

Abstract
Pseudomonas aeruginosa is an opportunistic Gram-negative pathogen that causes nosocomial infections for which there are limited treatment options. Penicillin-binding protein PBP3, a key therapeutic target, is an essential enzyme responsible for the final steps of peptidoglycan synthesis and is covalently inactivated by β-lactam antibiotics. Here we disclose the first high resolution cocrystal structures of the P. aeruginosa PBP3 with both novel and marketed β-lactams. These structures reveal a conformational rearrangement of Tyr532 and Phe533 and a ligand-induced conformational change of Tyr409 and Arg489. The well-known affinity of the monobactam aztreonam for P. aeruginosa PBP3 is due to a distinct hydrophobic aromatic wall composed of Tyr503, Tyr532, and Phe533 interacting with the gem-dimethyl group. The structure of MC-1, a new siderophore-conjugated monocarbam complexed with PBP3 provides molecular insights for lead optimization. Importantly, we have identified a novel conformation that is distinct to the high-molecular-weight class B PBP subfamily, which is identifiable by common features such as a hydrophobic aromatic wall formed by Tyr503, Tyr532, and Phe533 and the structural flexibility of Tyr409 flanked by two glycine residues. This is also the first example of a siderophore-conjugated triazolone-linked monocarbam complexed with any PBP. Energetic analysis of tightly and loosely held computed hydration sites indicates protein desolvation effects contribute significantly to PBP3 binding, and analysis of hydration site energies allows rank ordering of the second-order acylation rate constants. Taken together, these structural, biochemical, and computational studies provide a molecular basis for recognition of P. aeruginosa PBP3 and open avenues for future design of inhibitors of this class of PBPs.
Keywords: antibiotic resistance, cell wall, transpeptidase, covalent inhibitor
Infections caused by Gram-negative pathogens are a serious threat to public health. Multidrug-resistant Gram-negative organisms such as Pseudomonas aeruginosa, Klebsiella pneumoniae, Escherichia coli, and Acinetobacter baumannii are emerging as significant pathogens. Therapeutic options for their treatment are limited, in part due to more focused efforts over recent years to combat Gram-positive bacteria, such as Staphylococcus aureus and vancomycin-resistant enterococci (1, 2). Simultaneously, the emergence and spread of resistance mechanisms including β-lactamases have diminished the value of many marketed β-lactam antibiotics. New Gram-negative antibiotics for the treatment of nosocomial infections are urgently needed to tackle the high morbidity and mortality rates associated with multidrug resistant pathogens.
P. aeruginosa causes multiple types of infections including pneumonia, bacteremia, and urinary tract, ear, skin, and soft tissue infections. Current therapies for P. aeruginosa infections include broad-spectrum β-lactam antibiotics, carbapenems such as imipenem or meropenem, or a more Gram-negative selective monobactam such as aztreonam (3). In addition to producing β-lactamases, P. aeruginosa can exhibit or acquire additional resistance mechanisms including reduction of outer membrane permeability, expression of efflux pumps, or by mutating the target penicillin-binding proteins (4).
The macromolecular targets for β-lactam antibiotics are penicillin-binding proteins (PBPs), membrane-associated periplasmic enzymes that catalyze essential transpeptidase reactions involving peptidoglycan, a major component of bacterial cell walls. At least six PBPs of P. aeruginosa have been detected by their ability to form covalent adducts with radiolabeled penicillin G (5). Based on their structural features and enzyme activities, P. aeruginosa PBPs are categorized into three classes. High-molecular-weight class A PBPs (PBP1a and PBP1b) are bifunctional enzymes containing both transglycosylase and transpeptidase activities. High-molecular-weight class B PBP, of which Pseudomonas PBP3 is a member, possesses only transpeptidase activity. Low-molecular-weight class C PBPs, including PBP4 and PBP5, are soluble proteins and act as DD carboxypeptidases. The high-molecular PBPs are essential for cell growth and are validated and precedented targets for β-lactam antibiotics (6). PBP3, encoded by the ftsI gene, is the only known peptidoglycan synthase in E. coli and is essential for cell division. PBP3 inhibition by a β-lactam and gene deletion inhibit cell septation, resulting in filamentation (7). PBP2 from Neisseria gonorrhoeae (NgPBP2), is the only other known PBP3 ortholog from a Gram-negative pathogen that has been crystallized. It has two flexible loop regions that appear to block entry of inhibitors into the active site, resulting in unsuccessful soaking experiments despite numerous attempts to produce NgPBP2-inhibitor complexes (8).
One approach to circumvent permeability-mediated resistance of Gram-negative pathogens such as P. aeruginosa is to utilize the siderophore-mediated iron acquisition system to effectively deliver drug molecules to the periplasmic space (9). Bacteria require iron in order to survive, and the acquisition of iron depends on the production, release, and active reuptake of iron-scavenging molecules called siderophores. Our strategy relies on the incorporation of a hydroxypyridone moiety as a siderophore attached to a monocyclic β-lactam antibiotic, which is anticipated to result in increased concentrations of drug in the periplasm (10).
To help elucidate the molecular mechanism of P. aeruginosa PBP3 (PaPBP3) and its β-lactam recognition elements, we solved multiple X-ray structures of a soluble form of PaPBP3. High-resolution structures of the apo and ceftazidime-bound forms were obtained as well as complexes with meropenem, imipenem, and aztreonam. Extensive medicinal chemistry efforts led to the identification of MC-1, a siderophore-conjugated monocarbam that utilizes a carbonylaminosulfonyl species for β-lactam activation, and which shows remarkable antipseudomonal activity both in vitro and in vivo. The crystal structure of PaPBP3 complexed with MC-1 at 1.64-Å resolution reveals a unique binding mode of the siderophore. WaterMap analysis was carried out to evaluate the correlation between protein desolvation energies and the second-order acylation constants (k2/Kd) during ligand binding. A ligand-dependent water displacement effectiveness score (ϵi) provides a unique measure that rank orders the second-order acylation rate constants. Our biochemical, computational, and structural results provide key insight into recognition elements and avenues to advance the design of β-lactam inhibitors with improved efficiency of PBP3 interaction and effectiveness against Gram-negative pathogens.
Results and Discussion
Overall Structures of PaPBP3.
PaPBP3 has overall dimensions of ∼45 × 63 × 100 Å and consists of two domains, an N-terminal nonpenicillin-binding domain and a C-terminal transpeptidase (TP) domain (Fig. 1A). The N-terminal domain, resembling a pair of sugar tongs, extends with long β-strands ∼45 Å in length, which are unique to the high-molecular mass of PBPs of subclass B3 (11). The four conserved motifs of the N-terminal domain are located at the base of the sugar tongs and form important structural elements involved in interdomain interactions (Fig. S1). The N-terminal domain of the high-molecular-weight PBPs is required for the folding and stability of the C-terminal transpeptidase domain and has been suggested to provide recognition sites for formation of the multiprotein complex responsible for cell wall biosynthesis (12). The structure of the C-terminal domain (residues 225–579) is similar to other TP domains of PBPs, carrying the classical signature of the penicilloyl serine transferase superfamily. The active site including the nucleophilic Ser294 residue is located between two subdomains, the α-subdomain and α/β-subdomain. The α-subdomain containing α2, α4–α6, and α8 forms one side of the active site groove. A β-hairpin extension connecting α2 and α4 is composed of four short β-strands (β2a, β2b, β2c, and β2d) and forms two perpendicular antiparallel β-sheets (β2a/β2d and β2b/β2c) at the top of the active site. The α/β subdomain contains a central core of a five-stranded antiparallel β-sheet (β3/β4/β5/β1/β2) sandwiched by several helices on both sides (α1, α9, α10, and α11). In all six PaPBP3 structures, the two loop regions (residues 332–338 connecting β2c to β2d and residues 526–533 connecting α5 to α11) display substantial conformational changes upon binding different inhibitor classes and play an important role in substrate/inhibitor recognition.
Fig. 1.

Overall structure of the high-molecular-weight class B PBP3 from Gram-negative bacteria. (A) Stereoview of the PaPBP3 complexed with MC-1. The N-terminal domain is shown in orange and the C-terminal domain in cyan. The bound MC-1 is shown as spheres. (B) Chemical structures of antibiotics relevant to this study. The leaving group of ceftazidime is shown in red.
Induced-Fit-Conformational Changes of the PaPBP3 in Complex with Ceftazidime.
Ceftazidime, a third-generation cephalosporin, has good in vitro activity against P. aeruginosa and is clinically effective and safe in treating many nosocomial bacterial infections. Ceftazidime is composed of a β-lactam ring fused with a six-membered dihydrothiazine ring, a methyl pyridine at the C3 position, a carboxylic acid group at the C4 position, and a bulky aminothiazole-containing side chain at the C7 position (Fig. 1B). The crystal structure of PaPBP3 in complex with ceftazidime at 1.74-Å resolution clearly identifies ceftazidime covalently bound to Ser294 and the rearrangement of the dihydrothiazine ring to form an exocyclic methylene group at C3 position due to the departure of the pyridine moiety (Fig. S2) (13). The carbonyl oxygen of the covalent enzyme-ceftazidime ester is positioned into the oxyanion hole and hydrogen bonds with the main chain amides of Ser294 and Thr487. The C4 carboxylic acid group is anchored by two hydrogen bonds with the hydroxyl groups of Ser485 and Thr487 in the KSGT motif. The Lys297 in the conserved SXXK motif no longer hydrogen-bonds to Ser294 and is positioned to interact with SXN motif, residues Ser349 and Asn351 (Fig. 2A).
Fig. 2.

Interaction of ceftazidime, imipenem, and meropenem in the active site of the PaPBP3. (A) Active site in the ceftazidime-acyl PaPBP3 structure. Ceftazidime is shown in stick rendering with cyan carbons and hydrogen bonds as dashes. (B) Molecular surface of the PaPBP3 in the active site region in complex with ceftazidime (cyan). The apo PaPBP3 structure is shown in pink. (C) Active site of PaPBP3 bound to imipenem (orange). The loop connecting β5 and α11 undergoing significant conformational changes is shown in magenta. (D) Active site of PaPBP3 bound to meropenem (navy). The loop connecting β5 and α11 undergoing significant conformational changes is shown in magenta.
The bulky aminothiazole-containing group has extensive interactions with PaPBP3. The amide bond of the ceftazidime is wedged between the Thr487 carbonyl backbone and the Asn351 side chain. The aminothiazole moiety is stabilized by hydrogen bonds with the Glu291 side chain and Arg489 carbonyl backbone and by hydrophobic interaction with Tyr409. Importantly, the Tyr409 side chain blocking the aminothiazole binding site in the absence of ligand is swung out toward the solvent-exposed surface to avoid a steric clash upon ceftazidime binding. The Tyr409, flanked by two highly flexible glycine residues, is also present in high-molecular mass PBPs of subclass B3 from other Gram-negative bacteria including N. gonorrhoeae PBP2 and E. coli PBP3, and similar conformational changes involving tyrosine residues appear to occur upon the binding of aminothiazole moieties (8, 14). In contrast, the aminothiazole moiety makes relatively few direct interactions with β-lactamases and apparently adopts a different conformation in the binding site of low molecular mass PBPs, suggesting different recognition mechanisms for the aminothiazole moieties among different classes of PBPs and β-lactamases (15, 16).
Unexpectedly, Tyr532 and Phe533 adopt a completely different conformation upon ceftazidime binding, displacing the Tyr503 side chain toward the active site. These changes result in a unique aromatic wall composed of Tyr503, Tyr532, and Phe533 that accommodates the bulky gem-dimethyl group appended from the oxime by a hydrophobic interaction. The aromatic wall is further stabilized by forming a bridge with Val333, describing the boundary of the pocket for the gem-dimethyl group. Notably, upon ceftazidime binding, Val333 shifts by approximately 1.5 Å away from the active site, not only to interact with the gem-dimethyl group but also to allow van der Waals interaction with the six-membered dihydrothiazine ring in the active site (Fig. 2B). In addition, the carboxylic acid group forms a salt bridge with the guanidium group of Arg489. The key features of the aromatic wall, together with Val333 and Arg489, are likely to play a role in the efficient inhibition of PaPBP3.
Conformational Rearrangement of Tyr532 and Phe533 upon Carbapenem (Meropenem and Imipenem) Binding.
Carbapenems such as imipenem and meropenem possess a broad antibacterial spectrum and are used primarily to combat penicillin- and cephalosporin-resistant bacteria (17, 18). The electron density within the active site revealed each carbapenem molecule covalently bound to Ser294 (Fig. S2). Unlike the aminothiazole-containing ceftazidime complex, the Tyr409 side chain remains hydrogen-bonded to the Thr487 carbonyl backbone, where the Thr487 side chain is not close enough to make a direct hydrogen-bond interaction with the C3 carboxylic acid group. Instead, the C3 carboxylic acid is anchored by a water-mediated hydrogen bond with Thr487. In both the imipenem and meropenem complex structures, the formation of the complex was accompanied by substantial conformational change in the regions of β3, β4 and the loop connecting β5 and α11 at the mouth of the active site. The Cα atoms of Tyr532 and Phe533 in the imipenem complex were displaced 7.1 Å and 4.3 Å, respectively, toward the opening of the active site pocket compared to their positions in the apo structure (Fig. 2C). A simultaneous reorientation of Tyr503 and Arg489 results in formation of a stacking interaction with Tyr409. As a result, the hydrophobic pyrrolidine core of imipenem is completely buried by a hydrophobic pocket formed by residues Val333 and Tyr532.
Surprisingly, the additional methyl group of the pyrrolidine core in meropenem triggers a 180° flip of the Tyr532 side chain and moves its side chain away from the active site to interact with the Asn242 carbonyl backbone (Fig. 2D). Phe533 moves toward the active site forming a tunnel-like hydrophobic pillar with Val333 to stabilize the methyl group of the pyrrolidine core. The carbamoyl pyrrolidinyl group of meropenem is further stabilized by water-mediated hydrogen bonds with the Ile347 and Gly535 backbone. In contrast, in the imipenem complex structure, the electron density beyond the thioether sulfur atom is weak and discontinuous, suggesting that the C-2 side chain is not only flexible but also does not form strong interactions with PaPBP3. Furthermore, a competition assay using a labeled penicillin G shows that imipenem competes 16-fold less efficiently than meropenem for inactivation of PBP3 by a fluorescent penicillin analog (Table 1). Although the PaPBP3 crystal structures in complex with carbapenems are products of inactivation reactions, the hydrophobic interactions involving two different residues, Tyr532 and Phe533, and additional water-mediated hydrogen bonds likely play an important role in carbapenem recognition and positioning for acylation of Ser 294.
Table 1.
In vitro characterization of β-lactams in PaPBP3 and hydration site analysis
| Compound | Second-order acylation constant | PaPBP3 binding | Antibacterial activity | Thermal stability | Stable water, ΔG < 0 kcal/mol | Unstable water, ΔG > 0 kcal/mol | Water displacement effectiveness score | |||||
| k2/Kd (M-1 s-1) | EC50 (uM) | MIC90 (ug/mL) | Tm* (°C) | 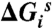 |
 |
 |
 |
 |
 |
ϵi | log(k2/Kd) M-1 s-1 | |
| Meropenem | 63,760 | 0.157 | > 64 | 42 | −3.563 | 10 | −0.356 | 6.035 | 9 | 0.671 | −0.531 | 4.805 |
| Imipenem | 712 | 2.5 | > 64 | 40 | −0.211 | 2 | −0.105 | 6.473 | 11 | 0.588 | −0.179 | 2.852 |
| Ceftazidime | 24,707 | 0.17 | > 64 | 54 | −2.597 | 6 | −0.433 | 24.238 | 23 | 1.054 | −0.411 | 4.393 |
| Aztreonam | 4,396,400 | 0.086 | > 64 | 56 | −3.935 | 4 | −0.984 | 10.789 | 12 | 0.899 | −1.094 | 6.643 |
| MC-1 | 45,108 | 0.109 | 0.5 | 58 | −5.996 | 9 | −0.666 | 30.395 | 24 | 1.266 | −0.526 | 4.654 |
*Tm, thermal stability (apo PaPBP3 = 46 °C);  , where (ΔG < 0 kcal/mol) for ligand i;
, where (ΔG < 0 kcal/mol) for ligand i;  , where (ΔG > 0 kcal/mol) for ligand i;
, where (ΔG > 0 kcal/mol) for ligand i;  , number of stable hydration sites affected by ligand i;
, number of stable hydration sites affected by ligand i;  , number of unstable hydration sites affected by ligand i;
, number of unstable hydration sites affected by ligand i;  , unstable water displacement efficiency index of ligand i;
, unstable water displacement efficiency index of ligand i;  , stable water displacement efficiency index of ligand i;
, stable water displacement efficiency index of ligand i;  , water displacement effectiveness score of ligand i.
, water displacement effectiveness score of ligand i.
The Efficient Binding of Monobactam Aztreonam to PaPBP3.
The monobactam aztreonam possesses very high affinity for PBP3 from a wide range of aerobic Gram-negative bacteria including P. aeruginosa, but is inactive against Gram-positive bacteria due to poor interaction with their essential PBPs (19). The structure activity relationship of monobactams suggests that the 2-aminothiazole moiety of aztreonam contributes to the activity against Gram-negative bacteria, and anti-Pseudomonal activity is enhanced by the addition of an iminopropyl carboxyl moiety. The sulfonic acid group attached to the nitrogen of the β-lactam ring is responsible for β-lactam activation, and methylation at the C4 position provides enhanced stability of the ring to β-lactamase attack (20). The crystal structure of PaPBP3 in complex with aztreonam reveals that the bulky aminothiazole-containing moiety is in a similar position to that adopted in the ceftazidime complex, and the same induced-fit-conformational changes occur involving residues Tyr409, Arg489, and the aromatic wall composed of Tyr503, Tyr532, and Phe533 (Fig. 3A). β-lactam structural differences ranging from an unfused ring in aztreonam, to a five-membered fused ring in the carbapenems, or six-membered fused ring in ceftazidime, do not affect the hydrophobic interaction with Val333. The C4 methyl group of aztreonam does hinder rotation in the ring-open form and is stabilized by the hydrophobic patch composed of Val333 and Phe533. In addition, the sulfonic acid group is tightly anchored by side chains of Lys484, Ser485, and Thr487. Although the lactam cores of aztreonam and ceftazidime are quite distinct, the bulky aminothiazole-containing group plays an important role in inducing conformational changes critical for efficient interaction with P. aeruginosa PBP3.
Fig. 3.

Aztreonam and the siderophore-conjugated monocarbam, MC-1 bound to PaPBP3. (A) Active site in aztreonam-acyl-PaPBP3 complex. Aztreonam is shown in stick rendering with magenta carbons. (B) Interaction of MC-1 with PaPBP3. MC-1 is shown in stick rendering with two alternate conformations (green and magenta). (C) Molecular surface of the PaPBP3 in the active site region in compex with MC-1 (two alternate conformation in green and magenta).
The Monocarbam Siderophore Conjugate MC-1 and its in Vitro Activity.
The monocarbams are monocyclic β-lactam antibacterial agents that have a carbonylaminosulfonyl activating group at the N-1 position. Significant efforts have been devoted to the modification of β-lactams by incorporating a siderophore moiety to promote uptake by Gram-negative bacteria by exploiting the siderophore active transport system (21, 22). MC-1 is a siderophore-conjugated monocarbam with a carbonylaminosulfonyl activating group at N-1 position and a hydroxypyridone siderophore connected by a triazolone-derived linker (Fig. 1B). Compared to aztreonam, MC-1 was found to have exquisite potency against P. aeruginosa clinical isolates with a MIC90 of 0.5 μg/mL (Table 1).
In the cocrystal structure of PaPBP3 with MC-1 at 1.64-Å resolution, the hydrophobic pillar composed of Val333 and Phe533 bisects the active site cleft opening with the iminopropyl carboxyl moiety on one side and the hydroxypyridone with triazolone linker on the opposite side (Fig. 3 B and C). The bulky aminothiazole-containing moiety displaces the Tyr409 side chain and induces similar conformational changes as seen in the ceftazidime- and aztreonam-bound structures. The gem-dimethyl group is stabilized by a hydrophobic interaction with residue Val333 and the aromatic wall composed of Tyr503, Tyr532, and Phe533. The carboxylic acid group forms salt bridges with Arg489. Notably, the triazolone linker adopts two alternative conformations in a perpendicular orientation, which allows the triazolone carbonyl group to interact either with Gly535 backbone or with Leu346 and Lys484 via a water molecule. The hydroxypyridone siderophore is oriented toward the solvent-exposed surface and involved in van der Waals interaction with Val333 and Phe533.
Unlike the sulfonic acid group directly attached to the N-1 position of the β-lactam ring in aztreonam, the MC-1 has a longer carbonylaminosulfonyl species for activation. The additional carbonylamino moiety pushes the sulfonyl group outside the active site by 1.3 Å. As a result, the sulfonyl group in the MC-1 acyl-enzyme structure forms two hydrogen bonds with backbone amides of Gly534 and Gly535, effectively stabilizing a helix dipole at the N-terminal end of the α11 helix. Interestingly, the α11 helix contains a proline residue (Pro540) that kinks the N-terminal end of helix α11 toward the active site and helps the Phe533-containing loop forming one side of active site cleft. Mutation of the equivalent Pro540 to serine in N. gonorrhoeae PBP2 leads to a decrease in acylation rate despite no discernible effect on the structure of the α11 helix (8). Importantly, in the MC-1 acyl-enzyme, the additional carbonylaminosulfonyl moiety forms an extensive hydrogen-bond network with conserved residues. The Ser349 of the SXN motif forms a hydrogen bond with the carbonyl oxygen of sulfonyl cabonylamino moiety, whereas Thr487, the fourth residue of the conserved KT/SGS/T motif, forms bidentate hydrogen bonds with the nitrogen atom and the other sulfonyl oxygen atom.
Solvent Rearrangement Effects and Second-Order Acylation Rate Constants.
A detailed molecular understanding of interactions between proteins and ligands involves many factors (23). Barriers to ligand binding and dissociation can arise from solvent rearrangement effects that can be rate determining when solvent transfer to and from the binding site is critical (24). A consequence of this for covalent inhibitors is that the second-order acylation rate constants, which are indicative of the efficiency of the ligand to inactivate the target, may be predicted by the protein desolvation effects that occur during ligand binding. This assumes that the energetics of covalent bond formation and protein conformational effects do not significantly change the rank ordering of the ligands.
A computational approach, called WaterMap, computes the thermodynamic profile and free energies of hydration sites in a protein active site relative to bulk solvent and has been successfully applied in other systems such as peptides binding to PDZ domains, kinase selectivity, and ranking of congeneric molecules binding to factor Xa (25–28). The approximation of free energy is based exclusively on the displacement of water molecules within the site upon binding of the ligand and ignores other terms such as protein–ligand van der Waals contacts, electrostatic interactions, internal strain (ligand and protein), protein conformational effects, and entropy changes. WaterMap energies also allow the categorization and classification of the computed hydration sites into stable and unstable waters based on the thermodynamic profile of each computed water cluster. As ligand binding involves the exchange of protein–water for protein-ligand contacts, the dissection of the thermodynamics of hydration sites could provide insights into ligand binding and thereby the efficiency of PBP3 inactivation by siderophore-conjugated monocarbams (24).
Hydration sites were categorized into two different types of waters—stable (ΔG < 0 kcal/mol) and unstable (ΔG > 0 kcal/mol). Protein desolvation energies for each type of water site were calculated for each ligand based on the respective crystal structures (Table 1). A normalization process, in which the ligand-affected total free energy of hydration sites ( ) was divided by the number of ligand-affected hydration sites (
) was divided by the number of ligand-affected hydration sites ( ), standardizes this measure for any size effect of the ligand. This yields two protein desolvation efficiency indices: an unstable water displacement efficiency index
), standardizes this measure for any size effect of the ligand. This yields two protein desolvation efficiency indices: an unstable water displacement efficiency index  and a stable water displacement efficiency index
and a stable water displacement efficiency index  . A composite water displacement effectiveness score (ϵi) is obtained by taking the ratio of
. A composite water displacement effectiveness score (ϵi) is obtained by taking the ratio of  over
over  . Both imipenem and meropenem binding do not involve induced conformational changes of Tyr409 relative to the apo form and displace stable hydration sites (ΔG < 0) to varying degrees. Meropenem displaces 10 stable hydration sites, whereas imipenem displaces only 2 stable hydration sites. Both these compounds displace similar numbers of unstable hydration sites (ΔG > 0). In addition to the solvent rearrangement effects, imipenem and meropenem induce significant protein conformational changes in Tyr532 and Phe533 that were observed in the covalent-adduct formation (vide supra). In contrast, ceftazidime, MC-1, and aztreonam displace a significant number of unstable hydration sites (Fig. 4) and their unstable water displacement efficiency index ranges from 0.90 to 1.27 compared to 0.59 to 0.67 for imipenem and meropenem. MC-1 and ceftazidime effectively displace a greater number of unstable hydration sites than other β-lactams, whereas aztreonam and MC-1 effectively displace a greater number of stable hydration sites. A plot between log(k2/Kd) and (ϵi) indicates a reasonable trend between second-order acylation constants and water displacement effectiveness scores (Table 1 and Fig. S3).
. Both imipenem and meropenem binding do not involve induced conformational changes of Tyr409 relative to the apo form and displace stable hydration sites (ΔG < 0) to varying degrees. Meropenem displaces 10 stable hydration sites, whereas imipenem displaces only 2 stable hydration sites. Both these compounds displace similar numbers of unstable hydration sites (ΔG > 0). In addition to the solvent rearrangement effects, imipenem and meropenem induce significant protein conformational changes in Tyr532 and Phe533 that were observed in the covalent-adduct formation (vide supra). In contrast, ceftazidime, MC-1, and aztreonam displace a significant number of unstable hydration sites (Fig. 4) and their unstable water displacement efficiency index ranges from 0.90 to 1.27 compared to 0.59 to 0.67 for imipenem and meropenem. MC-1 and ceftazidime effectively displace a greater number of unstable hydration sites than other β-lactams, whereas aztreonam and MC-1 effectively displace a greater number of stable hydration sites. A plot between log(k2/Kd) and (ϵi) indicates a reasonable trend between second-order acylation constants and water displacement effectiveness scores (Table 1 and Fig. S3).
Fig. 4.

WaterMap results of P. aeruginosa PBP3 in complex with MC-1 crystal structure. Hydration sites are depicted as spheres colored by ΔG. Stable waters, ΔG < 0, are blue and unstable waters, ΔG > 0, are red in color. MC-1 is shown in stick representation. Representative water sites are exemplified.
For a simple one-step binding process where covalent bond formation is rapid, the rate-determining step is the formation of the encounter complex: a complicated process involving protein–ligand recognition, protein conformational changes, and desolvation effects. Depending on the compound and the binding event, the slowest, rate-determining step can be defined by one of the many effects indicated above. It is interesting to note that despite the diversity of scaffolds involved in the core β-lactam of these five ligands and the various protein conformational changes observed, the composite water displacement effectiveness score (ϵi) rank orders the five ligands according to their second-order acylation constants (k2/Kd). This composite score balances two types of desolvation energies—unstable water displacement efficiency index  , which provides an indication of the boost that is obtained by displacing loosely held waters that need not be a specific recognition element, and the stable water displacement efficiency index
, which provides an indication of the boost that is obtained by displacing loosely held waters that need not be a specific recognition element, and the stable water displacement efficiency index  that captures specific pharmacophoric recognition elements by displacing tightly held waters. The index (ϵi) appears to provide a measure of how effectively the ligand binds in the initial interaction complex formed before the covalent bond formation, which is exclusively based on protein desolvation energies.
that captures specific pharmacophoric recognition elements by displacing tightly held waters. The index (ϵi) appears to provide a measure of how effectively the ligand binds in the initial interaction complex formed before the covalent bond formation, which is exclusively based on protein desolvation energies.
Compounds that have more positive  tend to stabilize the covalent–adduct complex, as measured by thermal stabilization assay (Table 1), whereas compounds that do not stabilize the covalent-adduct complex relative to the apo form have lower
tend to stabilize the covalent–adduct complex, as measured by thermal stabilization assay (Table 1), whereas compounds that do not stabilize the covalent-adduct complex relative to the apo form have lower  . On the other hand, the stable waters are critical for appropriate recognition of key functionalities in the protein. Hence the displacement of these waters with polar functional groups is critical for both recognition and biological activity values. Compounds that have good submicromolar EC50 values all appear to be replacing stable waters to a significant extent and have more negative
. On the other hand, the stable waters are critical for appropriate recognition of key functionalities in the protein. Hence the displacement of these waters with polar functional groups is critical for both recognition and biological activity values. Compounds that have good submicromolar EC50 values all appear to be replacing stable waters to a significant extent and have more negative  . Despite its small size, aztreonam binds efficiently by replacing most of the stable waters it is able to access with polar functionalities and has the most negative
. Despite its small size, aztreonam binds efficiently by replacing most of the stable waters it is able to access with polar functionalities and has the most negative  and the most potent EC50 value. Significant thermal stabilization is also gained from the covalent-adduct formation (in addition to the solvent rearrangement effects), and the thermodynamics of covalent bond formation are not discussed here.
and the most potent EC50 value. Significant thermal stabilization is also gained from the covalent-adduct formation (in addition to the solvent rearrangement effects), and the thermodynamics of covalent bond formation are not discussed here.
In conclusion, the unique conformation of PaPBP3 with β-lactam inhibitors represents a distinct high-molecular-weight class B PBP subfamily identifiable by the common features of the aromatic wall formed by Tyr503, Tyr532, and Phe533 and the structural flexibility of Tyr409 flanked by two glycine residues. The guanidium group of Arg 489 shows a specific interaction with the acid moiety of the inhibitors. This is the first structure of a siderophore-conjugated triazolone-linked monocarbam complexed with PBP and provides molecular insights into the efficient interaction of siderophore-conjugated monocarbams with the important therapeutic target PBP3. The analysis of energetics and rearrangement of waters suggests that protein desolvation effects contribute to PBP3 binding, and the water displacement effectiveness score (ϵi) is indicative of the second-order acylation constants. Our structural, biochemical, and computational studies provide a molecular basis for understanding the coupled activity and recognition specificity for P. aeruginosa PBP3 and for design of inhibitors of this class of compounds.
Experimental Procedures.
Complete details about the experimental procedures can be found in SI Text.
Ninety Percent of the Minimal Inhibitory Concentration (MIC90).
One hundred six recent P. aeruginosa clinical isolates were assayed using broth microdilution, according to the Clinical and Laboratory Standards Institute guidelines for antimicrobial susceptibility testing.
P. aeruginosa PBP3 Competition of Binding Assay.
Two hundred nanograms of purified PaPBP3 was assayed against various concentrations of β-lactam and 0.65 μM Bocillin FL (at its apparent Km value) at the same time (no preincubation), which allows for simple competitive binding to the PaPBP3.
Rapid Quench Flow Experiment.
Acylation rates of PaPBP3 and β-lactam antibiotics were determined using a RQF-3 Rapid Quench Flow instrument (Kintek), as discussed in detail previously (29).
Protein Cloning, Expression, and Purification.
A soluble form of PaPBP3 construct (residues 50–579) appended with an N-terminal His6 tag was cloned into pET28 (Novagen) and overexpressed in E. coli BL21(Gold) in Novagen’s autoinduction medium for 24 h at 25 °C. Purified PaPBP3 was concentrated to 10 mg/mL for crystallization trials.
Crystallization.
Crystals of apo-PaPBP3 were obtained with a reservoir solution containing 30% PEG 4000, 0.2 M MgCl2, and 0.1 M Tris pH 8.5. Each compound (ceftazidime, imipenem, meropenem, and aztreonam) was soaked into the crystals. Cocrystallization of PaPBP3 with MC-1 was achieved with well solutions containing 0.1 M citrate; pH 6.2, 30% PEG 3350, and 0.3 M (MgNO3)2. Crystals were cryoprotected by dragging the crystals through MiTeGen’s LV CryoOil™ (MiTeGen, LLC) and flash frozen in liquid nitrogen.
Structure Determination.
Diffraction data were collected from flash-frozen crystals at 100 K at the Advanced Photon Source of the Argonne National Laboratory on a ADSC Quantum 210 CCD detector. Data were processed using the HKL2000 suite of software (30). Data collection statistics are summarized in Table S1. The structure of apo-PaPBP3 was solved by molecular replacement methods with the CCP4 version of PHASER (31), using Neisseria gonorrhoeae PBP2 [Protein Data Bank (PDB) ID code 3EQU] as a search model. After molecular replacement, maximum likelihood-based refinement of the atomic position and temperature factors were performed with REFMAC (32) and autoBUSTER (33), and the atomic model was built with the program COOT (34). The refined PaPBP3-apo structure was then used as a starting model for all other complexes. The stereochemical quality of the final model was assessed by PROCHECK (35). Crystallographic statistics for the final models are shown in Table 1. Figures were prepared with PYMOL (36).
Thermal Stability Assay.
The thermal shift assay was conducted in the iCycler iQ Real Time Detection System (Bio-Rad), originally designed for PCR. The fluorescence intensity was measured with Ex/Em: 490/530 nm.
WaterMap Calculations of PBPs.
WaterMap calculations were performed on the five ligand-bound crystal structures (aztreonam, ceftazidime, meropenem, imipenem, and MC-1) and the apo PaPBP3 crystal structure.
Coordinates.
Coordinates for the PaPBP3 and inhibitor structures have been deposited with the Protein Data Bank.
Supplementary Material
Acknowledgments.
We thank Lisa Mullins for determining MIC90 and John Mueller, Mark Plummer, Mark Mitton-Fry, and Xiayang Qiu for insightful discussions.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
Data deposition: The crystallography, atomic coordinates, and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 3PBN, 3PBO, 3PBQ, 3PBR, 3PBS, and 3PBT).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1013092107/-/DCSupplemental.
References
- 1.Giske CG, Monnet DL, Cars O, Carmeli Y. Clinical and economic impact of common multidrug-resistant gram-negative bacilli. Antimicrob Agents Chemother. 2008;52:813–821. doi: 10.1128/AAC.01169-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boucher HW, et al. Bad bugs, no drugs: No ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis. 2009;48:1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 3.Giamarellou H, Kanellakopoulou K. Current therapies for pseudomonas aeruginosa. Crit Care Clin. 2008;24:261–278. doi: 10.1016/j.ccc.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 4.Siegel RE. Emerging gram-negative antibiotic resistance: Daunting challenges, declining sensitivities, and dire consequences. Respir Care. 2008;53:471–479. [PubMed] [Google Scholar]
- 5.Liao X, Hancock RE. Susceptibility to beta-lactam antibiotics of Pseudomonas aeruginosa overproducing penicillin-binding protein 3. Antimicrob Agents Chemother. 1997;41:1158–1161. doi: 10.1128/aac.41.5.1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spratt BG, Cromie KD. Penicillin-binding proteins of gram-negative bacteria. Rev Infect Dis. 1988;10:699–711. doi: 10.1093/clinids/10.4.699. [DOI] [PubMed] [Google Scholar]
- 7.Denome SA, Elf PK, Henderson TA, Nelson DE, Young KD. Escherichia coli mutants lacking all possible combinations of eight penicillin binding proteins: Viability, characteristics, and implications for peptidoglycan synthesis. J Bacteriol. 1999;181:3981–3993. doi: 10.1128/jb.181.13.3981-3993.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Powell AJ, Tomberg J, Deacon AM, Nicholas RA, Davies C. Crystal structures of penicillin-binding protein 2 from penicillin-susceptible and -resistant strains of Neisseria gonorrhoeae reveal an unexpectedly subtle mechanism for antibiotic resistance. J Biol Chem. 2009;284:1202–1212. doi: 10.1074/jbc.M805761200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Page MG, Dantier C, Desarbre E. In vitro properties of BAL30072, a novel siderophore sulfactam with activity against multiresistant gram-negative bacilli. Antimicrob Agents Chemother. 2010;54:2291–2302. doi: 10.1128/AAC.01525-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zurenko GE, Truesdell SE, Yagi BH, Mourey RJ, Laborde AL. In vitro antibacterial activity and interactions with beta-lactamases and penicillin-binding proteins of the new monocarbam antibiotic U-78608. Antimicrob Agents Chemother. 1990;34:884–888. doi: 10.1128/aac.34.5.884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goffin C, Ghuysen JM. Multimodular penicillin-binding proteins: An enigmatic family of orthologs and paralogs. Microbiol Mol Biol Rev. 1998;62:1079–1093. doi: 10.1128/mmbr.62.4.1079-1093.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sauvage E, Kerff F, Terrak M, Ayala JA, Charlier P. The penicillin-binding proteins: Structure and role in peptidoglycan biosynthesis. FEMS Microbiol Rev. 2008;32:234–258. doi: 10.1111/j.1574-6976.2008.00105.x. [DOI] [PubMed] [Google Scholar]
- 13.Faraci WS, Pratt RF. Interactions of cephalosporins with the Streptomyces R61 DD-transpeptidase/carboxypeptidase. Influence of the 3′-substituent. Biochem J. 1986;238:309–312. doi: 10.1042/bj2380309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wientjes FB, Nanninga N. On the role of the high molecular weight penicillin-binding proteins in the cell cycle of Escherichia coli. Res Microbiol. 1991;142:333–344. doi: 10.1016/0923-2508(91)90049-g. [DOI] [PubMed] [Google Scholar]
- 15.Powers RA, Caselli E, Focia PJ, Prati F, Shoichet BK. Structures of ceftazidime and its transition-state analogue in complex with AmpC beta-lactamase: Implications for resistance mutations and inhibitor design. Biochemistry. 2001;40:9207–9214. doi: 10.1021/bi0109358. [DOI] [PubMed] [Google Scholar]
- 16.Kuzin AP, Liu H, Kelly JA, Knox JR. Binding of cephalothin and cefotaxime to D-ala-D-ala-peptidase reveals a functional basis of a natural mutation in a low-affinity penicillin-binding protein and in extended-spectrum beta-lactamases. Biochemistry. 1995;34:9532–9540. doi: 10.1021/bi00029a030. [DOI] [PubMed] [Google Scholar]
- 17.Lee VJ, Miller GH, Yagisawa M. What’s new in the antibiotic pipeline. Curr Opin Microbiol. 1999;2:475–482. doi: 10.1016/s1369-5274(99)00003-x. [DOI] [PubMed] [Google Scholar]
- 18.Quinn JP. Clinical strategies for serious infection: A North American perspective. Diagn Micr Infec Dis. 1998;31:389–395. doi: 10.1016/s0732-8893(98)00023-6. [DOI] [PubMed] [Google Scholar]
- 19.Sykes RB, Bonner DP, Bush K, Georgopapadakou NH. Azthreonam (SQ 26,776), a synthetic monobactam specifically active against aerobic Gram-negative bacteria. Antimicrob Agents Chemother. 1982;21:85–92. doi: 10.1128/aac.21.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bonner DP, Sykes RB. Structure activity relationships among the monobactams. J Antimicrob Chemother. 1984;14:313–327. doi: 10.1093/jac/14.4.313. [DOI] [PubMed] [Google Scholar]
- 21.Miller MJ. Syntheses and therapeutic potential of hydroxamic acid based siderophores and analogues. Chem Rev. 1989;89:1563–1579. [Google Scholar]
- 22.Mollmann U, Heinisch L, Bauernfeind A, Kohler T, Ankel-Fuchs D. Siderophores as drug delivery agents: Application of the “Trojan Horse” strategy. Biometals. 2009;22:615–624. doi: 10.1007/s10534-009-9219-2. [DOI] [PubMed] [Google Scholar]
- 23.Mobley DL, Dill KA. Binding of small-molecule ligands to proteins: “What you see” is not always “what you get”. Structure. 2009;17:489–498. doi: 10.1016/j.str.2009.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pearlstein RA, et al. New hypotheses about the structure-function of proprotein convertase subtilisin/kexin type 9: Analysis of the epidermal growth factor-like repeat A docking site using WaterMap. Proteins. 2010;78:2571–2586. doi: 10.1002/prot.22767. [DOI] [PubMed] [Google Scholar]
- 25.Young T, Abel R, Kim B, Berne BJ, Friesner RA. Motifs for molecular recognition exploiting hydrophobic enclosure in protein-ligand binding. Proc Natl Acad Sci USA. 2007;104:808–813. doi: 10.1073/pnas.0610202104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abel R, Young T, Farid R, Berne BJ, Friesner RA. Role of the active-site solvent in the thermodynamics of factor Xa ligand binding. J Am Chem Soc. 2008;130:2817–2831. doi: 10.1021/ja0771033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beuming T, Farid R, Sherman W. High-energy water sites determine peptide binding affinity and specificity of PDZ domains. Protein Sci. 2009;18:1609–1619. doi: 10.1002/pro.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Robinson DD, Sherman W, Farid R. Understanding kinase selectivity through energetic analysis of binding site waters. ChemMedChem. 2010;5:618–627. doi: 10.1002/cmdc.200900501. [DOI] [PubMed] [Google Scholar]
- 29.Lu WP, Kincaid E, Sun Y, Bauer MD. Kinetics of beta-lactam interactions with penicillin-susceptible and -resistant penicillin-binding protein 2x proteins from Streptococcus pneumoniae. Involvement of acylation and deacylation in beta-lactam resistance. J Biol Chem. 2001;276:31494–31501. doi: 10.1074/jbc.M102499200. [DOI] [PubMed] [Google Scholar]
- 30.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 31.McCoy AJ, et al. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 33.Blanc E, et al. Refinement of severely incomplete structures with maximum likelihood in BUSTER-TNT. Acta Crystallogr D. 2004;60:2210–2221. doi: 10.1107/S0907444904016427. [DOI] [PubMed] [Google Scholar]
- 34.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 35.Laskowski RA, Moss DS, Thornton JM. Main-chain bond lengths and bond angles in protein structures. J Mol Biol. 1993;231:1049–1067. doi: 10.1006/jmbi.1993.1351. [DOI] [PubMed] [Google Scholar]
- 36.Delano WL. The PyMOL Molecular Graphics System. Palo Alto, CA: Delano Scientific LLC; 2002. www.pymol.org. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.