

Abstract
An efficient pyrene-assisted method has been developed for the photolysis of disulfide bonds, with 77% of disulfides cleaved after only 20 minutes of irradiation (0.3W) at 350nm. By employing a DNA framework, it was possible to observe both a distance-dependent cleavage pathway and a radical-forming photoreaction mechanism. To demonstrate the biomedical applications of such pyrene disulfide molecular assemblies, a DNA micelle structure and DNAzyme analog were further studied. Rapid photo-driven disassembly of DNA micelles was achieved, allowing the further design of controlled pharmaceutical release at the target region and at a specific time. The DNAzyme analog can carry out multiple turnover reactions that follow the Michaelis-Menton equation, with a kcat of 10.2 min-1 and a KM of 46.3μM (0.3W 350nm light source), comparable to that of common DNAzymes, e.g., 8-17 DNAzyme.
Keywords: disulfide bond photolysis, pyrene, DNA micelle, DNAzyme analog
INTRODUCTION
In the late 1950s, Walling and Rabinowitz (1) demonstrated that the homolysis of alkyl disulfides could be sensitized by aromatic hydrocarbons. To date, there have been few applications of this cleavage because of the relatively low cleavage efficiency (<30% after hours of irradiation), the need for UV irradiation, and the aqueous insolubility of aromatic hydrocarbons. However, disulfide linkages play an important role in the folding/unfolding structure switch of proteins (2), and chemical cleavage of the disulfide bonds can result in the inactivation of sulfur-containing proteins (3). At the same time, extensive efforts have been devoted to disulfide-based carrier systems (4) because disulfide bonds provide temporary high stability in the extracellular compartment, followed by relatively rapid cleavage inside cells, facilitating the release of therapeutic drugs or nucleic acids. However, the reduction of cellular disulfides is highly dependent on the expression level and location of redox enzymes (4a). Thus, some extraneous stimulus, such as heat, chemical reaction or irradiation, is needed. It is conceivable that when the cleavage is triggered by light, precise control of initiation and termination is possible, in preference to using an additional reducing reagent, such as dithiothreitol (DTT), which could be toxic.
In this work, we report an efficient pyrene-assisted disulfide photolysis strategy using 350nm light. Pyrene is a common, spatially sensitive fluorescent dye (5). Its high extinction coefficients (~2×104 M-1cm-1 at 350nm) and long-lived (~110ns), high-energy (~70kcal/mol) excited singlet state make pyrene useful for driving numerous photochemical reactions (6).
EXPERIMENTAL SECTION
Chemicals and Reagents
The materials for DNA synthesis, including C-6 disulfide phosphoramidite, 6-fluorescein (FAM) phosphoramidite, 5’-4-(4-dimethylaminophenylazo) benzoic acid (Dabcyl) phosphoramidite and CPG columns, were purchased from Glen Research (Sterling, VA). Unless otherwise stated, all the chemicals were used without further purification. The syntheses of pyrene phosphoramidite and lipid phosphoramidite were reported previously (5a, 11a). All reagents for buffer preparation and HPLC purification came from Fisher Scientific.
DNA Synthesis
All of the oligonucleotides were synthesized using an ABI 3400 DNA synthesizer (Applied Biosystems, Inc. Foster City, CA) at 1.0 micromolar scale. After complete cleavage and deprotection, most DNA sequences were purified on a ProStar HPLC system (Varian, Palo Alto, CA) with a C-18 reversed-phase column (Alltech, 5μm, 250mm × 4.6 mm). The DNA with 5’-end lipid modification was purified using a C-4 column (BioBasic-4, 200mm × 4.6mm, Thermo Scientific). In both cases, the eluent was 100mM triethylamine-acetic acid buffer (TEAA, pH 7.5) and acetonitrile (0-30min, 10-100%). All DNA concentrations were characterized with a Cary Bio-300UV spectrometer (Varian) using the absorbance of DNA at 260nm.
Synthesis of disulfide bridged pyrene derivative, PnSS
The conjugation of pyrene and the disulfide bond was realized by the amide bond formation via the reaction between 1-aminopyrene and 3, 3’-dithiodipropanoyl chloride. The product was characterized by 1H-NMR (CDCl3): δ 8.28-8.22 (m, 8H), δ 8.10-8.04 (m, 10H), δ 3.24 (s, 4H), δ 3.08 (d, 4H).
UV Irradiation
For the photoregulation of the pyrene-disulfide molecular assembly, all samples were irradiated in de-ionized water at 350 nm with a UV-B lamp centered at 302 nm (SANKYO DENKI, Japan) with a 352 nm optical filter (3 nm half bandwidth; Oriel Instruments, Stratford, CT, Newport).
Gel Electrophoresis and Data Analysis
Polyacrylamide gel electrophoresis (PAGE) was performed on a 20% denatured gel in TBE buffer (89mM Tris-HCl, 89mM boric acid, 2mM EDTA, pH 8.0) for 45min at 150V. After that, gels were stained using Stains-All (Sigma-Aldrich) to image the position of DNA. Photographic images were obtained under visible light with a digital camera and were then quantitatively analyzed with Image J software from NIH. Origin 8.0 was used for data analysis.
Micelle Characterization
A FluoroMax-4 Spectrofluorometer with a temperature controller (Jobin Yvon) was used for all steady-state fluorescence measurements. Fluorescence intensity was recorded using excitation at 350nm (spectral bandwidth=3nm). Agarose gel electrophoresis was performed using a 4% agarose gel in TBE buffer with constant 75V for about 90 min. The DNA bands were visualized by UV illumination (312nm) and photographed by a digital camera.
Gel-Based DNAzyme-Mimic Activity Assay
After a series of 350nm irradiation times, fluorescence intensities at 515nm were recorded using a FluoroMax-4 Spectrofluorometer with excitation at 488 nm. Then a 12% Native gel in TBE buffer was applied for 50min at 100V. The fluorescence images of gels were recorded by FAM fluorescence, with excitation at 488nm and emission at 526nm using a Typhoon 9410 variable mode imager.
RESULTS AND DISCUSSION
The irradiation of disulfide bridged pyrene derivative, PnSS, at 350nm (λmax of the pyrene ring) results in highly efficient disulfide bond cleavage, as demonstrated in ESI-MS experiments (Figure 1a). Photocleavage of disulfide bonds results in the intensity changes in two peaks: m/z=631 (pyrene derivative +Na+) decreases while m/z=305 (photolysis product) increases. For further studies of pyrene-assisted disulfide photolysis, the S-S linkage and pyrene were incorporated into a DNA framework, which also provided feasible detection using gel electrophoresis and fluorescence analysis. Other advantages of using DNA can also be identified: (a) DNA base stacking allows efficient long-range charge transfer, which has been reported to facilitate electron transfer in donor-acceptor systems (7); (b) the sequence structure can be modified to place the disulfide and pyrene at specific sites; and (c) oligonucleotides can be routinely synthesized by solid-phase synthesis using the phosphoramidite method.
FIGURE 1.
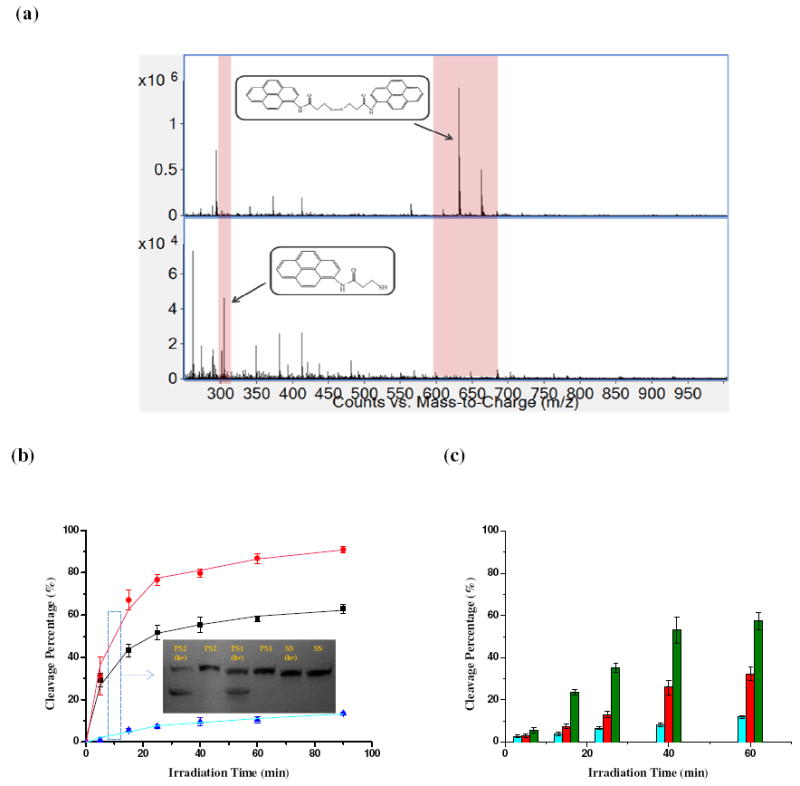
(a) Mass spectra of disulfide-bridged pyrene derivative PnSS before (top) and after (bottom) irradiation; a 10 μM sample was irradiated for 30 minutes at 350nm. (b)(c) Cleavage efficiency for 10μM PS2, PS1 and SS as a function of UV irradiation time. Denatured PAGE gel image was obtained with a digital camera for (b) PS2 (red), PS1 (black) and SS (blue) (c) P1S (green), P3S (red) and P5S (blue) after 5, 15, 25, 40 and 60 min of irradiation. Data were analyzed with Image J software from NIH.
Two groups of DNA molecules with pyrene and disulfide bridge assemblies were synthesized, and gel analysis was employed to quantify the cleavage efficiency. The base sequences are shown in Table 1. The first group, termed as PS2 and PS1, linked, respectively, double and single pyrene moieties to the disulfide bond. The same base sequence with no pyrene moiety, SS, functioned as a control molecule.
Table 1.
Sequences of DNA used in the paper a
| Name | Sequence |
|---|---|
| PS1 | 5’-AAAAAAAATC-/Pyr/- SS- A bCAGATGAGT-3’ |
| PS2 | 5’-AAAAAAAATC-/Pyr/-SS-/Pyr/-ACAGATGAGT-3’ |
| SS | 5’-AAAAAAAATC-SS-ACAGATGAGT-3’ |
| P1S | 5’-AAAAAAAAT-/Pyr/-C-SS-ACAGATGAGT-3’ |
| P3S | 5’-AAAAAAA-/Pyr/- ATC-SS-ACAGATGAGT-3’ |
| P5S | 5’-AAAAA-/Pyr/- AAATC-SS-ACAGATGAGT-3’ |
| P20 | 5’-/Pyr/-AAAAAAAATCACAGATGAGT-3’ |
| LP-20 | 5’-Lip-/Pyr/-AAAAAAAATCACAGATGAGT-3’ |
| LSP-20 | 5’-Lip-SS-/Pyr/-AAAAAAAATCACAGATGAGT-3’ |
Pyr, SS and Lip stand for the pyrene, disulfide and lipid modified nucleotide, respectively.
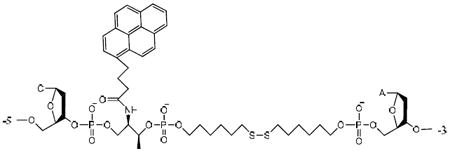
The actual structure of “/Pyr/- SS” molecular assembly in PS1 is:
The difference in cleavage efficiency is obvious when the gel results are compared (Figure 1b). The photocleavage of PS1 and PS2 was found to be more rapid, and had better yields, than that of SS. The yields of PS1 and PS2 photocleavage products increased rapidly with time, and after only 20 minutes (0.3W) the yields for PS1 and PS2 were 52% and 77%, respectively. The yield of SS products increased slowly, less than 10%, even after irradiation for 1h. The weakened photocleavage activity for PS1 and PS2 after 20 min perhaps stemmed from the equilibrium between the two disulfide molecular states, the oxidized form (S-S) and the reduced form (-SH).
To study the effect of distance between the pyrene moiety and the S-S link, a second group of DNA molecules, termed as P1S, P3S and P5S, was prepared with the same base sequence as group 1, but with the pyrene group separated from the S-S by 1, 3 or 5 nucleotides (Table 1). Under 350nm light irradiation, the photocleavage efficiency decreased in accordance with the extended distance from the pyrene moiety: 53%, 26% and 8.3% after 40 minutes (0.3W), respectively, for P1S, P3S and P5S. Such distance-dependent cleavage may potentially be useful for controllable photolysis of specific disulfide bonds, especially considering the sharp spatial control achievable by irradiation. Taking all these findings together, it may be concluded that the photolysis of disulfide bonds can be facilitated by nearby pyrene moieties in DNA frameworks.
Such efficient disulfide photocleavage at 350nm, to the best of our knowledge, has never been reported. We asked what properties of pyrene would account for this effect. The mechanism of disulfide cleavage in proteins via UV irradiation at 280nm (λmax of the tryptophan group) was investigated by Hayon and co-workers (8). The disulfide radical anion and tryptophan radical cation were believed to form by the electron transfer. It has also been widely accepted that pyrene-mediated photo-induced electrons can be injected into the DNA base stack (9), thus generating large amounts of pyrene radical cations. A possible mechanism is:
| (1) |
| (2) |
Because pyrene has a relatively high extinction coefficient (ε~2×104 M-1cm-1) (5b), a low concentration of pyrene can capture a significant fraction of the incident light. However, this property is not sufficient by itself. Two other dyes with relatively high extinction coefficients, Cy5 (cyanine 5, ε~2.5×105 M-1cm-1) and FAM (5-carboxyfluorescein, ε~7.6×104 M-1cm-1) were studied, and no obvious cleavage was detected with irradiation, either at the λmax of dyes (488nm for Cy5, 650nm for FAM) or at 350nm. To verify the possible radical cleavage mechanism, p-benzoquinone, a radical scavenger substrate, was added to capture any radicals formed. No photolysis of the disulfide bridge occurred, as indicated by the cleavage band having disappeared in gel analysis (Supporting Information Figure S1), thus proving that blocking the radical pathway prohibited the disulfide bond photocleavage. To further confirm this radical pathway, the radical indicator TEMPO-9-AC was employed. This compound fluoresces weakly at ~430nm in the absence of radicals (FI~8×105 in Supporting Information, Figure S2), but the fluorescence intensity increased to ~ 1.1×106 when SS was present and to ~1.4×106 in the presence of PS1. These results are consistent with a radical pathway for S-S cleavage, which is further enhanced in the presence of pyrene.
Micelle structures are of particular interest for biomedical applications (10), because of their biocompatibility, stability both in vitro and in vivo, and their ability to carry poorly soluble pharmaceuticals into intracellular areas. Our group has been investigating DNA-based micelle structures (11) composed of a single-stranded DNA corona and a hydrophobic diacyllipid core, which can be rapidly dissociated by UV/Vis irradiation. It is conceivable that photoinduced release of compounds carried in the micelle will allow precise spatial and temporal control of drug delivery. To achieve this goal, we have designed a pyrene-disulfide bridge assembly to separate the hydrophobic core-forming lipid block from the hydrophilic DNA block (Figure 2a). Upon irradiation at 350nm, disulfide bridge cleavage detaches the DNA from the lipid, leading to dissociation of the micelles. Here, the pyrene unit also acts as a fluorescence reporter; its unique excimer signal has been widely used to probe micelle formation and dissociation (12). After excitation at 350nm for 1 minute (band width=3nm), the broad emission of the pyrene excimer at 480nm shows strong π-π interactions of the pyrene chromophores inside the micelle systems, indicating the well-organized state of the assembled micelles (Figure 2b). After scanning the emission spectrum 6 times from 370nm to 600nm (λ ex=350nm), the excimer band at ~475nm disappeared, indicating that the micelles had completely dissociated. To provide direct evidence of micelle disaggregation, agarose gel electrophoresis experiments were conducted. (Supporting Information, Figure S3) Before irradiation, the slow-moving bands with green fluorescence (pyrene excimer) indicated the presence of only aggregated pyrene (micelles). After irradiation, the intensity of the green band decreased, but there was an additional violet, fast-moving band indicative of monomeric pyrene (dissociated micelles).
FIGURE 2.
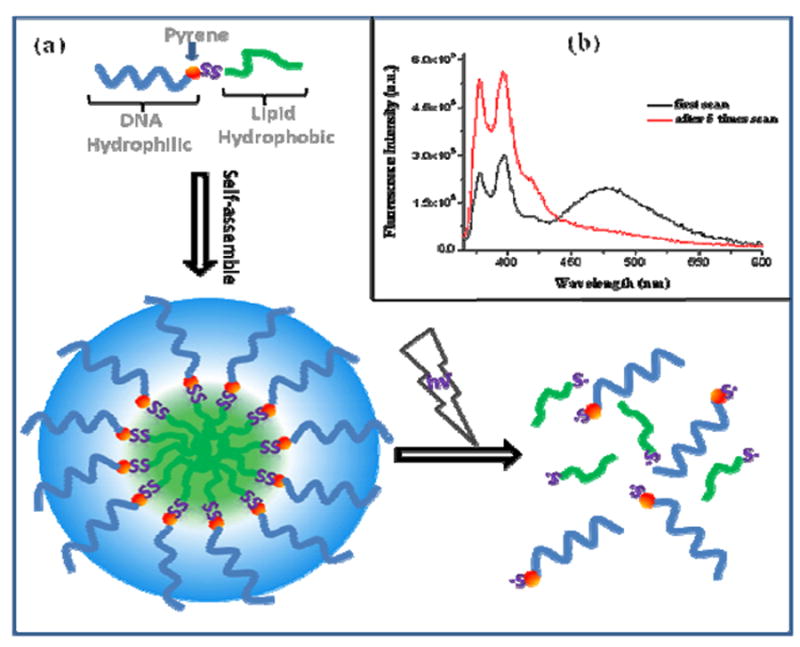
(a) The structure and photo-dissociation of the pyrene-disulfide modified DNA micelle. (b) Fluorescence spectra of a DNA micelle molecular assembly immediately after 350nm excitation for one minute (black line), and after 5 scans (red line) with λex=350nm.
DNAs having catalytic function, called deoxyribozymes or DNAzymes, have become important tools for numerous biochemical applications (13). The catalytic efficiency of DNAzymes depends on the existence of cofactors and specific cleavage sites. A photomodulated DNAzyme analog employing pyrene and disulfide bonds as cofactors and cleavage sites, respectively, was designed and tested (Figure 3). The pyrene DNAzyme analog catalyzed the cleavage of the complementary DNA substrate through the photo-induced breakage of active disulfide linkage. The relatively slow diffusion based DNA duplex formation, and subsequently rapid light-induced radical formation and cleavage formed the basis for the DNAzyme analog (14). In this application, light provides an effective way of intentionally inducing enzyme reactivity for studying complex biological processes. As shown in Figure 4a, the initial rate increased with increasing concentration of DNAzyme analog. Multiple-turnover reactions were also observed (Supporting Information Figure S4), and the data for the initial cleavage rate were fit to a Lineweaver - Burk plot (Figure 4b). Under these conditions, the calculated values of KM and kcat were 46.3μM and 10.2 min-1, respectively. The catalytic efficiency (kcat /KM=0.22 min-1·μM-1) of this photo-modulated DNAzyme analog was comparable to that for common DNAzymes (e.g., 1.1 min-1·μM-1 for 8-17 DNAzyme (15)), while the large kcat of 10.2 min-1 could result from the high efficiency of the photo-reaction. This result indicates that this enzyme analog is a real catalyst and can perform multiple-turnover reactions.
FIGURE 3.
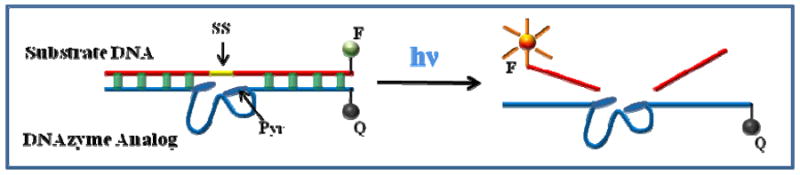
Design scheme for photo-modulated DNAzyme analog.
(Substrate DNA): 5’-FAM-ACTCACTATCT-SS-GGAAGAGATG-3’
(DNAzyme): 5’-CATCTCTTCT-/Pyr/-CCGAGCCGGTCGAA-/Pyr/-ATAGTGAGT-Dabcyl-3’
FIGURE 4.
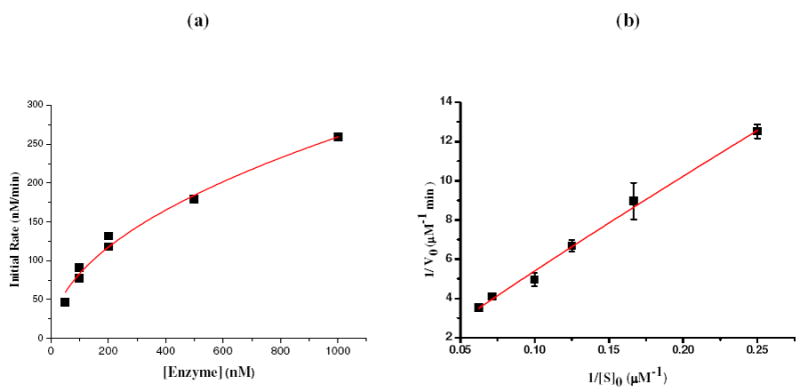
Catalytic activity of light-modulated DNAzyme analog: (a) Influence of DNAzyme concentration on the initial rate of cleavage of substrate DNA. Activity was measured at [substrate] =4.0 μM, 350nm, 0.3W light irradiation. (b) Lineweaver - Burk plot for initial cleavage rate of substrate by DNAzyme analog (100nM).
CONCLUSIONS
In conclusion, we have demonstrated high efficiency pyrene-assisted photolysis of disulfide bonds using 350nm irradiation. DNA provides a feasible matrix for quantitatively exploiting the cleavage efficiency and mechanism. As indicated by the examples of DNA micelle photo-disaggregation and catalytic function of DNAzyme analogs, numerous applications of pyrene-disulfide molecular assemblies are possible in biomedical and proteomics scenarios, with light-induced spatio-temporal control.
Supplementary Material
Acknowledgments
We would like to thank Dr. Valeria D. Kleiman and Dr. Gail E. Fanucci for their constructive discussion and suggestions. We also thank NIH for supporting this work.
Footnotes
Supporting information available: Radical cleavage mechanism study results, including gel analysis results after adding the radical scavenger p-benzoquinone, the fluorescence spectrum from radical indicator TEMPO-9-AC, agarose gel analysis of DNA micelle disaggregation after irradiation, and confirmation of multiple catalytic turnovers property by pyrene DNAzyme analog. This information is available free of charge via the Internet at http://pubs.acs.org/.
REFERENCES AND NOTES
- 1.Walling C, Rabinowitz RJ. Am Chem Soc. 1959;81:1137–1143. [Google Scholar]
- 2.(a) Riemer J, Bulleid N, Herrmann JM. Science. 2009;324:1284–1287. doi: 10.1126/science.1170653. [DOI] [PubMed] [Google Scholar]; (b) Park SW, Zhen GH, Verhaeghe C, Nakagami Y, Nguyenvu LT, Barczak AJ, Killeen N, Erle D. Proc Natl Acad Sci U S A. 2009;106:6950–6955. doi: 10.1073/pnas.0808722106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Matsumura M, Matthews BW. Science. 1989;243:792–794. doi: 10.1126/science.2916125. [DOI] [PubMed] [Google Scholar]; (b) Hogg PJ. Trends Biochem Sci. 2003;28:210–214. doi: 10.1016/S0968-0004(03)00057-4. [DOI] [PubMed] [Google Scholar]
- 4.See, for example: Saito G, Swanson JA, Lee KD. Adv Drug Deliver Rev. 2003;55:199–215. doi: 10.1016/s0169-409x(02)00179-5.Bauhuber S, Hozsa C, Breunig M, Gopferich A. Adv Mater. 2009;21:3286–3306. doi: 10.1002/adma.200802453.Chari RVJ. Acc Chem Res. 2008;41:98–107. doi: 10.1021/ar700108g.
- 5.(a) Conlon P, Yang JC, Wu YR, Chen Y, Martinez K, Kim Y, Stevens N, Marti AA, Jockusch S, Turro NJ, Tan W. J Am Chem Soc. 2008;130:336–342. doi: 10.1021/ja076411y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kumar CV, Buranaprapuk A, Opiteck GJ, Moyer MB, Jockusch S, Turro NJ. Proc Natl Acad Sci U S A. 1998;95:10361–10366. doi: 10.1073/pnas.95.18.10361. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Xu Z, Singh NJ, Lim J, Pan J, Kim HN, Park S, Kim KS, Yoon J. J Am Chem Soc. 2009;131:15528–15533. doi: 10.1021/ja906855a. [DOI] [PubMed] [Google Scholar]
- 6.See, for example: Mack ET, Carle AB, Liang JT, Coyle W, Wilson M. J Am Chem Soc. 2004;126:15324–15325. doi: 10.1021/ja0473788.
- 7.Boussicault F, Robert M. Chem Rev. 2008;108:2622–2645. doi: 10.1021/cr0680787. [DOI] [PubMed] [Google Scholar]
- 8.(a) Bent DV, Hayon E. J Am Chem Soc. 1975;97:2612–2619. doi: 10.1021/ja00843a004. [DOI] [PubMed] [Google Scholar]; (b) Fung YME, Kjeldsen F, Silivra OA, Chan TWD, Zubarev RA. Angew Chem Int Ed. 2005;44:6399–6403. doi: 10.1002/anie.200501533. [DOI] [PubMed] [Google Scholar]; (c) Ly T, Julian RR. Angew Chem Int Ed. 2009;48:2–10. doi: 10.1002/anie.200900613. [DOI] [PubMed] [Google Scholar]
- 9.(a) Tashiro R, Ohtsuki A, Sugiyama H. J Am Chem Soc. 2010;132:14361–14363. doi: 10.1021/ja106184w. [DOI] [PubMed] [Google Scholar]; (b) Daublain P, Thazhathveetil AK, Wang Q, Trifonov A, Fiebig T, Lewis FD. J Am Chem Soc. 2009;131:16790–16797. [Google Scholar]; (c) Kawai K, Takada T, Tojo S, Ichinose N, Majima T. J Am Chem Soc. 2001;123:12688–12689. doi: 10.1021/ja0158152. [DOI] [PubMed] [Google Scholar]
- 10.(a) Bae Y, Kataoka K. Adv Drug Delivery Rev. 2009;61:768–784. doi: 10.1016/j.addr.2009.04.016. [DOI] [PubMed] [Google Scholar]; (b) Discher DE, Eisenberg A. Science. 2002;297:967–973. doi: 10.1126/science.1074972. [DOI] [PubMed] [Google Scholar]
- 11.(a) Liu H, Zhu Z, Kang H, Wu Y, Sefan K, Tan W. Chem Eur J. 2010;16:3791–3797. doi: 10.1002/chem.200901546. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wu Y, Sefah K, Liu H, Wang R, Tan W. Proc Natl Acad Sci U S A. 2010;107:5–10. doi: 10.1073/pnas.0909611107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Behera GB, Mishra BK, Behera PK, Panda M. Adv Colloid Interface Sci. 1999;82:1–42. [Google Scholar]
- 13.(a) Silverman SK. Angew Chem Int Ed. 2010;49:2–24. [Google Scholar]; (b) Liu J, Cao Z, Lu Y. Chem Rev. 2009;109:1948–1998. doi: 10.1021/cr030183i. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Silverman SK. Acc Chem Res. 200942:1521–1531. doi: 10.1021/ar900052y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.A review about artificial photocatalysis system could be found: Stoll RS, Hecht S. Angew Chem Int Ed. 2010;49:5054–5075. doi: 10.1002/anie.201000146.
- 15.Yim T, Liu J, Lu Y, Kane RS, Dordick JS. J Am Chem Soc. 2005;127:12200–12201. doi: 10.1021/ja0541581. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.