

Abstract
Background
Use of alternative gene promoters that drive widespread cell-type, tissue-type or developmental gene regulation in mammalian genomes is a common phenomenon. Chromatin immunoprecipitation methods coupled with DNA microarray (ChIP-chip) or massive parallel sequencing (ChIP-seq) are enabling genome-wide identification of active promoters in different cellular conditions using antibodies against Pol-II. However, these methods produce enrichment not only near the gene promoters but also inside the genes and other genomic regions due to the non-specificity of the antibodies used in ChIP. Further, the use of these methods is limited by their high cost and strong dependence on cellular type and context.
Methods
We trained and tested different state-of-art ensemble and meta classification methods for identification of Pol-II enriched promoter and Pol-II enriched non-promoter sequences, each of length 500 bp. The classification models were trained and tested on a bench-mark dataset, using a set of 39 different feature variables that are based on chromatin modification signatures and various DNA sequence features. The best performing model was applied on seven published ChIP-seq Pol-II datasets to provide genome wide annotation of mouse gene promoters.
Results
We present a novel algorithm based on supervised learning methods to discriminate promoter associated Pol-II enrichment from enrichment elsewhere in the genome in ChIP-chip/seq profiles. We accumulated a dataset of 11,773 promoter and 46,167 non-promoter sequences, each of length 500 bp, generated from RNA Pol-II ChIP-seq data of five tissues (Brain, Kidney, Liver, Lung and Spleen). We evaluated the classification models in building the best predictor and found that Bagging and Random Forest based approaches give the best accuracy. We implemented the algorithm on seven different published ChIP-seq datasets to provide a comprehensive set of promoter annotations for both protein-coding and non-coding genes in the mouse genome. The resulting annotations contain 13,413 (4,747) protein-coding (non-coding) genes with single promoters and 9,929 (1,858) protein-coding (non-coding) genes with two or more alternative promoters, and a significant number of unassigned novel promoters.
Conclusion
Our new algorithm can successfully predict the promoters from the genome wide profile of Pol-II bound regions. In addition, our algorithm performs significantly better than existing promoter prediction methods and can be applied for genome-wide predictions of Pol-II promoters.
Background
Alternative promoter usage is known to affect more than half of all human and mouse genes, and has been proposed as a primary driver of varied transcriptional regulation in different cellular conditions or developmental stages [1-4]. Numerous genes displaying complex transcriptional regulation, because of the use of alternative promoters, have been studied thoroughly [5]. Recent annotations of the human and mouse genomes suggest that many differentiation and disease-associated genes contain alternative promoters. Annotation of all human and mouse gene promoters that are differentially used during development, in different cell/tissue types or aberrantly activated in disease conditions is still incomplete and is essential for defining the transcriptome and proteome of human and mouse genomes.
The development of chromatin immunoprecipitation methods coupled with DNA microarray (ChIP-chip) technology and massively parallel sequencing (ChIP-seq) has enabled genome-wide identification of promoters, using antibody against RNA polymerase II (Pol-II) in different cells or tissues [6,7]. The combined signatures of RNA Pol-II binding and histone modification marks like H3K4me3, H3K9Ac obtained by these high throughput technologies are being used to identify human and mouse transcriptional units [8]. However, there are some challenges in predicting promoter usage based on the enrichment regions/peaks observed in these ChIP-chip/seq experiments. The ChIP-chip/seq technology requires antibodies with extremely high affinity and specificity for the target transcription factors. Unfortunately, such antibodies are not available for most human transcription factors, including Pol-II, producing non-specific enrichment in the ChIP-chip/seq profiles. The non-specific enrichment regions could be eliminated from the analysis by performing a ChIP-chip/seq experiment on the same cell or tissue lacking the specific factor. However, in most cases this is not feasible and we have to rely on other methodologies like the use of non-specific IgG ChIP-chip/seq to decrease the non-specific enrichment background. The major challenge in annotating promoters based on RNA Pol-II enriched regions/peaks is the spread of the transcribing polymerase throughout the gene and as a result all genomic regions bound by RNA Pol-II are enriched in these experiments, producing significantly large number of enriched peaks after the initial statistical analysis [9]. Though the initiator form of RNA polymerase II (phosphorylated CTD at Ser5) is enriched at a higher level in promoter region of actively transcribed genes, it is not restricted to the promoter region. Moreover, the promoters with stalled RNA Pol-II do not show an enrichment for the Ser5 phosphorylated form of RNA Pol-II [10]. Similarly, the histone marks namely H3K4me3 and H3K9Ac, which are highly enriched in promoter regions, are not exclusively present in promoter regions [8,11]. Currently it is not possible to identify promoters with high confidence based on RNA Pol-II ChIP-chip/seq enrichment data alone, thus warranting development of better classification algorithms for accurate identification of promoter related Pol-II enriched regions.
Here, we developed a computational method to discriminate promoter associated RNA Pol-II enriched regions of length 500 bp from the enrichment at other genomic regions, using the rich source of existing promoter data and associated chromatin modification signatures and various DNA sequence features. We prepared a data-set consisting of 11,773 Pol-II enriched promoters and 46,167 Pol-II enriched non-promoter regions from our recent ChIP-seq experiments, using antibody against Pol-II on five mouse tissues. We systematically trained and evaluated recent ensemble classifiers on this data set, using both 10-fold cross validation and testing on independent test set, and selected Bagging and Random Forest classifier for the final algorithm.
Methods
Dataset of Pol-II enriched promoters and non-promoters for training the classification models
The training set was generated from RNA Pol-II ChIP-seq data of five mouse tissues (Brain, Kidney, Liver, Lung and Spleen) generated by our lab. The RNA Pol-II ChIP-seq data was first processed and Pol-II enrichment peaks were identified at an FDR of 0.001, by assuming that peaks in the random background would follow Poisson statistics. The identified peaks were compared with TSS of non-redundant gene list generated from four different sources: RefSeq, Vega, Ensembl and UCSC. The gene lists were downloaded from UCSC genome browser [12] for mm9. Any peak that falls within -300 bp to +200 bp of known TSS (from compiled non-redundant TSS) were taken as promoter peak. The Pol-II peaks which are inside a gene but do not overlap with any of the known TSS are candidate non-promoter peaks. For our negative set, we consider only those peaks which fall within transcripts that possess a Pol-II enriched peak at the corresponding promoter (-300 bp to +200 bp). Also, any peak which falls within the promoter region of homologous gene known in other organism and within the 5' end of compiled set of non-redundant expression sequence tags (ESTs) was removed from the negative set, because some of those could be undiscovered novel promoters in the mouse genome. The homologous gene track (xenoRefGene track) was downloaded from UCSC genome browser. After identification of promoter and non-promoter peaks, the Pol-II peak was annotated as actual TSS for the transcript in that particular tissue. For each annotated peak (in both promoter and non-promoter sets), sequences were generated each of length 500 bp (-300 bp to +200 bp around the peak). After performing this procedure for all the tissues, we again compared the records in promoter and non-promoter sets with each other and removed the overlapping records from the non-promoter set. Finally, our complete dataset has 11,773 records in the promoter set and 46,167 records in the non-promoter set. We used 8,793 and 34,686 records from promoter and non-promoter sets respectively for training the classification models. In addition to 10-fold cross-validation, we used the remaining records (2,980 promoter and 11,481 non-promoter cases) to test the performance of the fitted models. The data sets are available as supplementary information at [13].
Classification models
We tried different state-of-art ensemble and meta classifiers for identification of promoter and non-promoter classes. The WEKA data-mining toolbox [14] was used for building the classification models. The different classifiers tested on 39-dimensional feature vector are: LogitBoost [15], Bagging [16], Rotational Forest [17] and Random Forest [18]. The detailed description of the classification methods is provided in Supplementary Method.
Feature variables used in classification model
Each sequence record (500 bp window) of the promoter and non-promoter set is represented by a 39-dimensional feature vector. The feature values were calculated based on (a) DNA sequence composition; (b) DNA physico-chemical-structural properties, and (c) experimental data. Most of the conversion tables that are based on DNA physical-chemical-structural properties were downloaded from [19]. The feature variables used in the classification models are briefly described below.
Let a given DNA sequence be: S = s1s2s3s4⋯sL-1sL, where si ∈ {A, C, G, T}, 1 ≤ i ≤ L (length of sequence, here L = 500). Let Δ1 ≡ {A, C, G, T}, Δ2 ≡ {AA, AC, AG, AT, CA, CC, CG, CT, GA, GC, GG, GT, TA, TG, TC, TT}, Δ3 ≡ {AAA, AAC, AAG, AAT, ACA, ACC, ⋯, TTT}, Δ4 ≡ {AAAA, AAAC, AAAG, AAAT, AACA, AACC, ⋯, TTTT} represent single, di, tri, tetra nucleotide symbol set respectively.
(a) Properties based on DNA sequence composition
We calculate 10 different features in this category. The first 7 features are calculated from single nucleotide composition. Let nx represents the total number of times symbol x appeared in S and x ∈ Δ1 for single nucleotide features.
1. A_Fraction: nA/L
2. C_Fraction: nC/L
3. G_Fraction: nG/L
4. T_Fraction: nT/L
5. PurPyr_Fraction: (nA + nG - nC - nT)/L
6. AmKe_Fraction: (nA + nC - nG - nT)/L
7. WeSt_Fraction: (nA + nT - nC - nG)/L
The remaining 3 features are related to CpG island. One of the features is based on di-nucleotide composition, where x ∈ Δ2. And remaining 2 features are based on tri-nucleotide composition, where x ∈ Δ3. Similar CpG features were used in [19] for promoter prediction.
8. CpG1: (2*nCG + 2*nGC)/(L-1)
9. CpG2: (nACG + nAGC + nCAG + nCCG + nCGA + nCGC + 2* nCGG + nCGT + nCTG + nGAC + nGCA + 2* nGCC + nGCG + nGCT + 2* nGGC + nGTC + nTCG + nTGC)/(L-2)
10. CpG3: (4* nCAG + nCCG + nCGG + 4* nCTG + 4* nGAC + nGCC + nGGC + 4* nGTC)/(L-2)
(b) Properties based on physico-chemical-structural property of DNA sequences
In this category we calculate 22 features. Let φp(x) represent a mapping function for a property 'P', where x ∈ Δ1 or x ∈ Δ2 or x ∈ Δ3 or x ∈ Δ4 depending upon given property. The feature value for a given sequence 'S' based on property 'P' is given by 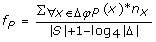 , where nx represents total number of times symbol x has appeared in S. And Δ ≡ Δ1 or Δ ≡ Δ2 or Δ ≡ Δ3 or Δ ≡ Δ4 depending up on the property 'P'.
, where nx represents total number of times symbol x has appeared in S. And Δ ≡ Δ1 or Δ ≡ Δ2 or Δ ≡ Δ3 or Δ ≡ Δ4 depending up on the property 'P'.
The DNA sequence properties 'P' from which the features are calculated is as follow:
1. A-philicity [20]
2. Base-stacking [21]
3. B-DNA twist [22]
4. DNA bending stiffness [23]
5. Di-nucleotide flexibility energy [24]
7. Duplex stability disrupt energy[27]
8. Duplex stability free energy [28]
9. Helical rise [29]
10. Helical twist [29]
11. Helical tilt [29]
12. Helical roll [29]
13. Helical shift [29]
14. Helical slide [29]
15. Propeller twist [30]
16. Protein induced deformability [31]
17. Protein-DNA twist [31]
18. Z-DNA stabilizing energy [32]
19. Tri-nucleotide bendability [33]
20. Nucleosome position preference [34]
21. Tetra-nucleotide flexibility [35] and
22. EIIP [36]
(c) Feature variables from experimental data
In this category we calculate 7 features. The feature values are calculated from CAGE tags, RNA Pol II Chip-seq and H3K4me3 Chip-seq data sets. The CAGE tags were downloaded from FANTOM4 project [37]. RNA Pol II and H3K4me3 ChIP-seq datasets taken up for our study were downloaded from NCBI GEO website. The accession numbers for the datasets are as follow: GSE14254 [38], GSE12241 [39], GSE11074 [40], GSM281696 [41], GSM307623 [39], GSE13511 [42]. Each ChIP-seq dataset was processed at an FDR of 0.001 using our program for significant region identification. The tags which are part of significant regions were considered for feature value calculation. In total 16 different samples of H3K4me3 ChIP-seq datasets and 12 different samples of Pol-II (including 5-different tissue data generated at our lab) were collected. Total number of CAGE tag count per million (TPM) that falls in 500 bp windows was taken as CAGE tag related feature. For both, H3K4me3 ChIP-seq and RNA Pol-II ChIP-seq data we calculated the following three different features:
1. Average Tag count per million (TPM)
2. Maximum TPM
3. Maximum TPM/average TPM
The performance of the fitted models and other best performing promoter prediction programs, which are publicly available for download to run on whole chromosomes on a local computer, was tested on an independent un-seen data set. When a predicted promoter overlaps with Pol-II enriched promoter, then the respective record is counted as true positive (TP). And when such case is missed it is termed as false negative (FN). When a predicted promoter overlaps with Pol-II enriched non-promoter, then such case is counted as false positive (FP). And if such case is predicted as non-promoter then it is termed as true negative (TN). The performance of classifier is evaluated based on the promoter prediction metrics suggested by Bajic et. al. [43]: sensitivity (SN), positive predictive value (PPV), correlation coefficient (CC) and true-positive cost (TPC). The equations for the performance metrics are as follow:
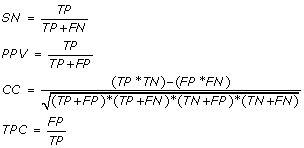 |
Results
Classification models to predict promoters using chromatin modification signatures and DNA sequence features
For selecting the best performing classifier, we trained four different ensemble and meta classification models on 3/4th of the dataset and tested on the remaining 1/4th of the dataset. The performance measures obtained by using 10-fold cross-validation and independent test set are presented in Tables 1 and 2. The performance measures, in terms of sensitivity and positive predictive accuracy, among the classifiers do not vary much over the four different models. Bagging and Random Forest models are slightly better than the other two models, showing lower error rates. Figure 1 allows for a more informative discussion on the relative predictive performance of the models. It is clear that Bagging, LogitBoost and Random Forest perform more or less similar and slightly better than Rotational Forest, with overall positive predictive value greaten than 95 and correlation coefficient greaten than 0.9. We then implemented the classification models given by Bagging and Random Forest methods in our algorithm which, will be applied to scan all the Pol-II enriched peaks in the mouse genome.
Table 1.
Performance statistics of classification models based on 10-fold cross validation
| Method | 10-fold cross-validation test result, 39 features, Promoters = 8793, NonPromoters = 34686 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| # of true positive | # of false negative | # of true negative | # of false positive | Sensitivity (%) | Positive predictive value (PPV) | Mathew correlation coefficient | True positive cost | ROC Area | |
| Bagging | 7603 | 1190 | 34354 | 332 | 86.47 | 95.82 | 0.89 | 0.04 | 0.97 |
| LogitBoost | 7638 | 1155 | 34252 | 434 | 86.86 | 94.62 | 0.88 | 0.06 | 0.97 |
| Random Forest | 7626 | 1167 | 34921 | 395 | 86.73 | 95.08 | 0.89 | 0.05 | 0.97 |
| Rotational Forest | 7153 | 1640 | 34198 | 488 | 81.35 | 93.61 | 0.84 | 0.07 | 0.96 |
Table 2.
Performance statistics of classification models and other existing programs based on independent test set
| Method | Promoters = 2980, NonPromoters = 11481 | |||||||
|---|---|---|---|---|---|---|---|---|
| # of true positive | # of false negative | # of true negative | # of false positive | Sensitivity (%) | Positive predictive value (PPV)% | Mathew correlation coefficient | True positive cost | |
| Bagging | 2593 | 387 | 11385 | 96 | 87.01 | 96.43 | 0.9 | 0.04 |
| LogitBoost | 2594 | 386 | 11356 | 125 | 87.05 | 95.4 | 0.89 | 0.05 |
| Random Forest | 2599 | 381 | 11349 | 132 | 87.21 | 95.17 | 0.89 | 0.05 |
| Rotational Forest | 2391 | 589 | 11332 | 149 | 80.23 | 94.13 | 0.84 | 0.06 |
| EP3 Program | 2493 | 487 | 11064 | 417 | 86.91 | 85.67 | 0.81 | 0.17 |
| Eponine Program | 2581 | 399 | 9633 | 1848 | 87.01 | 58.28 | 0.62 | 0.72 |
| ProSOM | 2563 | 417 | 8817 | 2664 | 86.01 | 49.03 | 0.53 | 1.04 |
| FirstEF | 1714 | 1226 | 11402 | 79 | 57.52 | 95.6 | 0.70 | 0.05 |
Figure 1.

ROC curve for four classification models. The ROC curve obtained by 10-fold cross-validation test for the four different classification methods.
While the classification methods used here are considered "black box" methods, with no interpretable classification model, the methods still provide useful information, such as variable importance. One of the measures of variable importance in Random Forest method is the mean decrease in accuracy, calculated using the out-of-bag sample. The difference between the prediction accuracy on the untouched out-of-bag sample and that on the out-of-bag sample permuted on one predictor variable is averaged over all trees in the forest and normalized by the standard error. This gives the mean decrease in accuracy of that particular predictor variable which has been permuted. Thus, the importance of the predictor variables can be ranked by their mean decrease in accuracy. Figure 2 shows the list of feature variables ranked according to mean decrease in accuracy of classification. It is interesting to note that feature variables based on experimental data such as CAGE tags, Pol-II enrichment, and H3K4me3 enrichment rank among the most discriminative variables from mean decrease in accuracy graph (Figure 2).
Figure 2.

Variable Importance table. Top ranking feature variables selected by Random Forest and their mean decrease in accuracy and mean decrease in Gini measure in discriminating Pol-II enriched promoter regions and Pol-II enriched non-promoter regions. The mean decrease in accuracy/Gini measure was an average of 100 runs of RF.
Comparison with other promoter prediction programs
We compared our algorithm with some of the existing best performing promoter prediction methods [44]: EP3 [19], Eponine [45], FirstEF [46], ProSOM 2.5 [47]. Table 2 and Figure 3 show that our classification model out-performs these existing programs based on independent (unseen) test set of Pol-II enriched promoters and Pol-II enriched non-promoters.
Figure 3.

ROC curve for comparison of our method with existing programs. The ROC curve obtained on the test set using our method and other existing programs: EP3, Eponine, FirstEF and ProSOM.
Annotation of promoters in mouse genome using Pol-II ChIP-seq data
Although extensive promoter annotations are available from the EBI and UCSC genome servers, most annotations do not contain information about tissue or cell-type information from experimental data. To demonstrate the efficacy of our new algorithm in finding promoters and to provide the annotation of potential novel promoters, we used the Pol-II enrichment peaks obtained from seven ChIP-seq datasets available on in vitro adipocyte differentiation of mouse 3T3-L1 cells and mouse ES cells, and the results are presented in Table 3. We applied the Random forest classification model (built using the training set) on a sequence of length 500 bp around each Pol-II enriched peak (-300 to +200 bp) in both strands. If 500 bp region from both strands are predicted as peak then they are merged and counted as one promoter. For each predicted promoter region, we annotated it to a nearby gene, if the Pol-II enriched peak is located within -2 Kbp to +500 bp around the corresponding gene TSS. We generated a non-redundant TSS file for coding genes from Refseq, Vega, ensembl and UCSC genes. For non-coding genes we used information available at RefSeq, Vega, ensembl, UCSC, miRBase [48] and recently discovered large non-coding RNAs (lincRNA) [49]. Table 3 presents the total number of peaks predicted by the model as promoters and also the number of annotated coding and non-coding genes in each sample. We then combined the results from all seven samples of predicted promoters in order to identify alternative promoters for each gene. For protein coding gene set, we found that there are 13413, 5064, and 4865 genes with one promoter, two promoters, and three or more promoters respectively. In other words, based on these annotations, 42.5% of the protein coding genes in mouse genome have two or more alternative promoters. For non-coding genes, we found that 4757, 1181, and 677 genes with one promoter, two promoters, and three or more promoters respectively.
Table 3.
Summary of prediction and annotation of Pol-II promoters from published ChIP-seq datasets
| Stage | D0 | D1 | D2 | D3 | D4 | D6 | ES Cell |
|---|---|---|---|---|---|---|---|
| Total tags | 5252311 | 5252311 | 5252311 | 5252311 | 5252311 | 5252311 | 2688589 |
| # of peaks | 108416 | 134674 | 153097 | 140137 | 159599 | 88606 | 13942 |
| # of peaks predicted as promoter | 24888 | 25179 | 24510 | 25101 | 22374 | 15838 | 5889 |
| # of predicted promoters assigned to known coding genes | 10645 | 10632 | 10349 | 10539 | 9701 | 8153 | 5034 |
| # of predicted promoters assigned to known non-coding genes | 1039 | 1095 | 1088 | 1101 | 1029 | 708 | 313 |
| # of unassigned predicted promoters (potential novel promotes) | 11684 | 13452 | 13673 | 13461 | 11644 | 6977 | 542 |
Future directions
We will use this program to annotate human gene Pol-II promoters by running on all the publicly available ChIP-seq Pol-II enrichment profiles. Our method successfully predicts 500 bp promoter regions (-300 bp to + 200 bp) and to better localize the core-promoter regions within the predicted promoters, we will apply recently developed CoreBoost_HM program published by Zhang Laboratory at CSHL [50].
Discussion
Chromatin modification and transcription factor binding profiles in the mammalian genomes is rapidly accumulating with the advent of next generation sequencing approaches. However, computational methods to effectively integrate these profiles to identify and annotate the promoter usage in specific cell/tissue types or developmental stages, are still limited. Recently, machine learning strategies have been applied to combine some of the wealth of published ChIP-seq data sets, such as chromatin modification signatures, to predict core promoter regions [50]. A logical step in analyzing the Pol-II enriched genomic regions is to scan those regions by existing promoter prediction methods to predict whether the enriched region is a promoter or non-promoter. However, we found that the performance of the existing methods is not satisfactory, and we speculate that the training set used in building the classifier was mostly responsible for their poor performance. We, therefore, build a bench-mark data set of Pol-II enriched promoters and Pol-II enriched non-promoters to train the classifiers, which shows significant improvement over the existing programs. Theoretical and empirical works using classification, regression trees, variable selection in linear and non-linear regression have shown that bagging and ensemble based methods can generate substantial prediction gain. In fact, based on the evaluation of 10-fold cross validation and testing on an independent data set, we found that both Bagging and Random Forest methods performed with highest accuracy (better than 95% prediction accuracy).
Conclusion
In conclusion, we have developed a novel algorithm based on Bagging and Random Forest based classification methods to predict Pol-II bound promoters from ChIP-seq profiles. The present algorithm will help the discovery of novel promoters and ongoing annotation of alternative promoters of human and mouse genes from different ChIP-seq experiments.
Supplementary material
Supplementary material is available at http://bioinfo.wistar.upenn.edu/promoterprediction/.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
RG designed the computational methods and performed the statistical analyses. PW performed some computational analysis, AB and FP performed database design and updates to MPromDb website. SP performed the biological experiments. RD formulated and directed the design of the study. All authors read and approved the final manuscript.
Contributor Information
Ravi Gupta, Email: rgupta@wistar.org.
Priyankara Wikramasinghe, Email: priyaw@wistar.org.
Anirban Bhattacharyya, Email: anirban@wistar.org.
Francisco A Perez, Email: fagosto@wistar.org.
Sharmistha Pal, Email: spal@wistar.org.
Ramana V Davuluri, Email: rdavuluri@wistar.org.
Acknowledgements
This work was supported by National Human Genome Research Institute grant R01HG003362 to RVD. RVD holds a Philadelphia Healthcare Trust Endowed Chair Position; research in his laboratory is supported by the Philadelphia Healthcare Trust, NHGRI/NIH and American Cancer Society. The use of computational resources in the Centre for Systems and Computational Biology and Bioinformatics Facility of Wistar Cancer Centre are gratefully acknowledged.
This article has been published as part of BMC Bioinformatics Volume 11 Supplement 1, 2010: Selected articles from the Eighth Asia-Pacific Bioinformatics Conference (APBC 2010). The full contents of the supplement are available online at http://www.biomedcentral.com/1471-2105/11?issue=S1.
References
- Sun H, Palaniswamy SK, Pohar TT, Jin VX, Huang TH, Davuluri RV. MPromDb: an integrated resource for annotation and visualization of mammalian gene promoters and ChIP-chip experimental data. Nucleic Acids Res. 2006. pp. D98–103. [DOI] [PMC free article] [PubMed]
- Baek D, Davis C, Ewing B, Gordon D, Green P. Characterization and predictive discovery of evolutionarily conserved mammalian alternative promoters. Genome Res. 2007;17(2):145–155. doi: 10.1101/gr.5872707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper SJ, Trinklein ND, Anton ED, Nguyen L, Myers RM. Comprehensive analysis of transcriptional promoter structure and function in 1% of the human genome. Genome Res. 2006;16(1):1–10. doi: 10.1101/gr.4222606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaji H, Severin J, Lizio M, Waterhouse A, Katayama S, Irvine KM, Hume DA, Forrest AR, Suzuki H, Carninci P. et al. The FANTOM web resource: from mammalian transcriptional landscape to its dynamic regulation. Genome Biol. 2009;10(4):R40. doi: 10.1186/gb-2009-10-4-r40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davuluri RV, Suzuki Y, Sugano S, Plass C, Huang TH. The functional consequences of alternative promoter use in mammalian genomes. Trends Genet. 2008;24(4):167–177. doi: 10.1016/j.tig.2008.01.008. [DOI] [PubMed] [Google Scholar]
- Singer GA, Wu J, Yan P, Plass C, Huang TH, Davuluri RV. Genome-wide analysis of alternative promoters of human genes using a custom promoter tiling array. BMC Genomics. 2008;9:349. doi: 10.1186/1471-2164-9-349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TH, Barrera LO, Zheng M, Qu C, Singer MA, Richmond TA, Wu Y, Green RD, Ren B. A high-resolution map of active promoters in the human genome. Nature. 2005;436(7052):876–880. doi: 10.1038/nature03877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129(4):823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Rozowsky J, Euskirchen G, Auerbach RK, Zhang ZD, Gibson T, Bjornson R, Carriero N, Snyder M, Gerstein MB. PeakSeq enables systematic scoring of ChIP-seq experiments relative to controls. Nat Biotechnol. 2009;27(1):66–75. doi: 10.1038/nbt.1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schones DE, Cui K, Cuddapah S, Roh TY, Barski A, Wang Z, Wei G, Zhao K. Dynamic regulation of nucleosome positioning in the human genome. Cell. 2008;132(5):887–898. doi: 10.1016/j.cell.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BM, Mahadevan LC. Stability of histone modifications across mammalian genomes: implications for 'epigenetic' marking. J Cell Biochem. 2009;108(1):22–34. doi: 10.1002/jcb.22250. [DOI] [PubMed] [Google Scholar]
- UCSC Genome Browser. http://hgdownload.cse.ucsc.edu/
- Center for Systems & Computational Biology, The Wistar Institute. http://bioinfo.wistar.upenn.edu/promoterprediction
- WEKA data-mining toolbox. http://www.cs.waikato.ac.nz/ml/weka/
- Friedman J, Hastie T, Tibshirani R. Additive logistic regression: a statistical view of boosting. Ann Stat. 1998;28:337–407. doi: 10.1214/aos/1016218223. [DOI] [Google Scholar]
- Breiman L. Bagging predictors. Mach Learn. 1996;24(2):123–140. [Google Scholar]
- Rodriguez JJ, Alonso CJ, Kuncheva LI. Rotation Forest: A New Classifier Ensemble Method. IEEE Trans Pattern Anal Mach Intell. 2006;28(10):1619–1630. doi: 10.1109/TPAMI.2006.211. [DOI] [PubMed] [Google Scholar]
- Breiman L. Random Forests. Mach Learn. 2001;45(1):5–32. doi: 10.1023/A:1010933404324. [DOI] [Google Scholar]
- Abeel T, Saeys Y, Bonnet E, Rouze P, Peer Y Van De. Generic eukaryotic core promoter prediction using structural features of DNA. Genome Research. 2008;18(2):310–323. doi: 10.1101/gr.6991408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov VI, Minchenkova LE. [The A-form of DNA: in search of the biological role] Mol Biol (Mosk) 1994;28(6):1258–1271. [PubMed] [Google Scholar]
- Ornstein RL, Rein R, Breen DL, Macelroy RD. OPTIMIZED POTENTIAL FUNCTION FOR CALCULATION OF NUCLEIC-ACID INTERACTION ENERGIES .1. BASE STACKING. Biopolymers. 1978;17(10):2341–2360. doi: 10.1002/bip.1978.360171005. [DOI] [PubMed] [Google Scholar]
- Gorin AA, Zhurkin VB, Olson WK. B-DNA twisting correlates with base-pair morphology. J Mol Biol. 1995;247(1):34–48. doi: 10.1006/jmbi.1994.0120. [DOI] [PubMed] [Google Scholar]
- Sivolob AV, Khrapunov SN. Translational positioning of nucleosomes on DNA: the role of sequence-dependent isotropic DNA bending stiffness. J Mol Biol. 1995;247(5):918–931. doi: 10.1006/jmbi.1994.0190. [DOI] [PubMed] [Google Scholar]
- Packer MJ, Dauncey MP, Hunter CA. Sequence-dependent DNA structure: dinucleotide conformational maps. J Mol Biol. 2000;295(1):71–83. doi: 10.1006/jmbi.1999.3236. [DOI] [PubMed] [Google Scholar]
- Blake RD, Delcourt SG. Thermal stability of DNA. Nucleic Acids Res. 1998;26(14):3323–3332. doi: 10.1093/nar/26.14.3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake RD, Bizzaro JW, Blake JD, Day GR, Delcourt SG, Knowles J, Marx KA, SantaLucia J Jr. Statistical mechanical simulation of polymeric DNA melting with MELTSIM. Bioinformatics. 1999;15(5):370–375. doi: 10.1093/bioinformatics/15.5.370. [DOI] [PubMed] [Google Scholar]
- Breslauer KJ, Frank R, Blocker H, Marky LA. Predicting DNA duplex stability from the base sequence. Proc Natl Acad Sci USA. 1986;83(11):3746–3750. doi: 10.1073/pnas.83.11.3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto N, Nakano S, Yoneyama M, Honda K. Improved thermodynamic parameters and helix initiation factor to predict stability of DNA duplexes. Nucleic Acids Res. 1996;24(22):4501–4505. doi: 10.1093/nar/24.22.4501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goni JR, Perez A, Torrents D, Orozco M. Determining promoter location based on DNA structure first-principles calculations. Genome Biol. 2007;8(12):R263. doi: 10.1186/gb-2007-8-12-r263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- el Hassan MA, Calladine CR. Propeller-twisting of base-pairs and the conformational mobility of dinucleotide steps in DNA. J Mol Biol. 1996;259(1):95–103. doi: 10.1006/jmbi.1996.0304. [DOI] [PubMed] [Google Scholar]
- Olson WK, Gorin AA, Lu XJ, Hock LM, Zhurkin VB. DNA sequence-dependent deformability deduced from protein-DNA crystal complexes. Proc Natl Acad Sci USA. 1998;95(19):11163–11168. doi: 10.1073/pnas.95.19.11163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho PS, Zhou GW, Clark LB. Polarized electronic spectra of Z-DNA single crystals. Biopolymers. 1990;30(1-2):151–163. doi: 10.1002/bip.360300115. [DOI] [PubMed] [Google Scholar]
- Brukner I, Sanchez R, Suck D, Pongor S. Sequence-dependent bending propensity of DNA as revealed by DNase I: parameters for trinucleotides. EMBO J. 1995;14(8):1812–1818. doi: 10.1002/j.1460-2075.1995.tb07169.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satchwell SC, Drew HR, Travers AA. Sequence periodicities in chicken nucleosome core DNA. J Mol Biol. 1986;191(4):659–675. doi: 10.1016/0022-2836(86)90452-3. [DOI] [PubMed] [Google Scholar]
- Packer MJ, Dauncey MP, Hunter CA. Sequence-dependent DNA structure: tetranucleotide conformational maps. J Mol Biol. 2000;295(1):85–103. doi: 10.1006/jmbi.1999.3237. [DOI] [PubMed] [Google Scholar]
- Cosic I. Macromolecular bioactivity: is it resonant interaction between macromolecules?--Theory and applications. IEEE Trans Biomed Eng. 1994;41(12):1101–1114. doi: 10.1109/10.335859. [DOI] [PubMed] [Google Scholar]
- FANTOM4 Project. http://fantom.gsc.riken.jp/4/
- Wei G, Wei L, Zhu J, Zang C, Hu-Li J, Yao Z, Cui K, Kanno Y, Roh TY, Watford WT. et al. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity. 2009;30(1):155–167. doi: 10.1016/j.immuni.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP. et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448(7153):553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen TS, Hanna J, Zhang X, Ku M, Wernig M, Schorderet P, Bernstein BE, Jaenisch R, Lander ES, Meissner A. Dissecting direct reprogramming through integrative genomic analysis. Nature. 2008;454(7200):49–55. doi: 10.1038/nature07056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, Zhang X, Bernstein BE, Nusbaum C, Jaffe DB. et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454(7205):766–770. doi: 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen R, Pedersen TA, Hagenbeek D, Moulos P, Siersbaek R, Megens E, Denissov S, Borgesen M, Francoijs KJ, Mandrup S. et al. Genome-wide profiling of PPARgamma:RXR and RNA polymerase II occupancy reveals temporal activation of distinct metabolic pathways and changes in RXR dimer composition during adipogenesis. Genes Dev. 2008;22(21):2953–2967. doi: 10.1101/gad.501108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajic VB, Tan SL, Suzuki Y, Sugano S. Promoter prediction analysis on the whole human genome. Nat Biotechnol. 2004;22(11):1467–1473. doi: 10.1038/nbt1032. [DOI] [PubMed] [Google Scholar]
- Abeel T, Peer Y Van de, Saeys Y. Toward a gold standard for promoter prediction evaluation. Bioinformatics. 2009;25(12):i313–320. doi: 10.1093/bioinformatics/btp191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Down TA, Hubbard TJ. Computational detection and location of transcription start sites in mammalian genomic DNA. Genome Res. 2002;12(3):458–461. doi: 10.1101/gr.216102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davuluri RV, Grosse I, Zhang MQ. Computational identification of promoters and first exons in the human genome. Nat Genet. 2001;29(4):412–417. doi: 10.1038/ng780. [DOI] [PubMed] [Google Scholar]
- Abeel T, Saeys Y, Rouze P, Peer Y Van de. ProSOM: core promoter prediction based on unsupervised clustering of DNA physical profiles. Bioinformatics. 2008;24(13):i24–31. doi: 10.1093/bioinformatics/btn172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008. pp. D154–158. [DOI] [PMC free article] [PubMed]
- Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, Huarte M, Zuk O, Carey BW, Cassady JP. et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458(7235):223–227. doi: 10.1038/nature07672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Xuan Z, Zhao X, Li Y, Zhang MQ. High-resolution human core-promoter prediction with CoreBoost_HM. Genome Res. 2009;19(2):266–275. doi: 10.1101/gr.081638.108. [DOI] [PMC free article] [PubMed] [Google Scholar]