

Abstract
The expression of MHC class II (MHC-II) on the surface of antigen-presenting cells, such as dendritic cells (DCs), is tightly regulated during cellular activation. Many cells, including DCs, are activated following stimulation of innate Toll-like receptors (TLRs) by products of microorganisms. In the resting (immature) state, MHC-II is ubiquitinated in immature DCs and is rapidly degraded; however, after activation of these cells with MyD88-dependent TLR ligands, MHC-II ubiquitination is blocked, and MHC-II survival is prolonged. We now show that DC activation using MyD88-dependent TLR ligands, MyD88-independent TLR ligands, and even infection with the intracellular parasite Toxoplasma gondii leads to identical changes in MHC-II expression, ubiquitination, and surface stability, revealing a conserved role for enhanced MHC-II stability after DC activation by different stimuli.
Keywords: Dendritic Cell, MHC, Protein Stability, Toll-like Receptors (TLR), Ubiquitination
Introduction
Dendritic cells (DCs)3 are antigen-presenting cells that play a vital role in the immune system. A major function of DCs is to capture, process, and present antigens to T cells (1). To perform this task, DCs possess MHC class II (MHC-II) proteins onto which processed antigens are loaded (2). The peptide-MHC-II (pMHC-II) complexes are subsequently transported to the cell surface, where they are presented to CD4+ T cells. In the resting (immature) state, DCs are relatively poor activators of T cells; however, T cell-stimulating activity dramatically improves when DCs receive a maturation signal. Upon receipt of such a signal, DCs go through profound changes, including increased surface expression of molecules important for T cell activation, such as pMHC-II and the co-stimulatory molecules CD40 and CD86 (1). Chemokine receptor expression pattern is also modified upon DC activation, resulting in efficient recruitment of DCs to secondary lymphoid tissues, where they encounter T cells and initiate appropriate immune responses.
Toll-like receptors (TLRs) are expressed on antigen-presenting cells, and these TLRs trigger DC activation after they bind to pathogen-associated molecular patterns present on microorganisms (3). There are 13 known TLRs found in mammals, and they can be found on the plasma membrane as well as in intracellular compartments. Pathogen-associated molecular patterns are typically conserved structures found in microorganisms, and when TLRs bind a pathogen-associated molecular pattern, a signaling cascade is initiated that leads to up-regulation of molecules and cytokines that will fight and eliminate the encountered microorganisms.
The ligands that bind TLRs vary and include cell wall components of Gram-negative bacterial LPS for TLR4 and Gram-positive bacterial peptidoglycan (PGN) for TLR2, single-stranded RNA for TLR7/8, double-stranded RNA (or poly(I:C)) for TLR3, and unmethylated bacterial CpG DNA for TLR9 (3). The immune response following TLR activation also varies depending on the nature of the ligand and results in a myriad of different immunological consequences depending on the particular TLR stimulated.
All TLR family members signal using adaptor molecules upon ligand binding. Four cytosolic adaptor molecules are thought to play a crucial role in the specificity of the individual TLR-mediated signaling pathway: MyD88, MAL (MyD88 adaptor-like), TRIF (Toll/IL-1 receptor domain-containing adaptor-inducing IFN-beta), and TRAM (TRIF-related adaptor molecule) (4). The signaling pathways activated by TLRs can be broadly classified as MyD88-dependent and MyD88-independent (3, 4). MyD88 is recruited and utilized by all of the TLRs except TLR3, which utilizes TRIF. Curiously, TLR4 can also signal via TRIF, increasing the diversity of responses from antigen-presenting cells stimulated with bacterial LPS.
MHC-II is known to be ubiquitinated in immature DCs (5, 6), and one of the many consequences of DC activation with LPS is that expression of the MHC-II E3 ubiquitin ligase MARCH-I is terminated and MHC-II ubiquitination abolished (7, 8). We have recently demonstrated that one result of impaired MHC-II ubiquitination in DCs is that the MHC-II half-life is prolonged (23); however, it remains to be determined whether activation of DCs with different TLR ligands, or even non-TLR ligands, leads to similar changes in MHC-II stability.
In this study, we activated monocyte-derived human DCs with ligands targeting different TLRs to investigate if there is a difference in MHC-II behavior depending on the signaling pathway utilized. We used LPS (TLR4), PGN (TLR2), and poly(I:C) (TLR3) as TLR ligands. We also infected DCs with the intracellular parasite Toxoplasma gondii, a pathogen that is known to stimulate DCs but whose mechanism of action on human DCs remains unknown. By assaying for up-regulation of MHC-II and CD86 on the cell surface, survival of MHC-II on the surface, and ubiquitination of pMHC-II, we found that regardless of the TLRs activated (both MyD88-dependent and MyD88-independent), the maturation of DCs followed the same pattern with up-regulation of pMHC-II and CD86 on the cell surface, cessation of MHC-II ubiquitination, and prolonged survival of plasma membrane MHC-II. These data strongly suggest that stabilization of MHC-II appears to be a highly conserved feature common to all forms of DC activation.
EXPERIMENTAL PROCEDURES
Cells and Antibodies
Human DCs were generated from elutriated human monocytes obtained from the National Institutes of Health Blood Bank as described previously (9). Briefly, monocytes were cultured in RPMI 1640 medium supplemented with 10% heat-inactivated FBS, 50 ng/ml human GM-CSF (PeproTech, Rocky Hill, NJ), 35 ng/ml human IL-4 (PeproTech), 2 mm l-glutamine, 50 μg/ml gentamycin, 1 mm sodium pyruvate, 55 μm β-mercaptoethanol, and nonessential amino acids (Sigma). Half of the medium was changed every 2 days, adding fresh medium containing twice the concentration of the cytokines mentioned above. After 6 days in culture, the cells were activated using 1 μg/ml LPS (Sigma), 10 μg/ml PGN (Sigma), or 5 μg/ml poly(I:C) (Sigma). DCs were incubated with irradiated T. gondii overnight at a multiplicity of infection of 1:10. Stimulated DCs were analyzed 24 h after the addition of each stimulus.
Anti-human pMHC-II mAb L243, anti-CD86 mAb, mouse IgG2a, and mouse IgG1 antibodies were from BD Biosciences. Biotinylated anti-ubiquitin mAb P4D1 was from Covance (Denver, PA). Anti-human MHC-II β-chain mAb XD5.A11 has been described previously (10). Alexa Fluor-conjugated secondary antibodies were from Molecular Probes (Eugene, OR), and HRP-conjugated reagents were from Southern Biotech (Birmingham, AL). Transgenic T. gondii tachyzoites expressing YFP or RFP (kindly provided by Boris Striepen (University of Georgia) and Michael Grigg (NIAID), respectively) were maintained by serial passage on human foreskin fibroblasts cultured at 37 °C in DMEM (Invitrogen) supplemented with 10% heat-inactivated FBS. Parasites were harvested from 80% lysed fibroblast monolayers, and non-replication tachyzoites were obtained by irradiation using 15,000 rads (JL Shepherd Mark I cesium irradiator).
FACS Staining
DCs cultured overnight in medium alone or in the presence of different stimuli were harvested, washed once with FACS buffer (Hanks' balanced salt solution (HBSS) containing 2% FCS), and incubated with primary mAb L243 or anti-CD86 mAb for 20 min on ice. The cells were washed twice with FACS buffer before being incubated with Alexa 488-labeled goat anti-mouse secondary antibody for 20 min on ice. The cells were subsequently washed twice with FACS buffer and finally fixed in HBSS containing 1% paraformaldehyde. The cells were analyzed in a FACSCalibur flow cytometer using mouse IgG2a as a negative control for mAb L243 and mouse IgG1 as a control for anti-CD86 mAb.
Immunoprecipitation and Immunoblotting
DCs cultured overnight in medium alone or in the presence of different stimuli were harvested and washed once with HBSS. The cells were lysed in 10 mm Tris and 150 mm NaCl (pH 7.4) containing 1% Triton X-100, protease inhibitors, and 25 mm N-ethylmaleimide to inhibit deubiquitination activity. pMHC-II complexes were isolated by immunoprecipitation using mAb L243 immobilized on protein A-Sepharose beads (Sigma) and analyzed by immunoblotting. Immunoblots were probed for ubiquitinated proteins using biotinylated anti-ubiquitin mAb P4D1 and HRP-streptavidin and probed for total MHC-II β-chain present in each immunoprecipitate using mAb XD5.A11 as described previously (11).
Surface MHC-II Survival
Plasma membrane proteins of immature and mature DCs were surface-biotinylated by incubating ∼10 × 106 cells/ml using the membrane-impermeable biotinylation reagent sulfo-NHS-biotin (1 mg/ml; Pierce) in a buffer of HBSS for 30 min on ice according to the manufacturers instructions. After biotinylation, cells were extensively washed with ice-cold HBSS, resuspended in complete medium, and incubated for various times on ice or at 37 °C. Cells were then harvested and lysed in Triton X-100 lysis buffer, and surface proteins were isolated using streptavidin-agarose beads as described previously (9). Proteins adsorbed to the streptavidin-agarose beads were analyzed by immunoblotting for the presence of surface MHC-II using mAb XD5.A11. The bands were revealed with Western Lightning Plus chemiluminescence reagent (PerkinElmer Life Sciences). The relative amount of MHC-II present in each sample was determined by quantitative densitometry using a Molecular Dynamics densitometer and was expressed as a percentage of the amount of MHC-II isolated after culture at 37 °C compared with the amount of MHC-II isolated from the biotinylated cell population after culture on ice.
RNA Extraction and Quantitative Real-time PCR
RNA extractions were carried out with the RNeasy minikit (Qiagen, Valencia, CA) according to the manufacturer's instructions. RNA was reverse-transcribed into cDNA using a standard procedure and reagents from Promega. After initial template melting, PCR was performed using 30 cycles at 95 °C for 45 s, 65 °C for 45 s, and 72 °C for 60 s with a 2-min product extension time at 72 °C. The human MARCH-I forward primer sequence was caggagccagtcaaggttgt, and the reverse primer sequence was gaggggtttgagcttggtct. Gel images were acquired using an AlphaImager HP system (Alpha Innotech Corp., San Leandro, CA) and analyzed using Multi Gauge V3.0 (Fujifilm, Cypress, CA). Expression of GAPDH was monitored as a loading control. Serial dilutions of each template were used to avoid saturating conditions. Real-time PCR was performed using an ABI Prism 7900HT sequence detection system and a QuantiTect SYBR Green PCR kit (Qiagen) according to the manufacturer's instructions. Real-time PCR primers for MARCH-I were as described above, and the primer set for GAPDH was obtained from Qiagen.
RESULTS AND DISCUSSION
Stimulation of Human DCs with Different TLR Ligands Promotes Surface pMHC-II and CD86 Expression
Activation of DCs results in their transformation to efficient antigen-presenting cells, and increases in expression of both signal 1 (pMHC-II complexes) and signal 2 (co-stimulatory molecules, such as CD40 and CD86) are typically used as markers of DC activation. Stimulation of DCs with either LPS (TLR4) or CpG (TLR9) activates DCs and terminates ubiquitination of MHC-II (5–8). To investigate how MHC-II ubiquitination is regulated during DC activation and whether it is solely regulated by MyD88-dependent TLR signaling, we stimulated human DCs with MyD88-dependent (TLR2 and TLR4) and MyD88-independent (TLR3) ligands for 24 h. Stimulation of TLR2 (PGN), TLR3 (poly(I:C)), and TLR4 (LPS) resulted in a robust increase in expression of both pMHC-II and CD86 on the DC surface that was similar under all conditions tested (Fig. 1).
FIGURE 1.

DCs are activated by different TLR ligands. Human DCs were left in medium alone (mock) or incubated for 24 h in the presence of LPS, PGN, or poly(I:C). The cells were analyzed by FACS analysis for expression of surface pMHC-II complexes using the conformation-sensitive mAb L243 (upper panel) and the co-stimulatory molecule CD86 (lower panel). The staining of an isotype control mAb is shown in light blue. Each experiment was performed at least three times, and representative histograms are shown.
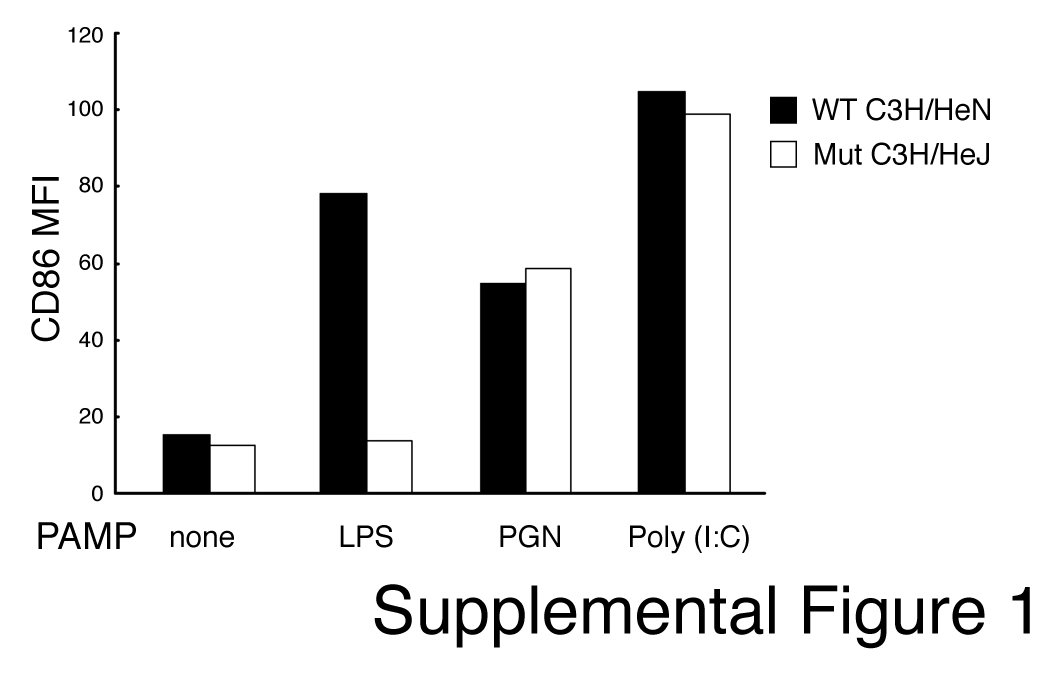
To rule out the possibility that the different DC stimuli used in this study were contaminated with the potent DC activator endotoxin/LPS, wild-type or LPS-resistant immature mouse DCs were incubated with PGN, poly(I:C), or LPS for 24 h, and DC activation was assessed by CD86 up-regulation. PGN and poly(I:C) activated both wild-type (C3H/HeN) and LPS-resistant (C3H/HeJ) immature DCs, whereas LPS activated only wild-type DCs but not LPS-resistant DCs (supplemental Fig. 1). These data demonstrate that the preparations of PGN and poly(I:C) used in this study were not contaminated with LPS and show that DC activation by TLR2, TLR3, and TLR4 results in similar profiles of MHC-II expression on DCs.
Ubiquitination of MHC-II Is Terminated upon DC Activation
MHC-II is oligoubiquitinated in immature DCs (5, 6, 12), and activation of these cells with the TLR4 ligand LPS prevents MHC-II ubiquitination at lysine 225 in the MHC-II β-chain (5, 6). Immunoprecipitation of pMHC-II from immature human DCs followed by anti-ubiquitin immunoblotting revealed a ladder of up to five 7-kDa ubiquitin chains on MHC-II (Fig. 2). Activation of human DCs by stimulation of TLR4 with LPS, TLR2 with PGN, or TLR3 with poly(I:C) prevented MHC-II ubiquitination despite the fact that these triggers actually augmented the amount of MHC-II present in each immunoprecipitate. These data demonstrate that regardless of the stimulus, DC activation leads to the termination of MHC-II ubiquitination.
FIGURE 2.

Ubiquitination is down-regulated upon DC activation. Human DCs were left in medium alone (mock) or incubated in the presence of LPS, poly(I:C), or PGN. After 24 h, the cells were lysed in Triton X-100, and aliquots of the lysate were subjected to immunoprecipitation (IP) using anti-pMHC-II mAb L243. The immunoprecipitates were analyzed by immunoblotting with antibodies recognizing ubiquitin (upper blot) or total MHC-II β-chain (lower blot). The ubiquitination of MHC-II in each condition was quantitated by densitometry and is expressed relative to that in the mock-treated sample. Each value was normalized for the total amount of MHC-II β-chain present in each immunoprecipitate. The data shown are the mean ± S.D. obtained from three independent experiments. *, p < 0.05.
Expression of MARCH-I Is Terminated upon DC Activation
The E3 ligase MARCH-I has been found to be the major E3 ligase responsible for the ubiquitination MHC-II in B cells and DCs (12), and MARCH-I mRNA and protein expression has been reported to be down-regulated upon DC maturation with LPS (7, 8). Unfortunately, we were unable to visualize MARCH-I protein by either immunofluorescence microscopy or immunoblot analysis, most likely due to the very low expression of this protein in DCs. Therefore, to determine whether MARCH-I expression is terminated by the same signals that lead to the cessation of MHC-II ubiquitination, we performed RT-PCR analysis of resting and stimulated human DCs to monitor MARCH-I mRNA expression. Using primers specifically targeting MARCH-I and the ubiquitously expressed GAPDH gene, we found that MARCH-I mRNA expression was markedly down-regulated when DCs were stimulated with LPS (TLR4), PGN (TLR2), or poly(I:C) (TLR3) (Fig. 3). Quantitative real-time PCR confirmed that activation with each of these ligands profoundly reduced MARCH-I expression. Together with previous work showing that stimulation of TLR9 on mouse DCs with CpG inhibits MHC-II ubiquitination (8), these data demonstrate that stimulation of distinct TLRs leads to a common pathway of reduced MARCH-I expression and blocked MHC-II ubiquitination in DCs.
FIGURE 3.

MARCH-I expression is reduced upon DC activation. A, human DCs were left in medium alone (mock) or incubated for 24 h in the presence of LPS, PGN, or poly(I:C). cDNA was synthesized from each culture condition and used as a template for RT-PCR. Specific primers were used to amplify human MARCH-I and GAPDH mRNAs. B, the relative amount of MARCH-I mRNA present in each condition was determined by real-time PCR. The data were normalized based on the amount of GAPDH mRNA present in the same cDNA sample and is expressed as a percentage of the amount of MARCH-I mRNA present in the mock (immature) DC sample. The data shown are the mean ± S.D. obtained from three independent experiments. *, p < 0.05.
The Half-life of Surface MHC-II Is Prolonged in Mature DCs
It is well established that activation of DCs prolongs the half-life of MHC-II molecules, thereby enhancing the probability that activated DCs can present stimulatory pMHC-II complexes to CD4 T cells (13). It has also been shown that ubiquitination by MARCH-I dramatically reduces MHC-II expression in B cells (12) and promotes lysosomal proteolysis of MHC-II (5), highlighting the importance of ubiquitination in MHC-II expression and/or stability. To investigate the fate of MHC-II in DCs activated with different stimuli, we examined the stability of surface MHC-II in immature DCs and DCs stimulated with LPS (TLR4), PGN (TLR2), and poly(I:C) (TLR3). Surface proteins on each DC population were biotinylated on ice, and the cells were then cultured at 37 °C or kept on ice. After 2 h, the cells were lysed, surface proteins were isolated using avidin-agarose beads, and the presence of biotinylated (surface) MHC-II in the lysate was determined by immunoblot analysis. More than 40% of the total pool of surface MHC-II lost immunoreactivity when immature DCs were cultured at 37 °C for 2 h; however, MHC-II stability was dramatically enhanced when the cells were pre-activated with LPS, PGN, or poly(I:C) (Fig. 4). These results demonstrate that surface MHC-II is stabilized against degradation regardless of whether DCs are activated by triggering TLR2, TLR3, or TLR4.
FIGURE 4.

Survival of cell-surface MHC-II is prolonged following activation of DC with different TLR ligands. Immature (mock) or LPS-, PGN-, or poly(I:C)-treated human DCs were surface-biotinylated on ice and returned to culture in complete medium at 37 °C. The amount of biotinylated MHC-II isolated after incubation for 2 h from each DC population was determined by immunoblotting and is expressed as a percentage of the total amount of biotinylated MHC-II present in aliquots of cells that remained on ice after biotinylation. The data shown are the mean ± S.D. obtained from three independent experiments. *, p < 0.05.
Infection with T. gondii Activates Human DCs and Promotes MHC-II Stability
We attempted to identify a physiologically relevant pathogen that activates DCs to determine whether pathogens affect MHC-II biology in a manner indistinguishable from purified TLR ligands. Furthermore, to test our hypothesis that DC activation promotes an “MHC-II stability cascade” that is ligand-independent, we wanted to specifically use a pathogen whose mechanism of DC activation remains unknown. T. gondii is an intracellular parasite that activates both human and mouse DCs (14–17), and although this pathogen is known to stimulate TLR11 in mice (18), the mechanism by which T. gondii activates human DCs remains unknown. Incubation of human DCs with T. gondii effectively stimulated pMHC-II and CD86 expression in a manner indistinguishable from that of LPS (Fig. 5A). DC activation by T. gondii is not a consequence of LPS contamination, as T. gondii preparations do not stimulate TLR11 mutant DCs that remain responsive to LPS (18). In the process of parasite invasion, T. gondii establishes a non-fusogenic parasitophorous vacuole that avoids fusion with host cell endocytic and exocytic vesicular trafficking pathways (19–21). Nevertheless, T. gondii-induced DC maturation caused redistribution of MHC-II from the intracellular compartments out to the cell surface (Fig. 5B), similar to that induced by the TLR4 ligand LPS as described previously (22). Furthermore, like LPS-treated DCs, T. gondii-infected DCs did not express MARCH-I (Fig. 5C), did not possess ubiquitinated MHC-II (Fig. 5D), and showed a remarkable increase in surface MHC-II stability after incubation at 37 °C (Fig. 5E). Although the underlying molecular mechanism remains unclear, these data demonstrate that infection of human DCs with T. gondii leads to similar changes in MHC-II biology as those induced by direct TLR signaling with purified ligands.
FIGURE 5.

Infection with T. gondii stimulates DC activation, prevents MHC-II ubiquitination, and promotes surface MHC-II survival. Human DCs were left in medium alone (mock) or incubated for 24 h in the presence of LPS or YFP- or RFP-tagged T. gondii. A, DCs were analyzed by FACS analysis for expression of surface pMHC-II complexes using the conformation-sensitive mAb L243 and the co-stimulatory molecule CD86. The staining of an isotype control mAb is shown in solid light blue. FACS analysis revealed that ∼85% of the cells in the T. gondii-infected population were YFP-tagged T. gondii-positive. B, cells were fixed, permeabilized, and stained with anti-pMHC-II mAb L243 (green). The presence of RFP-tagged T. gondii (red) can be seen in vacuoles in infected cells. C, cDNA was prepared from mock-, LPS-, or T. gondii-treated DCs, and expression of MARCH-I and GAPDH mRNAs present in each sample was determined by RT-PCR. D, DCs obtained after incubation for 24 h in medium alone (mock) or with T. gondii were lysed in Triton X-100 and subjected to immunoprecipitation (IP) using anti-pMHC-II mAb L243. The immunoprecipitates were analyzed by immunoblotting with antibodies recognizing ubiquitin (upper blot) or total MHC-II β-chain (lower blot). The ubiquitination of MHC-II in each condition was quantitated by densitometry and is expressed relative to that in the mock-treated sample. Each value was normalized for the total amount of MHC-II β-chain present in each immunoprecipitate. The data shown are the mean ± S.D. obtained from three independent experiments. *, p < 0.05. E, immature (mock) or LPS- or T. gondii-treated human DCs were surface-biotinylated on ice and returned to culture in complete medium at 37 °C. The amount of biotinylated MHC-II isolated after incubation for 4 h from each DC population was determined by immunoblotting and is expressed as a percentage of the total amount of biotinylated pMHC-II present in aliquots of cells that remained on ice after biotinylation. The data shown are the mean ± S.D. obtained from three independent experiments. *, p < 0.05.
It is well established that activation by TLR ligands, such as LPS, leads to increased MHC-II, CD40, and CD86 expression on DCs and re-localization of MHC-II from intracellular antigen-processing compartments to the cell surface (22), reduced expression of the E3 ubiquitin ligase MARCH-I (7, 8), reduced MHC-II ubiquitination (5, 6), and prolonged stability of surface-expressed pMHC-II complexes (13). In this study, we investigated whether different stimuli lead to similar changes in MHC-II biology in DCs. MyD88 is an important adaptor protein that links the cytoplasmic Toll/IL-1 receptor domain of different TLRs to intracellular signaling pathways (4). We now show that stimulation of human DCs with either MyD88-dependent or MyD88-independent TLRs or with a pathogen that utilizes a yet unidentified mechanism of DC activation (T. gondii) leads to essentially identical changes in MHC-II expression, ubiquitination, and surface stability. Each of these DC stimuli activates a wide variety of signaling pathways, and it remains a challenge for the future to determine the common (or distinct) molecular signaling pathway(s) that lead(s) to the MHC-II stabilization phenotype observed here. Nevertheless, the common consequence of all of the ligands used in this study is that regardless of the DC activation stimulus, MHC-II expression is stabilized by activation-induced changes in MARCH-I expression and MHC-II ubiquitination.
Supplementary Material
Acknowledgments
We thank David Segal for many helpful discussions and our colleagues who provided reagents during the course of this study.
This work was supported, in whole or in part, by the National Institutes of Health Intramural Research Programs of NCI and NIAID.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. 1.
- DC
- dendritic cell
- MHC-II
- MHC class II
- TLR
- Toll-like receptor
- PGN
- peptidoglycan
- pMHC-II
- peptide-loaded MHC-II
- HBSS
- Hanks' balanced salt solution.
REFERENCES
- 1.Banchereau J., Steinman R. M. (1998) Nature 392, 245–252 [DOI] [PubMed] [Google Scholar]
- 2.Trombetta E. S., Mellman I. (2005) Annu. Rev. Immunol. 23, 975–1028 [DOI] [PubMed] [Google Scholar]
- 3.Hemmi H., Akira S. (2005) Chem. Immunol. Allergy 86, 120–135 [DOI] [PubMed] [Google Scholar]
- 4.McGettrick A. F., O'Neill L. A. (2004) Mol. Immunol. 41, 577–582 [DOI] [PubMed] [Google Scholar]
- 5.Shin J. S., Ebersold M., Pypaert M., Delamarre L., Hartley A., Mellman I. (2006) Nature 444, 115–118 [DOI] [PubMed] [Google Scholar]
- 6.van Niel G., Wubbolts R., Ten Broeke T., Buschow S. I., Ossendorp F. A., Melief C. J., Raposo G., van Balkom B. W., Stoorvogel W. (2006) Immunity 25, 885–894 [DOI] [PubMed] [Google Scholar]
- 7.De Gassart A., Camosseto V., Thibodeau J., Ceppi M., Catalan N., Pierre P., Gatti E. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 3491–3496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Young L. J., Wilson N. S., Schnorrer P., Proietto A., ten Broeke T., Matsuki Y., Mount A. M., Belz G. T., O'Keeffe M., Ohmura-Hoshino M., Ishido S., Stoorvogel W., Heath W. R., Shortman K., Villadangos J. A. (2008) Nat. Immunol. 9, 1244–1252 [DOI] [PubMed] [Google Scholar]
- 9.Walseng E., Bakke O., Roche P. A. (2008) J. Biol. Chem. 283, 14717–14727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roche P. A., Teletski C. L., Stang E., Bakke O., Long E. O. (1993) Proc. Natl. Acad. Sci. U.S.A. 90, 8581–8585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Poloso N. J., Muntasell A., Roche P. A. (2004) J. Immunol. 173, 4539–4546 [DOI] [PubMed] [Google Scholar]
- 12.Matsuki Y., Ohmura-Hoshino M., Goto E., Aoki M., Mito-Yoshida M., Uematsu M., Hasegawa T., Koseki H., Ohara O., Nakayama M., Toyooka K., Matsuoka K., Hotta H., Yamamoto A., Ishido S. (2007) EMBO J. 26, 846–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cella M., Engering A., Pinet V., Pieters J., Lanzavecchia A. (1997) Nature 388, 782–787 [DOI] [PubMed] [Google Scholar]
- 14.Subauste C. S., Wessendarp M. (2000) J. Immunol. 165, 1498–1505 [DOI] [PubMed] [Google Scholar]
- 15.Chaussabel D., Semnani R. T., McDowell M. A., Sacks D., Sher A., Nutman T. B. (2003) Blood 102, 672–681 [DOI] [PubMed] [Google Scholar]
- 16.Aliberti J., Jankovic D., Sher A. (2004) Immunol. Rev. 201, 26–34 [DOI] [PubMed] [Google Scholar]
- 17.Kang H. K., Lee H. Y., Lee Y. N., Jo E. J., Kim J. I., Aosai F., Yano A., Kwak J. Y., Bae Y. S. (2004) Biochem. Biophys. Res. Commun. 322, 899–904 [DOI] [PubMed] [Google Scholar]
- 18.Yarovinsky F., Zhang D., Andersen J. F., Bannenberg G. L., Serhan C. N., Hayden M. S., Hieny S., Sutterwala F. S., Flavell R. A., Ghosh S., Sher A. (2005) Science 308, 1626–1629 [DOI] [PubMed] [Google Scholar]
- 19.Joiner K. A., Fuhrman S. A., Miettinen H. M., Kasper L. H., Mellman I. (1990) Science 249, 641–646 [DOI] [PubMed] [Google Scholar]
- 20.Mordue D. G., Desai N., Dustin M., Sibley L. D. (1999) J. Exp. Med. 190, 1783–1792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mordue D. G., Håkansson S., Niesman I., Sibley L. D. (1999) Exp. Parasitol. 92, 87–99 [DOI] [PubMed] [Google Scholar]
- 22.Pierre P., Turley S. J., Gatti E., Hull M., Meltzer J., Mirza A., Inaba K., Steinman R. M., Mellman I. (1997) Nature 388, 787–792 [DOI] [PubMed] [Google Scholar]
- 23.Walseng E., Furuta K., Bosch B., Weih K. A., Matsuki Y., Bakke O., Ishido S., Roche P. A. (2010) Proc. Natl. Acad. Sci. U.S.A. 107, 20465–20470 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}