

Abstract
Tonic inhibition in the brain is mediated largely by specialized populations of extrasynaptic receptors, γ-aminobutyric acid receptors (GABAARs). In the dentate gyrus region of the hippocampus, tonic inhibition is mediated primarily by GABAAR subtypes assembled from α4β2/3 with or without the δ subunit. Although the gating of these receptors is subject to dynamic modulation by agents such as anesthetics, barbiturates, and neurosteroids, the cellular mechanisms neurons use to regulate their accumulation on the neuronal plasma membrane remain to be determined. Using immunoprecipitation coupled with metabolic labeling, we demonstrate that the α4 subunit is phosphorylated at Ser443 by protein kinase C (PKC) in expression systems and hippocampal slices. In addition, the β3 subunit is phosphorylated on serine residues 408/409 by PKC activity, whereas the δ subunit did not appear to be a PKC substrate. We further demonstrate that the PKC-dependent increase of the cell surface expression of α4 subunit-containing GABAARs is dependent on Ser443. Mechanistically, phosphorylation of Ser443 acts to increase the stability of the α4 subunit within the endoplasmic reticulum, thereby increasing the rate of receptor insertion into the plasma membrane. Finally, we show that phosphorylation of Ser443 increases the activity of α4 subunit-containing GABAARs by preventing current run-down. These results suggest that PKC-dependent phosphorylation of the α4 subunit plays a significant role in enhancing the cell surface stability and activity of GABAAR subtypes that mediate tonic inhibition.
Keywords: Cell Surface, GABA Receptors, Protein Kinase C (PKC), Protein Phosphorylation, Protein Stability, alpha 4, Insertion
Introduction
γ-Aminobutyric acid type A receptors (GABAARs)2 constitute the major inhibitory ligand-gated receptors in the adult central nervous system and are responsible for both phasic and tonic forms of inhibition (1). These receptors are pentameric, anion-selective ion channels that can be assembled from eight subunit classes: α(1–6), β(1–3), γ(1–3), δ, ϵ, θ, π, and ρ(1–3)(2–3). This large number of receptor subunits provides the basis for a significant degree of heterogeneity of GABAAR structure and function. However, previous studies suggest that in the brain, the majority of phasic inhibition is dependent upon a few GABAARs subunits, namely the α, β, and γ2 subunits located within synaptic sites (2, 3). In the adult brain, these receptors are specific targets for brief exposures to high concentrations of GABA, resulting in short lived, but significant, hyperpolarization. In contrast, tonic inhibition is characterized by a sustained reduction in the cell's input resistance, effectively reducing the probability of action potential generation (1, 4). Tonic inhibition is the result of persistent activation by GABAARs consisting primarily of α, β, and δ subunits located within peri- or extrasynaptic sites (1). With respect to specific brain regions, extrasynaptic GABAARs that mediate tonic inhibition in the thalamus and dentate gyrus of the hippocampus are composed of the α4 and β2/3 subunits with or without the δ subunit (5–9). Verification of the role that the α4 subunit plays in mediating tonic inhibition comes from α4 subunit knock-out mice, which have substantially lower levels of tonic inhibition in these brain areas (10, 11).
Changes in tonic inhibition associated with GABAARs containing the α4 subunit have been implicated in a number of normal and pathological states in which the thalamus and the hippocampus play a role. It is apparent that tonic inhibition is essential for dynamically regulating the neuronal output, frequency of firing, and gain control of neurotransmission (12–19). In addition, extrasynaptic GABAARs have been further shown to be targets for a wide range of endogenous and pharmacological agents, such as neurosteroids, anesthetics, ethanol, and anticonvulsants (20–23). Finally, modifications in the efficacy of tonic inhibition arise under pathological conditions including stress, fragile X syndrome, aberrant brain activity associated with menstrual cycle, postpartum depression, schizophrenia, and temporal lobe epilepsies (20, 24–28).
Little is known about the endogenous mechanism by which neurons control the functional properties of GABAAR subtypes that mediate tonic inhibition. It has long been established that a direct relationship exists between the number of synaptic GABAARs at the cell surface and the strength of inhibition at the synapse (29, 30). Therefore, modulating the insertion and removal rate of GABAARs into or from the cell membrane has a marked affect on the amplitude of inhibitory synaptic currents (31). One way in which modulation occurs is via posttranslational modifications of the synaptic GABAAR. Specifically, the phosphorylation of key residues on synaptic GABAAR subunits regulates the extent to which the GABAAR will interact with protein complexes responsible for endocytosis from and insertion to the cell membrane; however, the significance of these regulatory processes for subtypes that mediate tonic inhibition remains largely unknown (32, 33). Phosphorylation plays a role in regulating the functional expression of GABAARs containing α4 subunits. Our results reveal that the α4 subunit is phosphorylated on Ser443 within the intracellular loop between transmembrane domains 3 and 4 (TM3 and TM4) in a protein kinase C (PKC)-dependent manner. Activating PKC also resulted in higher steady state cell surface accumulation of GABAARs containing the α4 subunit that was dependent on enhanced insertion into the plasma membrane when expressed in a mammalian cell line. Consistent with this, PKC-dependent phosphorylation of Ser433 produced a robust enhancement in GABA-induced currents in this expression system. Finally, we also observed that PKC activity increased both the phosphorylation and cell surface stability of the α4 subunit in hippocampal slices, a phenomenon that should be correlated with an increase in tonic inhibition. Together, these experiments establish a crucial role for PKC in regulating the functional expression of GABAAR subtypes that mediate tonic inhibition via direct phosphorylation of the α4 subunit.
EXPERIMENTAL PROCEDURES
Antibodies and Expression Constructs
Polyclonal rabbit anti-α4 and anti-δ antibody was graciously provided to us by Dr. Verena Tretter and Dr. Werner Sieghart from Medical University Vienna. β3 and phospho-β3(phospho-S408A/S409A) antibodies were designed by the Moss laboratory (34). Peroxidase-conjugated IgG secondary antibody was from Jackson ImmunoResearch Laboratories. Fluorescently labeled α-bungarotoxin (α-Bgt) was purchased from Invitrogen. Wild-type and mutant α4 subunit and wild-type β3 cDNAs were cloned into the mammalian cytomegalovirus (CMV) promoter for transgene expression. For fluorescence experiments, α4 subunit protein was conjugated with red fluorescent protein and the Bgt binding site (BBS) (35). Briefly, dsRed monomer fluorescent protein (RFP) was introduced after the 4th amino acid of the mature α4 subunit followed by the BBS sequence (WRYYESSLEPYPD). The RFP and the BBS were separated by a 12-alanine/1-proline linker (RFP-BBSα4 and RFP-BBSα4S443A (mutant described below)). All constructs were generated using standard molecular biology cloning techniques and sequenced fully.
Site-directed Mutagenesis
Mutation of the α4 subunit was carried out using the QuikChange site-directed mutagenesis kit (Stratagene). The mutagenesis primers used to introduce an alanine in place of a serine at site 443 were CCTTTGCGGTCGGCGGCTGCTCGCCCGGCATTT and AAATGCCGGGCGCAGCCGCCGACCGCAAAGG. All mutations were verified by DNA sequencing.
Expression of GABAAR Subunit Constructs in COS-7 Cells
α4 subunit cDNAs (mutant and tagged versions) and β3 subunit cDNA (where specified) were transfected into COS-7 cells using electroporation with 2 μg of plasmid DNA per construct. COS-7 cells incubated in 60-mm dishes with 4 ml of Dulbecco's modified Eagle's medium (DMEM)/F-12 (Invitrogen) plus 10% fetal bovine serum (FBS) at 37 °C with a 95% O2, 5% CO2 gas mixture and were utilized 48 h after transfection (36).
HEK293 Cell Culture and Transfection
Human embryonic kidney (HEK293) cells were cultured in medium composed of Dulbecco's modified Eagle's medium supplemented with 10% calf serum and 1% penicillin/streptomycin at 37 °C in a humidified 5% CO2 atmosphere. Cells were electroporated (110 V, Bio-Rad Gene Pulser Xcell) with equal ratios of cDNA encoding for GABAA receptor subunits along with GFP cDNAs (in pCDM8). Cells were used 24–72 h after transfection. Successful transfection of the cells was determined by fluorescence microscopy to identify GFP-labeled cells.
Hippocampal Slice Preparation
Hippocampal slices (350 μm thick) from 10–11-week-old C57BL/6 mice were prepared with a microslicer (Leica VT1000S) and pooled in ice-cold oxygenated artificial cerebral spinal fluid (ACSF). ACSF solution contents differed depending on the experiment being conducted. For 32P labeling experiments (described below), ACSF contained the following: 125 mm NaCl, 4 mm KCl, 26 mm NaHCO3, 1.5 mm MgSO4, 1.5 mm CaCl2, and 10 mm glucose, pH 7.4. For slice biotinylation experiments (described below), ACSF contained the following: 124 mm NaCl, 3 mm KCl, 25 mm NaHCO3, 2 mm MgSO4, 2 mm CaCl2, 1.1 mm NaH2PO4, and 10 mm glucose, pH 7.4. The slices were transferred individually to a solution containing fresh ACSF, gassed with a mixture of 95% O2, 5% CO2, and equilibrated in a 30 °C water bath for 1 h. Afterward, slices were utilized for either 32P labeling or slice biotinylation.
Cell Lysis and Immunoprecipitation
Samples collected from either COS-7 cell cultures or hippocampal slices were lysed in lysis buffer containing the following: 20 mm Tris-HCl (pH 8.0), 150 mm NaCl, 5 mm EDTA, 10 mm NaF, 2 mm Na3VO4, 10 mm sodium pyrophosphate, 1% Triton X-100, and 0.1% SDS. In addition, the following protease inhibitors were added: 250 μg/ml 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride; 10 μg/ml leupeptin, 1 μg/ml pepstatin, and 10 μg/ml antipain. Samples were then sonicated and spun at 16,000 × g. The supernatant was collected and then subjected to a protein assay using a standard Bradford protocol. 100–200 μg of protein were loaded per immunoprecipitation sample along with 3 μg of indicated antibody and 40 μl of protein A-Sepharose beads (1:1 slurry) (GE Healthcare). Samples were allowed to conjugate for 18–24 h at 4 °C with constant agitation. The beads were precipitated by centrifugation at 500 × g and washed once with ice-cold Buffer A (20 mm Tris-HCl (pH 8.0), 150 mm NaCl, 5 mm EDTA, 10 mm NaF, 2 mm Na3VO4, 10 mm sodium pyrophosphate, and 1% Triton X-100 and protease inhibitors), two times with Buffer B composed of Buffer A supplemented with 500 mm NaCl, and once again with Buffer A. After the final wash, the beads were resuspended in 25 μl of sample buffer and subjected to SDS-PAGE.
Whole-cell COS-7 Cell and Hippocampal Slice Metabolic 32P Labeling
COS-7 cells were transfected and incubated as described above. Cells were initially incubated in 2 ml of phosphate-free DMEM (Invitrogen) for 30 min at 37 °C. Following this incubation, cells were labeled with 0.5 mCi/ml [32P]orthophosphoric acid for 4 h in phosphate-free DMEM. Hippocampal slices were prepared as described above. Slices were individually transferred to polypropylene tubes containing 2 ml of fresh ACSF; gassed with a mixture of 95% O2, 5% CO2; and maintained in a 30 °C water bath. Labeling was performed by adding 0.5 mCi/ml [32P]orthophosphoric acid for 1 h. For both COS-7 cells and hippocampal slices, samples were treated with drugs where indicated after the labeling period, followed by the cell lysis and immunoprecipitation procedure described above. Results were attained by SDS-PAGE followed by autoradiography.
Phosphopeptide Mapping and Phosphoamino Acid Analysis
To perform phosphopeptide mapping, gel slices from 32P labeling experiments were excised from SDS-polyacrylamide gels and washed and digested with 0.1 mg/ml trypsin and subjected to two-dimensional mapping, first by electrophoresis and then by thin layer chromatography (TLC). The resulting plate was then visualized by autoradiography (37). For phosphoamino acid analysis, phosphoproteins from gel slices were hydrolyzed using 6 n HCl. The resulting phosphoamino acids, along with phosphoamino acid standards, were separated by TLC and visualized by autoradiography (37).
Metabolic [35S]Methionine Labeling
Transfected COS-7 cells were incubated in methionine-free DMEM for 20 min and then pulsed with 0.5 mCi/ml [35S]methionine (PerkinElmer Life Sciences) for 30 min. Cells were washed and incubated in complete DMEM/F-12 with an excess amount of unlabeled methionine for the indicated time periods (chase). Cells were lysed and subjected to immunoprecipitation as described above.
COS-7 Cell and Hippocampal Slice Cell Surface Biotinylation Assay
For transfected COS-7 cells, cultures were washed once with ice-cold PBS and then incubated in 2 ml of ice-cold PBS containing 1 mg/ml NHS-SS-biotin (Pierce) for 20 min in order to label surface proteins with biotin. After labeling, the biotin was quenched by incubating cells in PBS containing 25 mm glycine and 10 mg/ml bovine serum albumin (BSA) (38, 39). Cells were then lysed in lysis buffer and sonicated. For hippocampal slice experiments, slices were incubated in ACSF described above at 30 °C for 1 h for recovery before experimentation. Slices were then placed on ice and incubated for 30 min with 1 mg/ml NHS-SS-biotin. Excess biotin was removed by washing slices three times in ice-cold ACSF and lysed as described above (40). For both COS-7 cells and hippocampal slices, insoluble material was removed by centrifugation. The supernatant lysates were incubated with NeutrAvidin beads (Pierce) for 18–24 h at 4 °C. Bound material was eluted with sample buffer and subjected to SDS-PAGE and then immunoblotted with indicated antibodies. Blots were then quantified using the CCD-based FujiFilm LAS 300 system.
Fluorescent BBS Cell Membrane Insertion Assay
COS-7 cells were transfected with RFP-BBSα4 or RFP-BBSα4S443A and the β3 subunit. All surface proteins expressing the BBS were blocked with 10 μg/ml unlabeled α-Bgt for 15 min at 18 °C. The cells were then washed extensively to remove unbound α-Bgt. Newly inserted RFP-BBSα4 or RFP-BBSα4S443A was labeled with 2 μg/ml Alexa 647-conjugated α-Bgt and fixed immediately with 4% paraformaldehyde after the indicated time points (35). Confocal images of fluorescently labeled COS-7 cells were collected using a ×60 objective, acquired with Nikon acquisition software, and analyzed with MetaMorph.
Patch Clamp Electrophysiology
Cells were superfused, at a rate of 2 ml/min, with an extracellular solution containing 140 mm NaCl, 5 mm KCl, 1.2 mm MgCl2, 2.5 mm CaCl2, 10 mm HEPES, 11 mm glucose and adjusted to pH 7.4 with NaOH. Borosilicate glass patch pipettes (resistance 2–5 megaohms) were filled with an internal solution containing 140 mm KCl, 2 mm MgCl2, 0.1 mm CaCl2, 1.1 mm EGTA, 10 mm HEPES, 2 mm ATP (Mg2+ salt), adjusted to pH 7.4 with KOH. GABA was applied once every 120 s via a fast step perfusion system (Warner Instruments, Hamden, CT). All experiments were carried out at 32–33 °C using a recording chamber and in-line perfusion heaters (Warner Instruments). Phorbol esters were applied to the cell either internally via the electrode solution or superfused into the recording chamber.
Data Acquisition and Analysis
For biochemical and immunofluorescent experiments, data are presented as means ± S.E. Statistical analysis was performed by using Student's t test where a p value of <0.5 is considered significant. For electrophysiological experiments, currents were recorded with an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA), filtered at 2 kHz and digitized at 20 kHz with a Digidata 1320A data acquisition system (Molecular Devices), and analyzed using either Clampfit (pClamp, Molecular Devices) or GraphPad Prism version 4 software (GraphPad Software, Inc., San Diego, CA). Statistical analysis was performed by using one-way ANOVA with a Bonferroni post-test with statistical significance set at p < 0.05. All data are expressed as mean ± S.E.
RESULTS
Basal Phosphorylation of the GABAA Receptor α4 Subunit Is Enhanced by PKC When Expressed in COS-7 Cells
Immunoprecipitation was used to examine the phosphorylation of GABAA receptors in COS-7 cells transiently transected with the GABAA receptor α4 and β3 subunits. Immunoprecipitation with anti-α4 from transfected COS-7 cells that had been prelabeled with [32P]orthophosphoric acid under basal conditions yielded a major phosphoprotein at ∼64 kDa, demonstrating that the recombinant α4 subunit is basally phosphorylated (Fig. 1A). A corresponding band was not observed in untransfected COS-7 cells. Previous studies have shown that GABAA receptor subunits are the target of PKC (41–43). To determine whether the α4 subunit is a substrate of PKC, specific kinase activators and inhibitors were utilized. Activation of PKC with 500 nm phorbol 12,13-dibutyrate (PDBu) for 10 min produced a significant increase (p < 0.05) in α4 subunit phosphorylation of 223.7 ± 25.17% (n = 3) compared with control cells treated with DMSO for 10 min (Fig. 1A). An inhibitor of PKC, GF 109203X (GFX) (10 μm for 20 min), had little effect on the basal phosphorylation of the α4 subunit (74.39 ± 15.38% of control, n = 3). However, treating transfected COS-7 cells with GFX 10 min prior to PDBu treatment prevented the increase in α4 subunit phosphorylation (81.1 ± 26.98% of control, n = 3) observed with PDBu treatment alone (Fig. 1A). Peptide mapping and phosphoamino acid analysis revealed that the PKC-dependent phosphorylation of the α4 subunit primarily occurs on serine residues (Fig. 1C) within one major phosphopeptide (Fig. 1B, circled). Together these results strongly suggest that PKC enhances basal levels of phosphorylation on the α4 subunit.
FIGURE 1.

α4 subunit phosphorylation is increased by PDBu, a specific PKC activator. A, untransfected COS-7 cells (UT) or COS-7 cells transfected with GABAA receptor α4 and β3 subunits were labeled with 0.5 mCi/ml [32P]orthophosphoric acid and then treated with either PDBu (500 nm for 10 min) alone or following pretreatment with GFX (1 μm for 10 min), a PKC inhibitor. The α4 subunit was immunoprecipitated, subjected to SDS-PAGE, and visualized with autoradiography (top). The level of phosphorylation was normalized to the amount observed in vehicle-treated samples (bottom) (dashed line represents vehicle set at 100%, p < 0.05). B, phosphopeptide map of the α4 subunit. [32P]α4 immunopurified from transfected COS-7 cells was digested with trypsin, and the resulting phosphopeptides were blotted onto TLC plates and subjected to electrophoresis followed by ascending chromatography. The small arrow indicates the origin. C, the α4 subunit was subjected to phosphoamino acid analysis followed by autoradiography. The migration of phosphoserine (pS), phosphothreonine (pT), and phosphotyrosine (pY) standards is indicated. Error bars, S.E.
Ser443 Is a Major Site for PKC-dependent Phosphorylation of the α4 Subunit When Expressed in COS-7 Cells
To further analyze α4 subunit phosphorylation, site-directed mutagenesis was utilized to convert candidate serine residues within the α4 subunit intracellular domain to alanines. Based on the consensus PKC motif of (R/K)X1–4(S/T)X1–3(R/K) (44–45), a mutant version of the α4 subunit was produced in which Ser443 was changed to an alanine (α4S443A subunit) (Fig. 2A). COS-7 cells transfected with wild-type α4 or α4S443A and β3 subunits were subjected to [32P]orthophosphoric acid labeling and treated with either DMSO or PDBu. PDBu significantly enhanced (p < 0.05) levels of phosphorylation of wild-type α4 336 ± 50.03% of control (Fig. 2B). In contrast to wild type, PDBu did not significantly enhance the phosphorylation of the α4S443A subunit (Fig. 2B). These results strongly suggest that the major site for PKC phosphorylation within the intracellular domain of the α4 subunit is Ser443.
FIGURE 2.

PKC-dependent phosphorylation of the α4 subunit occurs within a PKC consensus motif. A, schematic depicting the protein structure of the α4 subunit. Examination of the intracellular domain (ICD) between transmembrane domains 3 and 4 (TM3 and TM4) reveals a serine that is located in a PKC consensus motif. B, untransfected COS-7 cells (UT) or COS-7 cells co-transfected with either wild-type (α4-WT) or S443A mutant (α4-S443A) GABAA receptor α4 and β3 subunits were labeled with [32P]orthophosphoric acid and treated with PDBu (500 nm for 10 min). Detergent-soluble extracts were immunoprecipitated with anti-α4, resolved by SDS-PAGE, and then visualized by autoradiography (top). The histogram is presented as 32P incorporation expressed as a percentage of vehicle-treated control (bottom) (dashed line represents vehicle set at 100%, p < 0.05). Error bars, S.E.
PKC-dependent Phosphorylation of Ser443 Enhances α4 Subunit Cell Surface Expression Levels in COS-7 Cells
Previous studies have shown that GABAA subunit phosphorylation dynamically regulates GABAAR cell surface expression (38, 46, 47). To address the functional consequences of α4 subunit phosphorylation, we first measured the effect PKC activation has on α4 subunit cell surface expression in transfected COS-7 cells using a biotinylation assay (38, 39). This revealed that a 10-min treatment with 500 nm PDBu significantly increased (p < 0.05) the cell surface levels of the α4 subunit by 144.9 ± 21.78%, but the total levels of protein were not altered (Fig. 3A). Treatment with GFX alone had no significant effect, and co-treatment with GFX and PDBu resulted in a modest increase in α4 subunit cell surface levels (Fig. 3A). Insignificant amounts of actin protein were pulled down in our biotinylated samples, ensuring that only cell surface proteins were being collected (Fig. 3A). In contrast to these results, PDBu treatment did not significantly increase (p < 0.05) the cell surface expression level of α4S443A (Fig. 3B). Together, these biochemical experiments indicate that the PKC-dependent phosphorylation of the α4 subunit on Ser443 increases receptor cell surface expression.
FIGURE 3.

PKC-dependent phosphorylation on Ser443 regulates the cell surface expression of the α4 subunit in COS-7 cells. A, COS-7 cells transfected with GABAA receptor α4 and β3 subunits were treated with either PDBu (500 nm for 10 min) alone or following pretreatment with GFX (1 μm for 10 min) and then labeled with NHS-SS-biotin. Detergent-soluble extracts were then purified on NeutrAvidin. The purified cell surface (Surface) and 10% of the total fraction (Total) were immunoblotted with α4 antibodies (top). Surface and total fractions were also blotted with actin to ensure the integrity of the cell surface assay. The amount of α4 subunit on the cell surface was then measured for each condition and normalized to the amount observed in vehicle-treated samples (lower panel) (dashed line represents vehicle set at 100%, p < 0.05). B, COS-7 cells co-transfected with either wild-type (α4-WT) or S443A mutant (α4-S443A) GABAA receptor α4 and β3 subunits were treated with either vehicle or PDBu (500 nm for 10 min) and then subjected to biotinylation. Histograms show the proportion of cell surface α4 protein expressed as a percentage of vehicle-treated controls (dashed line represents vehicle set at 100%; p < 0.05). Error bars, S.E.
Analyzing the Phosphorylation of the β3 and δ Subunits
In the brain, the α4 subunit assembles with the β2/3 subunit with or without the δ subunit (5–9, 48). Thus, we examined if these subunits are also subject to PKC-dependent phosphorylation. To do so, the α4, β3, and δ subunits were co-expressed in COS-7 cells and then labeled with [32P]orthophosphoric acid. After treatment with 500 nm PDBu for 10 min, the δ subunit was isolated via immunoprecipitation after denaturing lysis. Under these conditions, minimal levels of phosphorylation of the δ subunit were seen under basal conditions or after the activation of PKC (Fig. 4B). However, robust immunoprecipitation of the δ subunit was seen as measured via immunoblotting (Fig. 4B).
FIGURE 4.
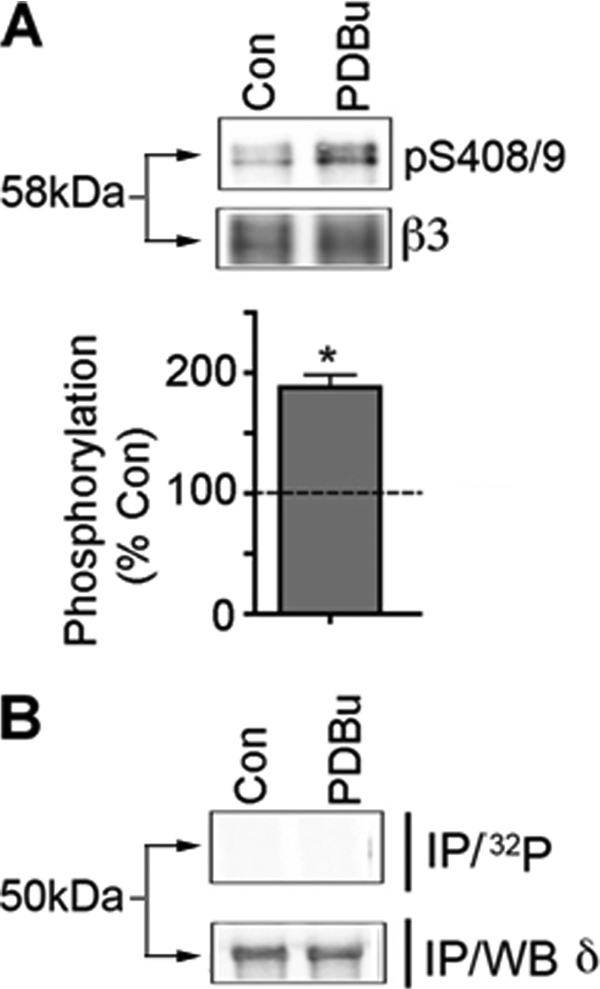
Analyzing PKC phosphorylation of GABAAR subunits that mediate tonic inhibition. A, COS-7 cells expressing the α4 and β3 subunits were treated with 500 nm PDBu for 10 min and then immunoblotted with phospho-S408A/S409A (pS408/9) or β3 antibodies, and the ratio of pS408/9/β3 immunoreactivity was determined and normalized to control (dashed line represents vehicle set at 100%; p < 0.05). B, COS-7 cells expressing α4, β3, and δ subunits were labeled with 0.5 mCi/ml [32P]orthophosphoric acid and treated with 500 nm PDBu for 10 min. The δ subunit was isolated by denaturing immunoprecipitation followed by SDS-PAGE (IP/32P). Parallel cultures were immunoprecipitated and immunoblotted with δ antibodies (IP/WB δ).
Previous studies have revealed that the β3 subunit is predominantly phosphorylated on serines 408/409 (Ser408/409) in neurons upon activation of PKC (34, 49). To examine if these residues are phosphorylated in COS-7 cells, lysates were immunoblotted with a phosphospecific antibody against these residues, phosphoserines 408/409 (phospho-Ser408/409). PDBu treatment produced a robust enhancement of Ser408/409 phosphorylation as measured via immunoblotting with Ser(P)408/409 antibody (Fig. 4A). Thus, this experiment suggests that the δ subunit is not a PKC substrate at least when expressed in COS-7 cells and suggests that the principle sites for PKC phosphorylation within GABAAR subtypes that mediate tonic inhibition are Ser443 in the α4 subunit and Ser408/409 in β3.
Mutation of Ser443 Increases the Rate of Insertion of the α4 Subunit on the Cell Membrane
To further evaluate the mechanism underlying PKC-dependent modulation of the α4 subunit cell surface stability, we determined what effect mutating the PKC site on the α4 subunit has on the level of insertion of α4 subunit-containing receptors. To do so, we utilized a Bgt binding assay that has previously been used to measure the rates of insertion of various receptor types (35, 50). To analyze the insertion of the α4 subunit, we engineered the α4 subunit with the BBS peptide, WRYYESSLEPYPD. The BBS is derived from the α subunit of the muscle nicotinic receptor and has been established to bind Bgt with an affinity of ∼3 nm (51, 52). The BBS together with a red fluorescent protein reporter were added to the N-terminal region of both the α4 and α4S443A subunit.
Before conducting our insertion assay, we verified that the BBS-tagged versions of our α4 constructs were capable of forming a functional GABAA R. To do so, we expressed RFP-BBSα4 and non-tagged α4 separately with the β3 subunit in HEK293 cells and measured the current responses to 1 μm and 1 mm GABA. Utilizing this method, we demonstrated that the RFP-BBSα4β3 GABAAR subtype forms a functional receptor and has GABA-mediated currents similar to those of the α4β3 GABAAR subtype (Fig. 5).
FIGURE 5.

The RFP-BBSα4 subunit forms a functional channel when expressed with the β3 subunit. Examples of GABA-mediated currents recorded from HEK293 cells expressing wild-type α4β3 and RFP-BBSα4β3 GABAARs. A solid line above the trace represents the application of either 1 mm (black line) or 1 μm (gray line) GABA.
To measure the role that Ser443 plays in regulating the cell surface accumulation of α4, RFP-BBSα4 and RFP-BBSα4S443A cDNAs were separately transfected into COS-7 cells along with the GABAA β3 subunit and subjected to the BBS insertion assay. To perform this assay, we first masked the surface RFP-BBSα4 and RFP-BBSα4S443A with unlabeled Bgt by incubating the transfected COS-7 cells with native Bgt at 18 °C for 15 min. Under these conditions, the unlabeled Bgt completely blocked the existing cell surface population of RFP-BBSα4 and RFP-BBSα4S443A subunits. Next, the cells were incubated at 37 °C with Alexa 647-Bgt in order to fluorescently label newly inserted RFP-BBSα4 and RFP-BBSα4S443A. Visually, it is apparent that there is a higher amount of RFP-BBSα4S443A inserted after 10 min compared with RFP-BBSα4 (Fig. 6A). To quantify these results, we calculated the ratio of the level of Alexa 647-Bgt staining to the level of RFP fluorescence. This ratio was significantly higher (p < 0.05) in COS-7 cells transfected with RFP-BBSα4S443A than in cells transfected with RFP-BBSα4 after 10 min of labeling (1.35 ± 0.33 versus 0.78 ± 0.16, n = 3; Fig. 6B). We also tested the rate of endocytosis of the RFP-BBSα4 and RFP-BBSα4S443A in a similar BBS assay and found no significant difference between the wild-type and mutant α4 subunit (data not shown). Together, these results strongly suggest that phosphorylation of Ser443, the major site of PKC-dependent phosphorylation within the α4 subunit, regulates the rate of insertion of the α4 subunit in COS-7 cells.
FIGURE 6.

S443A point mutation increases the rate of insertion of the α4 subunit into the cell membrane. A, α4-WT and α4-S443A DNA constructs were made containing an RFP tag as well as a BBS. COS-7 cells were then co-transfected with either wild-type (α4-WT-BBS) or S443A mutant (α4-S443A-BBS) and β3 GABAA receptor subunits. Transfected COS7 cells were then incubated with unlabeled Bgt (10 nm for 10 min) at 12 °C to block insertion. Cells were then incubated with Alexa 647-conjugated Bgt (10 nm for 10 min) at 37 °C. Cells were then fixed, and the level of newly inserted Alexa 647-tagged α4 was determined using confocal microscopy and quantified using MetaMorph. B, the graph is presented as a ratio of Alexa 647 fluorescent intensity (newly inserted protein) to RFP fluorescence (total protein) over specific time periods. Ratios for α4-WT and α4-S443A were then compared with one another (p < 0.05). Error bars, S.E.
Mutation of Ser443 Increases the Protein Stability of the α4 Subunit
To determine the role phosphorylation plays in the production and stability of the α4 subunit, COS-7 cells transfected with α4 or α4S443A alone were subjected to a [35S]methionine pulse-chase assay. Transfected COS-7 cells were labeled with 100 μCi/ml [35S]methionine for 30 min and chased for 0 and 4 h with excess cold methionine. Cell lysates were then prepared and subjected to immunoprecipitation with anti-α4 and resolved on SDS-polyacrylamide gels and quantified on a Bio-Rad isotope imager (Fig. 7). Data at 4 h are presented as a percentage of [35S]methionine-labeled protein existing at time 0. α4 subunit protein does not reach the cell surface unless a β3 subunit is also present (supplemental Fig. 1); therefore, under these conditions, we are measuring the stability of protein that is retained in the endoplasmic reticulum. Using this technique, we determined that 47.40 ± 8.18% of newly synthesized α4 subunit protein remained after 4 h (Fig. 7). Interestingly, this reduction was markedly less robust for the α4S443A subunit protein, reducing to 80.20 ± 7.00% of newly synthesized protein after 4 h (Fig. 7). Therefore, the α4S443A subunit is more stable than the wild-type α4 subunit, suggesting that PKC phosphorylation of the Ser443 site regulates the α4 subunit protein half-life.
FIGURE 7.

S443A point mutation reduces turnover of the α4 subunit in transfected COS-7 cells. Untransfected COS-7 cells (UT) or COS-7 transfected with either wild-type (α4-WT) or S443A mutant (α4-S443A) GABAA receptor subunits subjected to a pulse-chase with [35S]methionine. Cells were lysed and immunoprecipitated with anti-α4 subunit antibody and then subjected to SDS-PAGE. Bands were then analyzed by autoradiography (top). Turnover levels are presented as a percentage of levels at time 0 (bottom) (p < 0.05). Error bars, S.E.
Protein Kinase C Activation Reverses Current Run-down
The functional effects of protein kinase C activation were determined by whole-cell patch clamp recording of HEK293 cells. Transient expression of wild-type α4β3 receptors and mutant α4S443Aβ3 receptors resulted in functional channels that had GABA-evoked EC50 values of 1.7 ± 0.7 and 2.6 ± 0.9 μm (n = 3–10), respectively, with both Hill coefficients of 0.8 ± 0.2 (data not shown). To examine the run-down of GABA-mediated currents, 1 μm GABA was applied to the cells once every 2 min.
In the absence of PKC activation, the GABA-mediated current amplitude decreased over time but appeared to plateau after 16 min of recording. After 20 min of recording, the GABA-mediated current was 37 ± 12% (n = 5) of the initial response. Inclusion of 100 nm PDBu in the recording pipette solution prevented the GABA-mediated current amplitude run-down. At 20 min after the start of the experiment, the GABA-mediated current amplitude was 97 ± 9% (n = 3) of the initial GABA-mediated response. Similarly, external application of 100 nm PDBu also reverses GABA-mediated current amplitude run-down with current amplitude being 107 ± 12% (n = 4) compared with that at the start of the experiment (Fig. 8).
FIGURE 8.

Run-down of GABAA receptor α4β3-mediated responses are prevented with protein kinase C activation. A, 1 μm GABA-activated currents recorded at 0, 10, and 20 min after the start of the experiment (defined as t = 0 min and 100%), recorded 3–5 min after achieving the whole-cell configuration. Whole-cell currents were recorded from HEK293 cells expressing α4 and β3 subunits in the absence (control, upper currents) and presence (+[PDBu]i; lower currents) of 100 nm internal PDBu. Holding potential was −60 mV at 32 °C. B, time dependence relationship for 1 μm GABA-activated currents recorded from α4β3 receptors without (open squares) or with (solid squares) 100 nm PDBu internally perfused or with 100 nm PDBU externally perfused (solid diamonds). All data points are mean ± S.E. (error bars).
When the PKC-inactive phorbol ester, 4-α-phorbol 12,13-didecanoate (100 nm), was included in the intracellular solution of the recording pipette, the GABA-mediated current amplitude run-down was no different from control. Cells rarely remained healthy past 16 min of recording. After 16 min of recording, the GABA-evoked currents were 17 ± 6% (n = 3) of the first current in the presence of 100 nm 4-α-phorbol 12,13-didecanoate compared with 33 ± 10% (n = 7) in control (Fig. 9). At this time point, minimal run-down was observed in cells internally perfused with 100 nm PDBu (98 ± 9% of control, n = 3).
FIGURE 9.

Inclusion of the inactive phorbol ester, 4-α-Phorbol 12,13-didecanoate, does not prevent run-down of GABAA receptor α4β3-mediated responses. A, overlaid GABA-evoked currents from HEK293 cells expressing α4β3 receptors recorded at t = 0 (gray) and t = 16 (black) min after the start of the experiment. Significant run-down of current amplitude at t = 16 compared with t = 0 is observed in control and in the presence of 4-α-phorbol 12,13-didecanoate (4α-phorbol). In comparison, the current at t = 16 min in the presence of internal 100 nm PDBU was not different from that at t = 0 min. B, bar graph of the relative current at t = 16 min compared with current at t = 0 min for cells in control conditions (n = 8), perfused internally with 100 nm PDBU (n = 3) or 100 nm 4α-phorbol (n = 3). Values are mean ± S.E. (error bars).
The α4S443A Subunit Mutation Prevents Run-down
GABA-mediated currents in cells expressing α4S443Aβ3 receptors did not show the typical time-dependent run-down phenomena. Unlike that observed with wild-type α4-containing receptors in control conditions, after 20 min of recording, the GABA-mediated current from α4S443A containing receptors was 84 ± 13% (n = 4) of the initial GABA-mediated response. In the presence of internal 100 nm PDBu, GABA-mediated current was 98 ± 9% (n = 4) of the initial response after 20 min of recording (Fig. 10).
FIGURE 10.

Run-down is prevented with the inclusion of the α4S443A mutation. A, whole-cell currents recorded from HEK293 cells expressing α4S443A β3 receptors. 3–5 min after achieving the whole cell configuration (defined as t = 0 and 100%), mediated by 1 μm GABA were recorded at 0, 10, and 20 min after the start of the experiment and recorded. Current was recorded in the absence (control, upper currents) and presence of 100 nm PDBu (lower currents). B, time dependence relationship for 1 μm GABA-activated currents recorded from α4β3 receptors without (open squares) or with (solid squares) 100 nm PDBu internally perfused. All data points are mean ± S.E. (error bars).
PKC Activity Modulates the Phosphorylation and Cell Surface Stability of Endogenous α4 Subunit in Hippocampal Slices
To determine the relevance of our recombinant studies, we examined the phosphorylation of the endogenous α4 subunit and its effects on cell surface stability of this protein in hippocampal slices. We began by examining the PKC-dependent phosphorylation of the α4 subunit. Hippocampal slices were cut from brains dissected from 10–11-week-old C57BL/6 mice. Slices were labeled with [32P]orthophosphoric acid for 4 h in ACSF continuously being bubbled with a 95% O2, 5% CO2 gas mixture. Toward the end of the 4 h, control slices were treated with DMSO for 10 min, whereas PKC slices were treated with 500 nm PDBu for 10 min. Afterward, slices were lysed and subjected to immunoprecipitation with anti-α4. Under control conditions, a very faint band at ∼64 kDa was detected, representing the basal level of α4 subunit phosphorylation (Fig. 11A). Treatment of slices with PDBu significantly increased (p < 0.05) α4 subunit phosphorylation to 2016 ± 1260% of control (Fig. 11A).
FIGURE 11.

PKC increases the level of phosphorylation and cell surface expression of the α4 subunit in hippocampal slices. A, hippocampal slices from 10–11-week-old C57BL/6 male mice were labeled with [32P]orthophosphoric acid and treated with either vehicle or PDBu (500 nm for 10 min). Detergent-soluble extracts were immunoprecipitated with either rabbit IgG or anti-α4, resolved by SDS-PAGE, and then visualized by phosphorimaging (top). Histograms are presented as 32P incorporation expressed as a percentage of vehicle-treated control (bottom) (dashed line, p < 0.05). B, the immunoprecipitated α4 subunit from [32P]orthophosphoric acid-treated hippocampal slices was subjected to phosphoamino acid analysis followed by autoradiography. The migration of phosphoserine (pS), phosphothreonine (pT), and phosphotyrosine (pY) standards is indicated. C, hippocampal slices from 10–11-week-old C57BL/6 male mice treated with either vehicle or PDBu (500 nm for 10 min) were labeled with NHS-SS-biotin and detergent-soluble extracts were purified on NeutrAvidin. Cell surface (Surface) and 10% of total fractions (Total) were analyzed by immunoblotting with anti-α4 (top). Histograms show the proportion of cell surface α4 protein expressed as a percentage of vehicle-treated controls (bottom) (dashed line, vehicle set at 100%; p < 0.05). Error bars, S.E.
We further evaluated phosphorylation of the α4 subunit in hippocampal slices by performing phosphoamino acid analysis. We determined that PKC activation with PDBu resulted in the phosphorylation of the α4 subunit principally on serine resides (Fig. 11B), similar to what was observed in transfected COS-7 cells (Fig. 1C).
We also examined the effects of PKC activation on the cell surface stability of the α4 subunit in hippocampal slices using biotinylation. This revealed that activation with PDBu produced a significant increase (p < 0.05) in the amount of α4 subunit protein on the surface of cells in the hippocampal slice to 293.7 ± 90.70% of DMSO-treated slices (Fig. 11C). Similar to what was observed in our recombinant studies, these results demonstrate that the α4 subunit, in its native environment, is phosphorylated in a PKC-dependent fashion and that this post-translational modification increases the targeting of the α4 subunit to the cell surface.
DISCUSSION
The hippocampus is responsible for the formation of various types of memory in mammals. Pivotal to this role, the dentate gyrus of the hippocampus acts as an information-processing center between afferent connections from the entorhinal cortex and efferent connections to cornu ammonis region 3 of the hippocampus (53). Specifically, the dentate gyrus is responsible for filtering out stimulus-related high frequency firing from the entorhinal cortex and organizing it into a coherent signal that the rest of the hippocampus can utilize (54). Compromising filtering function of the dentate gyrus leads to epileptiform activity in downstream hippocampal structures (55). The filter-like function of the dentate gyrus is due to the intrinsic low excitability of granule cells residing in this region. This low excitability is a result of the high levels of protein expression of the GABAA α4 subunit, creating a significant degree of tonic inhibition (10). Therefore, understanding the cellular mechanisms that control the activity of the α4 subunit is crucial for better comprehending the complex mechanisms of memory formation as well as other higher brain functions where α4 subunit-mediated tonic inhibition has been shown to play a role.
Here we have begun to analyze the endogenous mechanisms that neurons utilize to regulate the efficacy of tonic inhibition, focusing on the possible role that direct phosphorylation may play in these processes. For these studies, we used receptors composed of α4β3 and α4β3δ subunits because both of these combinations have been demonstrated to be present within the dentate gyrus of the hippocampus as measured using biochemical and electrophysiological experiments (5–9). We initiated these studies by determining if the α4 subunit is phosphorylated by expressing this protein in COS-7 cells and measuring the amount of radiolabeled phosphate that covalently bonds to the α4 subunit. This revealed that the α4 subunit is phosphorylated basally, and activation of PKC enhances subunit phosphorylation. Peptide mapping and phosphoamino acid analysis showed that PKC-dependent phosphorylation of the α4 subunit occurs on a serine residue in one distinct phosphopeptide region. Using site-directed mutagenesis, we determined that the α4 subunit is phosphorylated at Ser443, an amino acid located in the major intracellular domain between TM3 and TM4. In addition to our recombinant studies, we also showed that PKC leads to high levels of α4 subunit serine phosphorylation in hippocampal slices from adult male mice. We also analyzed the phosphorylation of the δ and β3 subunits in our study. Consistent with studies on GABAAR subtypes that mediate phasic inhibition, Ser408/409 in the β3 subunit were phosphorylated by PKC activity when expressed with α4. However, at least in COS-7 cells, only low levels of δ subunit phosphorylation were seen. Thus, these results suggest that the primary PKC substrates with GABAAR subtypes that mediate tonic inhibition are Ser443 in the α4 subunit and Ser408/409 in β3.
To begin ascertaining the functional consequences of phosphorylation on tonic inhibition, we looked at the effect PKC activation had on the cell surface stability of the α4 subunit. The activation of PKC leads to a dramatic increase in the amount of α4 subunit protein at the cell surface in both transfected COS-7 cells and hippocampal slices, as measured by biotinylation. The Ser443 phosphorylation site plays a crucial role in this enhancement because mutation of this residue did not result in elevated levels of α4 subunit protein at the cell surface of COS-7 cells. At this point, it was clear that Ser443 is essential in mediating the effects of PKC activity on α4 subunit cell surface accumulation. To begin to address the underlying mechanism, we measured the rate of insertion of the α4 subunit into the cell membrane using a BBS fluorescent insertion assay. Here we discovered that over a 10-min period, more mutant α4 was being inserted into the COS-7 cell membrane than wild type α4 subunit. This increased rate of insertion was also paralleled with an increase in stability of newly translated α4S433A subunit compared with wild type α4 when expressed alone in COS-7 cells. Given that the α4 subunit is retained within the endoplasmic reticulum in homomeric expression, this result suggests that phosphorylation of Ser443 acts to regulate the stability of the α4 subunit in this intracellular compartment, which would be predicted to increase receptor assembly, leading to increased insertion into the plasma membrane.
To investigate this possibility, we measured the amount of protein degradation using an [35S]methionine pulse-chase assay and found that the α4 subunit Ser443 mutant was more stable in the endoplasmic reticulum over a 4-h period. Taking these results together, we see a situation in which the mutant version of the α4 subunit is not only degraded less but is inserted faster into the cell membrane. At first glance, this seems at odds with our results showing that Ser443 is a critical residue for the PKC-dependent phosphorylation of the α4 subunit that leads to higher levels of this protein on the cell surface. How is it then that ablating this phosphorylation site prevents α4 from being phosphorylated but still causes the mutant protein to be inserted at a faster rate? One answer to this question is that mutation of Ser443 to an alanine results in phosphorylation mimic of the α4 subunit. That is to say that masking the hydroxyl group that is normally found in a serine residue by removing it, which is what we do when we replace this residue with an alanine, is tantamount to masking it by covalently attaching a phosphate group. Both situations may lead to similar protein conformational changes that result in the α4 subunit being more stable and therefore being inserted at a faster rate. We can further draw this conclusion from our electrophysiological studies, which suggest a similar occurrence. In these studies, we found that PKC activation reverses the run down that normally occurs in non-treated HEK293 cells expressing the wild-type α4 subunit, as is expected from our biotinylation studies. Interestingly, HEK293 cells expressing the α4 phosphomutant do not exhibit any run down, either in the presence or absence of PKC activation. That is to say that the mutant by itself is protected by the run-down effect that is normally observed in the wild-type α4 subunit. If this is the case, it is logical to conclude that the reason PKC does not exert an effect on the mutant α4 subunit is that the protein is already acting as if it is constitutively phosphorylated and is being inserted into the membrane at a maximal rate.
Here we have for the first time a description of a PKC-dependent mechanism that regulates the activity of a GABAAR subunit that is primarily expressed in extrasynaptic sites. Our laboratory has shown in the past that synaptic GABAARs are highly regulated by kinase and phosphatase activity (32). Due to the plethora of kinases and phosphatases that exert an effect on synaptic GABAARs and the different brain regions and cell types in which this activity has been observed, phosphorylation regulates synaptic inhibition in a multitude of ways. However, one common facet of phosphoregulation of synaptic GABAARs, with respect to cell surface stability, is that it modulates the endocytosis of the receptor. In contrast, we have shown in this study that kinase activity affects the insertion of extrasynaptic GABAAR subtypes. Synaptic and extrasynaptic inhibition are two fundamentally different ways a neuron can regulate its excitability; therefore, it is important that the neuron be able to regulate each form by modulating different cellular mechanisms.
In summary, our studies demonstrate that the α4 subunit, a protein critical for tonic inhibition in the dentate gyrus of the hippocampus, is phosphorylated by PKC on Ser443. This phosphorylation leads to an increase in the functional expression of the α4 subunit-containing GABAAR by increasing its stability and enhancing the rate at which this receptor is inserted into the plasma membrane. Therefore, PKC-dependent phosphorylation of the α4 subunit may have profound effects on the efficacy of tonic inhibition mediated by GABAARs.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health/NINDS Grants NS048045, NS051195, NS056359, and NS054900 (to S. J. M.). S. J. M. serves as a consultant for Pfizer Pharmaceuticals, a relationship that is regulated by Tufts University and does not impact on this work.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. 1.
- GABAAR
- γ-aminobutyric acid type A receptor
- GABA
- γ-aminobutyric acid
- TM
- transmembrane domain
- Bgt
- bungarotoxin
- BBS
- bungarotoxin binding site
- RFP
- red fluorescent protein
- COS-7
- CV-1 monkey cell line in origin containing SV40 genetic material
- ACSF
- artificial cerebral spinal fluid
- NHS-SS-biotin
- succinimidyl 2-(biotinamido)-ethyl-1,3′ dithiopropionate
- PDBu
- phorbol 12,13-dibutyrate
- GFX
- GF 109203X.
REFERENCES
- 1.Farrant M., Nusser Z. (2005) Nat. Rev. Neurosci. 6, 215–229 [DOI] [PubMed] [Google Scholar]
- 2.Rudolph U., Crestani F., Möhler H. (2001) Trends Pharmacol. Sci. 22, 188–194 [DOI] [PubMed] [Google Scholar]
- 3.Sieghart W., Sperk G. (2002) Curr. Top. Med. Chem. 2, 795–816 [DOI] [PubMed] [Google Scholar]
- 4.Mody I. (2005) J. Physiol. 562, 37–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brünig I., Scotti E., Sidler C., Fritschy J. M. (2002) J. Comp. Neurol. 443, 43–55 [DOI] [PubMed] [Google Scholar]
- 6.Sun C., Sieghart W., Kapur J. (2004) Brain Res. 1029, 207–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jia F., Pignataro L., Schofield C. M., Yue M., Harrison N. L., Goldstein P. A. (2005) J. Neurophysiol. 94, 4491–4501 [DOI] [PubMed] [Google Scholar]
- 8.Bencsits E., Ebert V., Tretter V., Sieghart W. (1999) J. Biol. Chem. 274, 19613–19616 [DOI] [PubMed] [Google Scholar]
- 9.Mortensen M., Smart T. G. (2006) J. Physiol. 577, 841–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chandra D., Jia F., Liang J., Peng Z., Suryanarayanan A., Werner D. F., Spigelman I., Houser C. R., Olsen R. W., Harrison N. L., Homanics G. E. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 15230–15235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liang J., Suryanarayanan A., Chandra D., Homanics G. E., Olsen R. W., Spigelman I. (2008) Alcohol Clin. Exp. Res. 32, 19–26 [DOI] [PubMed] [Google Scholar]
- 12.Hamann M., Rossi D. J., Attwell D. (2002) Neuron 33, 625–633 [DOI] [PubMed] [Google Scholar]
- 13.Mitchell S. J., Silver R. A. (2003) Neuron 38, 433–445 [DOI] [PubMed] [Google Scholar]
- 14.Semyanov A., Walker M. C., Kullmann D. M. (2003) Nat. Neurosci. 6, 484–490 [DOI] [PubMed] [Google Scholar]
- 15.Chadderton P., Margrie T. W., Häusser M. (2004) Nature 428, 856–860 [DOI] [PubMed] [Google Scholar]
- 16.Cope D. W., Hughes S. W., Crunelli V. (2005) J. Neurosci. 25, 11553–11563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Park J. B., Skalska S., Stern J. E. (2006) Endocrinology 147, 3746–3760 [DOI] [PubMed] [Google Scholar]
- 18.Bright D. P., Aller M. I., Brickley S. G. (2007) J. Neurosci. 27, 2560–2569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rothman J. S., Cathala L., Steuber V., Silver R. A. (2009) Nature 457, 1015–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maguire J., Mody I. (2008) Neuron 59, 207–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Caraiscos V. B., Newell J. G., You-Ten K. E., Elliott E. M., Rosahl T. W., Wafford K. A., MacDonald J. F., Orser B. A. (2004) J. Neurosci. 24, 8454–8458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wallner M., Hanchar H. J., Olsen R. W. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 15218–15223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheng V. Y., Bonin R. P., Chiu M. W., Newell J. G., MacDonald J. F., Orser B. A. (2006) Anesthesiology 105, 325–333 [DOI] [PubMed] [Google Scholar]
- 24.Maguire J., Mody I. (2007) J. Neurosci. 27, 2155–2162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.D'Hulst C., De Geest N., Reeve S. P., Van Dam D., De Deyn P. P., Hassan B. A., Kooy R. F. (2006) Brain Res. 1121, 238–245 [DOI] [PubMed] [Google Scholar]
- 26.Maldonado-Avilés J. G., Curley A. A., Hashimoto T., Morrow A. L., Ramsey A. J., O'Donnell P., Volk D. W., Lewis D. A. (2009) Am. J. Psychiatry 166, 450–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Feng H. J., Kang J. Q., Song L., Dibbens L., Mulley J., Macdonald R. L. (2006) J. Neurosci. 26, 1499–1506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang N., Wei W., Mody I., Houser C. R. (2007) J. Neurosci. 27, 7520–7531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nusser Z., Cull-Candy S., Farrant M. (1997) Neuron 19, 697–709 [DOI] [PubMed] [Google Scholar]
- 30.Nusser Z., Hájos N., Somogyi P., Mody I. (1998) Nature 395, 172–177 [DOI] [PubMed] [Google Scholar]
- 31.Kittler J. T., Delmas P., Jovanovic J. N., Brown D. A., Smart T. G., Moss S. J. (2000) J. Neurosci. 20, 7972–7977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tretter V., Moss S. J. (2008) Front. Mol. Neurosci. 1, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tang X., Hernandez C. C., Macdonald R. L. (2010) J. Neurophysiol. 103, 1007–1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jovanovic J. N., Thomas P., Kittler J. T., Smart T. G., Moss S. J. (2004) J. Neurosci. 24, 522–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bogdanov Y., Michels G., Armstrong-Gold C., Haydon P. G., Lindstrom J., Pangalos M., Moss S. J. (2006) EMBO J. 25, 4381–4389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Connolly C. N., Kittler J. T., Thomas P., Uren J. M., Brandon N. J., Smart T. G., Moss S. J. (1999) J. Biol. Chem. 274, 36565–36572 [DOI] [PubMed] [Google Scholar]
- 37.McDonald B. J., Moss S. J. (1994) J. Biol. Chem. 269, 18111–18117 [PubMed] [Google Scholar]
- 38.Kittler J. T., Thomas P., Tretter V., Bogdanov Y. D., Haucke V., Smart T. G., Moss S. J. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 12736–12741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fairfax B. P., Pitcher J. A., Scott M. G., Calver A. R., Pangalos M. N., Moss S. J., Couve A. (2004) J. Biol. Chem. 279, 12565–12573 [DOI] [PubMed] [Google Scholar]
- 40.Terunuma M., Jang I. S., Ha S. H., Kittler J. T., Kanematsu T., Jovanovic J. N., Nakayama K. I., Akaike N., Ryu S. H., Moss S. J., Hirata M. (2004) J. Neurosci. 24, 7074–7084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brandon N. J., Delmas P., Kittler J. T., McDonald B. J., Sieghart W., Brown D. A., Smart T. G., Moss S. J. (2000) J. Biol. Chem. 275, 38856–38862 [DOI] [PubMed] [Google Scholar]
- 42.Krishek B. J., Xie X., Blackstone C., Huganir R. L., Moss S. J., Smart T. G. (1994) Neuron 12, 1081–1095 [DOI] [PubMed] [Google Scholar]
- 43.McDonald B. J., Amato A., Connolly C. N., Benke D., Moss S. J., Smart T. G. (1998) Nat. Neurosci. 1, 23–28 [DOI] [PubMed] [Google Scholar]
- 44.Kennelly P. J., Krebs E. G. (1991) J. Biol. Chem. 266, 15555–15558 [PubMed] [Google Scholar]
- 45.Senawongse P., Dalby A. R., Yang Z. R. (2005) J. Chem. Inf. Model 45, 1147–1152 [DOI] [PubMed] [Google Scholar]
- 46.Brandon N. J., Uren J. M., Kittler J. T., Wang H., Olsen R., Parker P. J., Moss S. J. (1999) J. Neurosci. 19, 9228–9234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kittler J. T., Moss S. J. (2003) Curr. Opin. Neurobiol. 13, 341–347 [DOI] [PubMed] [Google Scholar]
- 48.Pirker S., Schwarzer C., Wieselthaler A., Sieghart W., Sperk G. (2000) Neuroscience 101, 815–850 [DOI] [PubMed] [Google Scholar]
- 49.Jacob T. C., Wan Q., Vithlani M., Saliba R. S., Succol F., Pangalos M. N., Moss S. J. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 12500–12505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sekine-Aizawa Y., Huganir R. L. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 17114–17119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Scherf T., Kasher R., Balass M., Fridkin M., Fuchs S., Katchalski-Katzir E. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 6629–6634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Katchalski-Katzir E., Kasher R., Balass M., Scherf T., Harel M., Fridkin M., Sussman J. L., Fuchs S. (2003) Biophys. Chem. 100, 293–305 [DOI] [PubMed] [Google Scholar]
- 53.Carlson G. C., Coulter D. A. (2008) Nat. Protoc. 3, 249–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Behr J., Lyson K. J., Mody I. (1998) J. Neurophysiol. 79, 1726–1732 [DOI] [PubMed] [Google Scholar]
- 55.Heinemann U., Beck H., Dreier J. P., Ficker E., Stabel J., Zhang C. L. (1992) Epilepsy Res. Suppl. 7, 273–280 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.