and telomeres {{at}, {ah, bt}, {bh, ch}, {ct, at}, {ah, bt}, {bh, dh}, {dt, at}, {ah}, {eh, fh}, {et, ft}}. Because of the duplicated genes, there is no one-to-one correspondence between genomes and multisets of genes, adjacencies, and telomeres. For example, the genome composed of one linear chromosome, (+a, +b, -d, +a, +b, -c, +a) and one circular one (+e, -f) would have the same multisets of genes, adjacencies and telomeres as that in Figure 1.
Figure 1.

A very small genome G.
Preliminaries on the evolutionary model
We use two parameters: the probability of occurrence of a gene duplication, pd, and the probability of occurrence of a gene loss, pl; the probability of occurrence of a rearrangement is then just pr = 1 - pd - pl. The next event is chosen from the three categories according to these parameters.
For rearrangements, we will select two adjacencies or telomeres with replacement uniformly from the multiset of all adjacencies and telomeres and then decide which rearrangement event we apply. The four cases are as follows.
Select two different adjacencies, or one adjacency and one telomere, in the same chromosome
For example, select two different adjacencies 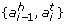 and
and 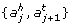 on one linear chromosome C = (a1 ... ai-1ai ... ajaj+1 ... an). Reversing all genes between ai and aj yields (a1 ... ai-1 - aj ... -aiaj+1 ... an). Two adjacencies, and , are replaced by two others,
on one linear chromosome C = (a1 ... ai-1ai ... ajaj+1 ... an). Reversing all genes between ai and aj yields (a1 ... ai-1 - aj ... -aiaj+1 ... an). Two adjacencies, and , are replaced by two others, 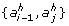 and
and 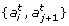 . This operation causes an inversion.
. This operation causes an inversion.
Select two adjacencies or one adjacency and one telomere in two different chromosomes
For example, select two adjacencies, 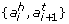 from one linear chromosome C = (a1 ... aiai+1 ... an) and
from one linear chromosome C = (a1 ... aiai+1 ... an) and 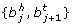 from another linear chromosome D = (b1 ... bjbj+1 ... bn). Now exchange the two segments between these two chromosomes C and D. There are two possible outcomes, (a1 ... aibj+1 ... bn) and (b1 ... bjai+1 ... an) or (a1 ... ai - bj ... -b1) and (-bn ... -bj+1ai+1 ... an). Two adjacencies, and , are replaced by
from another linear chromosome D = (b1 ... bjbj+1 ... bn). Now exchange the two segments between these two chromosomes C and D. There are two possible outcomes, (a1 ... aibj+1 ... bn) and (b1 ... bjai+1 ... an) or (a1 ... ai - bj ... -b1) and (-bn ... -bj+1ai+1 ... an). Two adjacencies, and , are replaced by 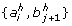 and
and 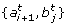 or
or 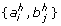 and
and 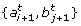 . This operation causes a translocation (or, if at least one chromosome is circular, a fusion).
. This operation causes a translocation (or, if at least one chromosome is circular, a fusion).
Select the same adjacency twice
For example, select the adjacency twice from linear chromosome C = (a1 ... aiai+1 ... an). Then split C into two new linear chromosomes, (a1 ... ai) and (ai+1 ... an). The adjacency is replaced by two telomeres 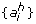 and
and 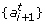 . This operation causes a fission for a linear chromosome, a linearization for a circular one.
. This operation causes a fission for a linear chromosome, a linearization for a circular one.
Select two telomeres
(Selecting one telomere twice is assimilated to selecting both telomeres of the linear chromosome.) For example, select telomeres and 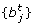 from two different linear chromosomes. Then concatenate these two linear chromosomes into a single new chromosome. Two telomeres, and , are replaced by two other telomeres, and . This operation causes a fusion on two linear chromosomes or a circularization on one linear chromosome.
from two different linear chromosomes. Then concatenate these two linear chromosomes into a single new chromosome. Two telomeres, and , are replaced by two other telomeres, and . This operation causes a fusion on two linear chromosomes or a circularization on one linear chromosome.
For gene duplication, we uniformly select a position to start duplicating a short segment of chromosomal material and place the new copy to a new position within the genome. We set Lmax as the maximum number of genes in the duplicated segment and assume that the number of genes in that segment is a uniform random number between 1 and Lmax. For example, select one segment ai+1 ... ai+L to duplicate and insert the copy between one adjacency . Such an operation duplicates L genes and L - 1 adjacencies, removes one adjacency, and adds two new adjacencies; thus genes ai+1,..., ai+L-1 and ai+L are added to the multiset of genes, the adjacency is removed, and L + 1 new adjacencies, 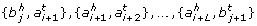 , are added.
, are added.
For gene loss, we restrict deletions to genes with at least two copies in the genome and we delete one gene at a time. We uniformly select one gene from the set of all candidate genes and delete it. For example, if we delete gene ai in the chromosome (... ai-1aiai+1 ...), one copy of ai is removed from the multiset of genes, while two adjacencies, and , are replaced by one adjacency, 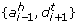 .
.
Methods
An overview of our technique for estimating the true evolutionary distance
The problem of estimating the true evolutionary distance is defined as follows:
Input: The original genome G and the final genome F.
Output: An estimate of the actual number of evolutionary events that took place in the evolutionary history to transform G into F.
Based on the multisets of genes and of adjacencies and telomeres of G, for any genome G* of N* genes and l* linear chromosomes, we can build the vector 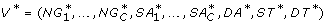 , where C is the upper bound for the number of copies of one gene,
, where C is the upper bound for the number of copies of one gene, 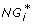 (i = 1,..., C) is the number of genes with exactly i copies in the genome G*,
(i = 1,..., C) is the number of genes with exactly i copies in the genome G*, 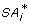 (i = 1,..., C) is the number of adjacencies with exactly i copies in G* that also appear in G, DA* is the number of adjacencies in G* that do not appear in G, ST * is the number of telomeres in G* that also appear in G, and DT* is the number of telomeres in G* that do not appear in G. We can write
(i = 1,..., C) is the number of adjacencies with exactly i copies in G* that also appear in G, DA* is the number of adjacencies in G* that do not appear in G, ST * is the number of telomeres in G* that also appear in G, and DT* is the number of telomeres in G* that do not appear in G. We can write
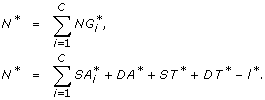 |
Let Gk be the genome obtained from G = G0 by applying k randomly selected evolutionary operations--under our model, the (i + 1)st evolutionary operation is selected from all possible rearrangements, gene duplications, and gene losses on genome Gi according to the parameters pd and pl. We can compute the vector 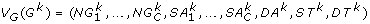 to represent the genome Gk with respect to G.
to represent the genome Gk with respect to G.
In the section, we show that, given G, we can also produce the estimate 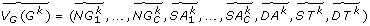 for the expected vector E(VG(Gk)), for any integer k > 0. Our approach for estimating the true evolutionary distance is then to return the integer k that minimizes the 1-norm distance between
for the expected vector E(VG(Gk)), for any integer k > 0. Our approach for estimating the true evolutionary distance is then to return the integer k that minimizes the 1-norm distance between 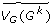 and VG(F).
and VG(F).
Estimation of the expected vector after some number of random evolutionary events
Given the original genome G, the complete vector for genome Gk is defined as 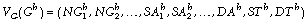 , where
, where 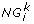 is the number of genes with exactly i copies in the genome Gk,
is the number of genes with exactly i copies in the genome Gk, 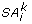 (shared adjacencies) is the number of adjacencies with exactly i copies in Gk that also appear in G, DAk (distinct adjacencies) is the number of adjacencies in Gk that do not appear in G, ST k (shared telomeres) is the number of telomeres in Gk that also appear in G, and DT k (distinct telomeres) is the number of telomeres in Gkthat do not appear in G.
(shared adjacencies) is the number of adjacencies with exactly i copies in Gk that also appear in G, DAk (distinct adjacencies) is the number of adjacencies in Gk that do not appear in G, ST k (shared telomeres) is the number of telomeres in Gk that also appear in G, and DT k (distinct telomeres) is the number of telomeres in Gkthat do not appear in G.
Assume the original genome G has N genes, where each gene has at most C = O(1) copies, and l linear chromosomes, with l = O(1). We thus ignore items and for (i >C). The initial vector VG(G0) is then 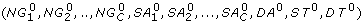 , where
, where 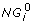 is the number of genes with exactly i copies,
is the number of genes with exactly i copies, 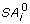 is the number of adjacencies with exactly i copies, DA0 = 0, ST 0 = 2l, and DT0 = 0. We now show how to update this vector under rearrangements, gene duplications and gene losses, respectively.
is the number of adjacencies with exactly i copies, DA0 = 0, ST 0 = 2l, and DT0 = 0. We now show how to update this vector under rearrangements, gene duplications and gene losses, respectively.
Rearrangements
We select two adjacencies or telomeres uniformly with replacement, from the multiset of all adjacencies or telomeres.
Theorem 1 Assume all genomes have O(1) linear chromosomes, each gene has at most C = O(1) copies, and represents the current genome Gk based on the original genome G. For conciseness, write 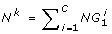 (the total number of genes) and lk = (STk +DTk)/2 (the number of linear chromosomes). Then we can write the expected vector for Gk+1 after one rearrangement operation:
(the total number of genes) and lk = (STk +DTk)/2 (the number of linear chromosomes). Then we can write the expected vector for Gk+1 after one rearrangement operation: 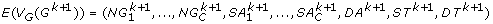 where we have
where we have
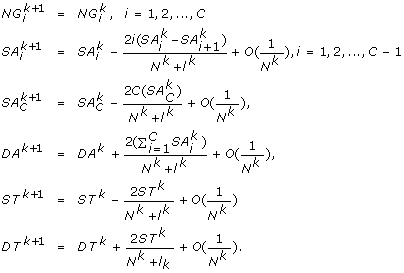 |
Proof In our evolutionary model, each rearrangement operation replaces old adjacencies or telomeres with new ones. Obviously, any rearrangement operation will not change the gene content, so 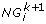 (i = 1,2,..., C) will be the same.
(i = 1,2,..., C) will be the same.
We first ignore the adjacencies or telomeres in the original genome G created after a rearrangement event. Remember two adjacencies or telomeres are selected with replacement uniformly from the multiset of all adjacencies and telomeres, and the number of all adjacencies and telomeres for genome Gk is (Nk +lk). Consider the multi-set Ai of adjacencies with exactly i copies in Gk that also appear in G. The probability that exactly one of the two selected adjacencies is in Ai is 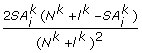 , the probability that two different adjacencies from Ai are selected is
, the probability that two different adjacencies from Ai are selected is 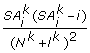 , the probability that equivalent adjacencies from Ai at different sites are selected is
, the probability that equivalent adjacencies from Ai at different sites are selected is 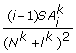 , and the probability that some adjacency from Ai is selected twice is
, and the probability that some adjacency from Ai is selected twice is 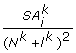 . For the time-being we ignore adjacencies and telomeres in G that may be created incidental. With probability
. For the time-being we ignore adjacencies and telomeres in G that may be created incidental. With probability 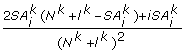 the number of adjacencies with exactly i copies decreases by i, and with probability the number of adjacencies with exactly i copies decreases by 2i. With probability
the number of adjacencies with exactly i copies decreases by i, and with probability the number of adjacencies with exactly i copies decreases by 2i. With probability 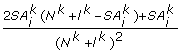 the number of adjacencies with exactly (i -1) copies increases by (i -1), with probability the number of adjacencies with exactly (i - 1) copies increases by 2(i - 1), and with probability the number of adjacencies with exactly (i -2) copies increases by (i -2). Considering i = 1,2,..., C and C = O(1), we have
the number of adjacencies with exactly (i -1) copies increases by (i -1), with probability the number of adjacencies with exactly (i - 1) copies increases by 2(i - 1), and with probability the number of adjacencies with exactly (i -2) copies increases by (i -2). Considering i = 1,2,..., C and C = O(1), we have
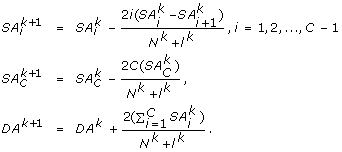 |
Now, we show that the correction for ignoring incidental creation of adjacencies or telomeres in G after a rearrangement event is 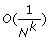 for each item. Consider any adjacency (a, b) in G: we might recover it only if we select two adjacencies or telomeres containing two genes a and b. Since each gene has at most C copies in the genome, there are at most C2 pairs of adjacencies or telomeres that may lead to recovery of the adjacency (a, b). So, with probability at most
for each item. Consider any adjacency (a, b) in G: we might recover it only if we select two adjacencies or telomeres containing two genes a and b. Since each gene has at most C copies in the genome, there are at most C2 pairs of adjacencies or telomeres that may lead to recovery of the adjacency (a, b). So, with probability at most 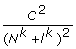 , one specific adjacency in G might be created by the rearrangement. Summing up all the N - l adjacencies in G, we see that the correction for ignoring the possible newly created adjacencies or telomeres in G is .
, one specific adjacency in G might be created by the rearrangement. Summing up all the N - l adjacencies in G, we see that the correction for ignoring the possible newly created adjacencies or telomeres in G is .
Similarly, we can get 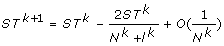 and
and 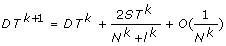 .
.
Gene duplication
We select uniformly at random an integer between 1 and Lmax (the maximum number of genes in the duplicated segment), then select uniformly at random a position in the genome where to start the duplication, then insert the copy at another position selected uniformly at random.
Theorem 2 Assume all genomes have O(1) linear chromosomes, each gene has at most C = O(1) copies, no two duplicate genes or adjacencies are within the segment to be duplicated, and represents the current genome Gk based on the original genome G. For conciseness, write (the total number of genes), lk = (STk + DTk)/2 (the number of linear chromosomes) and L = (Lmax + 1)/2 (the average number of genes in a duplicated segment). Then we approximate the expected vector for Gk+1 after one duplication operation with 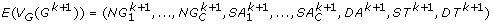 where we have
where we have
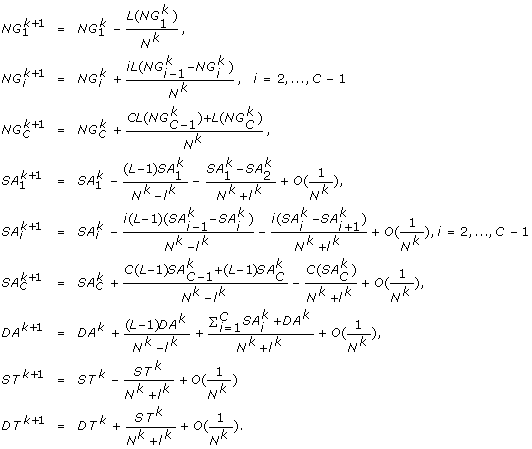 |
Proof In our model, we uniformly select a position to start duplicating L genes and transpose it to one new uniformly chosen position within the genome. The expected number of genes or adjacencies with exactly i copies within the duplicated segment is 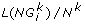 or
or 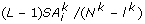 . The probability that the placement of the duplicated segment breaks one adjacency at any specific site is 1/(Nk + lk).
. The probability that the placement of the duplicated segment breaks one adjacency at any specific site is 1/(Nk + lk).
We again first ignore the adjacencies or telomeres in the original genome G created after a duplication event. Since we assume that no two genes or adjacencies are the same within the duplicated segment, we have
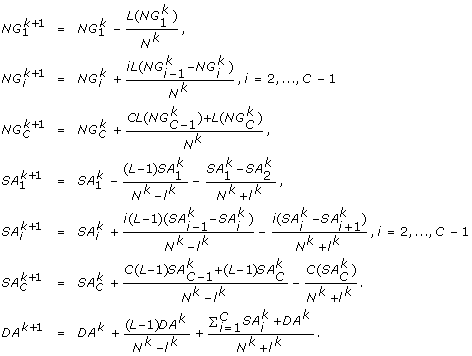 |
Now, we show that the correction for ignoring adjacencies or telomeres after a duplication event is to each item 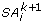 . Consider any adjacency (a, b) in G: we might recover it if we move gene a next to gene b after the duplication. Since each gene has at most C copies in the genome, there are at most 2LC2 possible duplication operations to recover that adjacency (a, b). There are altogether Ω (L(Nk + lk)2) different duplication operations. So, with probability
. Consider any adjacency (a, b) in G: we might recover it if we move gene a next to gene b after the duplication. Since each gene has at most C copies in the genome, there are at most 2LC2 possible duplication operations to recover that adjacency (a, b). There are altogether Ω (L(Nk + lk)2) different duplication operations. So, with probability 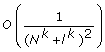 , one specific adjacency in G might be created by the duplication event. Summing up all the N - l adjacencies in G, we see that the correction for ignoring the newly created adjacencies or telomeres in G is .
, one specific adjacency in G might be created by the duplication event. Summing up all the N - l adjacencies in G, we see that the correction for ignoring the newly created adjacencies or telomeres in G is .
Similarly, we can get 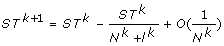 and
and 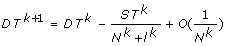 .
.
Gene loss
We uniformly select one gene with at least two copies and delete it.
Theorem 3 Assume each gene has at most C = O(1) copies and 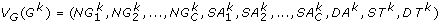 represents the current genome Gk based on the original genome G. For conciseness, write
represents the current genome Gk based on the original genome G. For conciseness, write 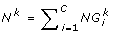 (the total number of genes) and lk = (STk+ DTk)/2 (the number of linear chromosomes). Then we can write the expected vector for Gk+1after one rearrangement operation as , where we have
(the total number of genes) and lk = (STk+ DTk)/2 (the number of linear chromosomes). Then we can write the expected vector for Gk+1after one rearrangement operation as , where we have
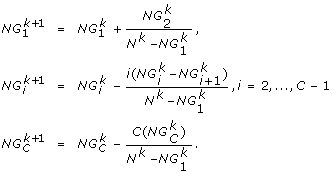 |
Proof In our model of gene loss, one gene with at least two copies is uniformly selected. The number of all possible genes to be deleted is 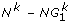 . For
. For 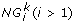 genes with exactly i copies in Gk, the probability that one of them is selected and deleted is
genes with exactly i copies in Gk, the probability that one of them is selected and deleted is 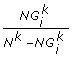 . So with probability , the number of genes with exactly i copies decreases by i and the number of genes with exactly (i -1) copies increases by (i -1).
. So with probability , the number of genes with exactly i copies decreases by i and the number of genes with exactly (i -1) copies increases by (i -1).
We ignore the adjacencies or telomeres in the original genome G to be created after one gene loss. For (i > 2) adjacencies with exactly i copies in Gk which also appears in G, it is difficult to compute the number fi(delj) of such adjacencies that each single deletion delj (j = 1,..., Nk - 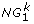 ) would affect. But we know that each adjacency with exactly i (i > 2) copies must relate to two genes with more than 2 copies, so we have
) would affect. But we know that each adjacency with exactly i (i > 2) copies must relate to two genes with more than 2 copies, so we have 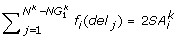 . Considering i = 2,..., C and C = O(1), we have
. Considering i = 2,..., C and C = O(1), we have
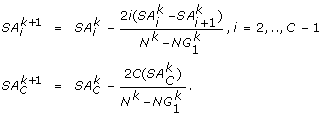 |
For 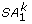 adjacencies with exactly 1 copy in Gk that also appears in G, it is also difficult to compute the number f1(delj) of such adjacencies that each single deletion delj (j = Nk - ) would affect. Assume
adjacencies with exactly 1 copy in Gk that also appears in G, it is also difficult to compute the number f1(delj) of such adjacencies that each single deletion delj (j = Nk - ) would affect. Assume 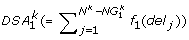 is the count of genes with at least two copies but related to those adjacencies with exactly 1 copy in Gk that also appear in G. We consider the effect of rearrangements, gene duplications and losses, and we approximate as follows:
is the count of genes with at least two copies but related to those adjacencies with exactly 1 copy in Gk that also appear in G. We consider the effect of rearrangements, gene duplications and losses, and we approximate as follows:
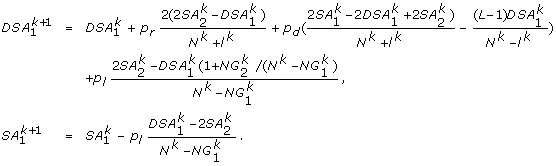 |
For telomeres, we simply assume ST k+1 = ST k and DT k+1 = DT k.
Finally, we also approximate the number of adjacencies RSAk+1 that we could thus ignore under rearrangements, gene duplications, and gene losses, and distribute it to the correction of as follows:
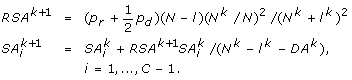 |
Now, given G0, we estimate E(VG(Gk)) for k > 0 by iterating k times the above formulas (using with pd and pl); at every step we identify E(VG(Gk-1)) with the actual vector VG(Gk-1).
Corollary 1 The estimated vector 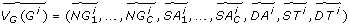 for all integers i (0 ≤ i ≤ k) can be computed in O(kC) time.
for all integers i (0 ≤ i ≤ k) can be computed in O(kC) time.
Results and discussion
We now present experimental results on the accuracy of our estimation of the expected vector after a given number of random evolutionary events and on the quality of our estimator for the true evolutionary distance (in terms of the actual number of evolutionary events). Our experiments all start with one genome with no duplicated genes and some chosen number of linear and circular chromosomes of various sizes. We first apply some number (usually 10) of duplication events (Lmax = 10 in all cases) to generate the original genome G with some initial duplicated genes. Then this genome is subjected to a prescribed number k of evolutionary events chosen according to pd and pl to obtain a final genome Gk. We vary k from 0 to twice the number of genes. We ran tests on any types of initial genomes designed to resemble actual organismal genomes; we tested different choices of parameters on different genomes; and in each case we generated 10,000 runs to obtain a tight estimate of variance.
We compute the vector representations for all intermediate genomes and then use our method to estimate the evolutionary distance. Due to space limitations, we present results on just three initial genomes: 25,000 genes and 25 linear chromosomes (pd = 0.05, pl = 0.15); 10, 000 genes and 5 linear chromosomes (pd = 0.1, pl = 0.2); and 1, 000 genes and 1 circular chromosome (pd = 0.2, pl = 0.6). The first two examples match large and smaller metazoan genomes, the last matches a small bacterial genome.
Accuracy of the expected vector after k random evolutionary events
We study the behavior of our estimator by comparing its prediction to the sample mean for VG(Gk), as computed from our 10,000 trials. In all of our experiments, we find that is very close to the sample mean for VG(Gk). Figure 2 shows the values in the vector as a function of the actual number of evolutionary events. 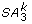 and
and 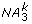 represent the number of adjacencies and genes with at least 3 copies in the original genome G, respectively. The figure shows that our estimation and the sample mean for VG(Gk) are always very close.
represent the number of adjacencies and genes with at least 3 copies in the original genome G, respectively. The figure shows that our estimation and the sample mean for VG(Gk) are always very close.
Figure 2.

The vector values as a function of the actual number of evolutionary events. (a) the color and shape code for the values, (b) Gene # = 1,000, Linear Chromosome # = 0, Circular Chromosome # = 1, (c) Gene # = 10,000, Linear Chromosome # = 10, Circular Chromosome # = 0, (d) Gene # = 25,000, Linear Chromosome # = 25, Circular Chromosome # = 0.
Accuracy of the estimation of the actual number of evolutionary events
We want to study the accuracy of our estimator for the actual number of evolutionary events; in order to do that, we create simulations with controlled numbers of evolutionary events and set up a threshold for correction in the estimation procedure. Specifically, we vary the actual number of evolutionary events from 0 to twice the number of genes in the original genome and we set 4 times the number of genes as an upper limit on the maximum number of evolutionary events. C is set to 10. Thus our estimated number k is chosen to minimize | - VG(F)|1, the 1-norm distance between and VG(F).
Figure 3 shows the mean and standard deviation for the actual number of evolutionary events estimated by our approach. Our approach provides accurate estimates, with very small variance.
Figure 3.

The actual number of evolutionary events (x axis)vs. our estimator (y axis). (a) Gene # = 1000, Linear Chromosome # = 0, Circular Chromosome # = 1, (b) Gene # = 10000, Linear Chromosome # = 10, Circular Chromosome # = 0, (c) Gene # = 25000, Linear Chromosome # = 25, Circular Chromosome # = 0. Mean is indicated by × and standard deviation is indicated by vertical bar.
We also study the mean absolute difference between the actual number of evolutionary events and our estimator, shown in Figure 4.
Figure 4.

The mean absolute difference between actual number of different evolutionary events and our estimation. The actual number of evolutionary events (x axis) vs. The mean absolute difference between actual number of different evolutionary events and our estimation (y axis). (o: Rearrangements, +: Duplications, ×: Losses). (a) Gene # = 1000, Linear Chromosome # = 0, Circular Chromosome # = 1, (b) Gene # = 10000, Linear Chromosome # = 10, Circular Chromosome # = 0, (c) Gene # = 25000, Linear Chromosome # = 25, Circular Chromosome # = 0.
Table 1 shows that the estimates are quite accurate up to very large numbers of events. Rearrangements, gene duplications, and gene losses fall under the category of "rare genomic events" (in the terminology of [14]), yet our estimator works well even for numbers that would instead indicate common events.
Table 1.
Relative error of our estimator as a function of the actual number of evolutionary events
| # genes | actual number of evolutionary events | |||||
|---|---|---|---|---|---|---|
| # genes × 1 | # genes × 2 | |||||
| Rearrangements | Duplications | Losses | Rearrangements | Duplications | Losses | |
| 1000 | 7.4% | 3.4% | 7.4% | 6.9% | 3.4% | 6.9% |
| 10,000 | 1.7% | 1.4% | 2.7% | 2.6% | 1.4% | 3.1% |
| 25,000 | 1.3% | 1.5% | 2.0% | 2.6% | 1.5% | 2.9% |
Robustness to unknown model parameters
Up to now we have fixed pd and pl. We now consider the case in which these parameters are unknown--clearly the more common case in practice. We generate 10,000 cases with randomly chosen parameters pd and pl (at 1% resolution, pd < 4pl) and with actual numbers of evolutionary events varying from 0 to twice the number of genes, setting an upper limit of 4 times the number genes for the maximum number of evolutionary events.
Given the original genome, our estimated vector 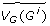 is in fact a function of i, pd, and pl. We enumerate all possible values for pd and pl (at 1% resolution, pd < 4 pl). For each different pair of parameters pd and pl, we compute all (i from 0 to 4 times the number of genes, C is set to 10). Our estimated number k is still chosen to minimize | - VG(F)|1, the 1-norm distance between and VG(F).
is in fact a function of i, pd, and pl. We enumerate all possible values for pd and pl (at 1% resolution, pd < 4 pl). For each different pair of parameters pd and pl, we compute all (i from 0 to 4 times the number of genes, C is set to 10). Our estimated number k is still chosen to minimize | - VG(F)|1, the 1-norm distance between and VG(F).
Figure 5 shows the comparison of our estimates to the actual number of evolutionary events. Our approach still provides accurate estimates in absence of known values for pd and pl and thus is quite robust. The mean absolute difference between the actual number of evolutionary events and our estimator becomes larger, especially when there are few common adjacencies left between the original and final genomes. (The duplications and losses may also partially cancel each other.)
Figure 5.
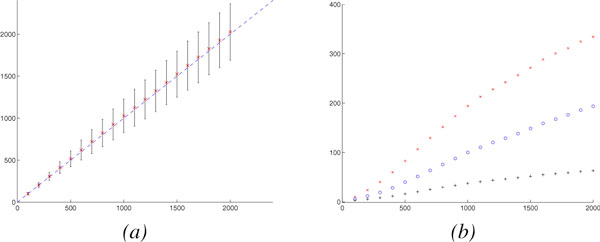
The actual number of evolutionary events vs. our estimator for unknown model parameters. (a): Mean (indicated by ×) and standard deviation (indicated by vertical bar) plots for the actual number of evolutionary events vs. our estimator (Gene # = 1000, Linear Chromosome # = 0, Circular Chromosome # = 1). (b): The mean absolute difference between actual number of different evolutionary events and our estimation (o: Rearrangements, +: Duplications, ×: Losses).
Conclusion
We propose a new evolutionary model for rearrangements, gene duplications and losses, and a corresponding method for estimating true evolutionary distance. The model is, to our knowledge, the first to preserve the structural dichotomy in genomic organization between most prokaryotes and most eukaryotes, and one of the few to unite rearrangements, duplications, and losses. Experimental results on a wide variety of genome structures exemplify the high accuracy and robustness of our estimator. This large gain in accuracy should translate into much better phylogenetic reconstructions as well as more accurate genomic alignments.
Competing interest
The authors declare that they have no competing interests.
Authors' contributions
YL conceived the idea, performed the analysis. YL, VR and KMS discussed and conducted the experiments. BMEM directed the project. YL and BMEM wrote the manuscript. All authors read and approved the final manuscript.
Contributor Information
Yu Lin, Email: yu.lin@epfl.ch.
Vaibhav Rajan, Email: vaibhav.rajan@epfl.ch.
Krister M Swenson, Email: krister.swenson@epfl.ch.
Bernard ME Moret, Email: bernard.moret@epfl.ch.
Acknowledgements
This article has been published as part of BMC Bioinformatics Volume 11 Supplement 1, 2010: Selected articles from the Eighth Asia-Pacific Bioinformatics Conference (APBC 2010). The full contents of the supplement are available online at http://www.biomedcentral.com/1471-2105/11?issue=S1.
References
- Fertin G, Labarre A, Rusu I, Tannier E, Vialette S. Combinatorics of Genome Rearrangements. MIT Press; 2009. [Google Scholar]
- Swofford D, Olsen G, Waddell P, Hillis D. In: Molecular Systematics. Hillis D, Mable B, Moritz C, editor. Sinauer Assoc., Sunderland, MA; 1996. Phylogenetic Inference; pp. 407–514. [Google Scholar]
- Moret B, Tang J, Wang LS, Warnow T. Steps toward accurate reconstructions of phylogenies from gene-order data. J Comput Syst Sci. 2002;65(3):508–525. doi: 10.1016/S0022-0000(02)00007-7. [DOI] [Google Scholar]
- Sankoff D, Blanchette M. Proc 3rd Conf Research Comput Mol Biol (RECOMB'99) ACM Press, New York; 1999. Probability models for genome rearrangement and linear invariants for phylogenetic inference; pp. 302–309. full_text. [Google Scholar]
- Wang LS. Proc 33rd ACM Symp Theory of Comput (STOC'01) ACM Press, New York; 2001. Exact-IEBP: a new technique for estimating evolutionary distances between whole genomes; pp. 637–646. full_text. [Google Scholar]
- Wang LS, Warnow T. Proc 1st Workshop Algs in Bioinfor-matics (WABI'01) Vol. 2149. Lecture Notes in Comp. Sci., Springer Verlag, Berlin; 2001. Estimating true evolutionary distances between genomes; pp. 176–190. [Google Scholar]
- Lin Y, Moret B. Estimating true evolutionary distances under the DCJ model. Proc 16th Conf Intelligent Systems for Mol Biol (ISMB'08), Bioinformatics. 2008;24(13):i114–i122. doi: 10.1093/bioinformatics/btn148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yancopoulos S, Attie O, Friedberg R. Efficient sorting of genomic permutations by translocation, inversion and block interchange. Bioinformatics. 2005;21(16):3340–3346. doi: 10.1093/bioinformatics/bti535. [DOI] [PubMed] [Google Scholar]
- Bergeron A, Mixtacki J, Stoye J. Proc 6th Workshop Algs in Bioinformatics (WABI'06) Vol. 4175. Lecture Notes in Comp. Sci., Springer Verlag, Berlin; 2006. A unifying view of genome rearrangements; pp. 163–173. [Google Scholar]
- El-Mabrouk N. Proc 11th Symp Combin Pattern Matching (CPM'00) Vol. 1848. Lecture Notes in Comp. Sci., Springer Verlag, Berlin; 2000. Genome rearrangement by reversals and insertions/deletions of contiguous segments; pp. 222–234. full_text. [Google Scholar]
- Yancopoulos S, Friedberg R. Proc 6th RE-COMB Workshop Comp Genomics (RECOMBCG'08) Vol. 5267. Lecture Notes in Comp. Sci., Springer Verlag, Berlin; 2008. Sorting genomes with insertions, deletions and duplications by DCJ; pp. 170–183. [Google Scholar]
- Swenson K, Marron M, Earnest-DeYoung J, Moret B. Proc 7th SIAM Workshop Alg Engin & Experiments (ALENEX'05) SIAM Press, Philadelphia; 2005. Approximating the true evolutionary distance between two genomes; pp. 121–129. [Google Scholar]
- Ouangraoua A, Boyer F, McPherson A, Tannier E, Chauve C. Proc 5th Int'l Symp on Bioinformatics Research and Applications, (ISBRA'09) Vol. 5542. Lecture Notes in Comp. Sci., Springer Verlag, Berlin; 2009. Prediction of contiguous regions in the amniote ancestral genome; pp. 173–185. [Google Scholar]
- Rokas A, Holland P. Rare genomic changes as a tool for phylogenetics. Trends in Ecol and Evol. 2000;15:454–459. doi: 10.1016/S0169-5347(00)01967-4. [DOI] [PubMed] [Google Scholar]