

Abstract
Aquifex aeolicus, a hyperthermophilic and microaerophilic bacterium, obtains energy for growth from inorganic compounds alone. It was previously proposed that one of the respiratory pathways in this organism consists of the electron transfer from hydrogen sulfide (H2S) to molecular oxygen. H2S is oxidized by the sulfide quinone reductase, a membrane-bound flavoenzyme, which reduces the quinone pool. We have purified and characterized a novel membrane-bound multienzyme supercomplex that brings together all the molecular components involved in this bioenergetic chain. Our results indicate that this purified structure consists of one dimeric bc1 complex (complex III), one cytochrome c oxidase (complex IV), and one or two sulfide quinone reductases as well as traces of the monoheme cytochrome c555 and quinone molecules. In addition, this work strongly suggests that the cytochrome c oxidase in the supercomplex is a ba3-type enzyme. The supercomplex has a molecular mass of about 350 kDa and is enzymatically functional, reducing O2 in the presence of the electron donor, H2S. This is the first demonstration of the existence of such a respirasome carrying a sulfide oxidase-oxygen reductase activity. Moreover, the kinetic properties of the sulfide quinone reductase change slightly when integrated in the supercomplex, compared with the free enzyme. We previously purified a complete respirasome involved in hydrogen oxidation and sulfur reduction from Aquifex aeolicus. Thus, two different bioenergetic pathways (sulfur reduction and sulfur oxidation) are organized in this bacterium as supramolecular structures in the membrane. A model for the energetic sulfur metabolism of Aquifex aeolicus is proposed.
Keywords: Bacterial Metabolism, Energy Metabolism, Membrane Enzymes, Metalloenzymes, Respiratory Chain, Hyperthermophile, Oxygen Respiration, Sulfur Metabolism, Supercomplex
Introduction
Aerobic respiration in the mitochondria of eukaryotes involves electron transfer through membrane-embedded enzyme complexes that use NADH to reduce O2 and generate a transmembrane proton gradient. The electron transport chain consists of the following four complexes: NADH and succinate dehydrogenases (complexes I and II), a bc1 complex (complex III), and a cytochrome c oxidase (complex IV). In prokaryotes, however, a variety of electron acceptors and donors may be used by the same organism. The composition of bioenergetic chains can thus differ significantly from that of mitochondria. These prokaryotic pathways are far more complex than that of mitochondria and are thus flexible to adapt to environmental changes.
Aquifex aeolicus is a hyperthermophilic, chemolithoautotrophic, and hydrogen-oxidizing bacterium isolated from a hydrothermal system at 102 °C at Vulcano Island, Italy (1–3). It obtains energy for growth from an unusual electron transfer, from H2 to O2, but only in the presence of a source of sulfur (thiosulfate or elemental sulfur S0), which is intriguing (4). We have proposed the existence of unusual aerobic respiratory chains in this bacterium (electron transfer from H2S to O2 as well as from H2 to O2), at least when it is grown with S0 (5, 6). We have also shown that it can reduce elemental sulfur with H2, producing H2S, in these growth conditions (4). All three pathways might function simultaneously in the cell (5). Moreover, we have demonstrated that the reduction of elemental sulfur involves a multiprotein supercomplex carrying the sulfur reducing activity (4).
The idea that sequential enzymes within a metabolic pathway interact with each other to form highly organized enzymes complexes was formulated a long time ago for soluble enzymes (7). In mitochondria, much evidence suggests that the different respiratory complexes are assembled into supermolecular structures (supercomplexes) to perform their role as respirasomes. The composition, abundance, and stability vary between different organisms (8–10). Some macromolecular complexes containing respiratory or photosynthetic proteins have been found in bacteria and archaea. In most cases, they contain a bc complex and a cytochrome c oxidase or a reaction center (5, 11–16). A supramolecular assembly with NADH oxidase activity in the membranes of the bacterium Paracoccus denitrificans constitutes a complete respirasome and includes complexes I, III, and IV (17). Several roles for association between proteins of a respiratory pathway have been proposed in the literature, including substrate channeling, optimization of electron transfer, and protein stabilization (10).
Previous studies on A. aeolicus membranes have revealed the possible existence of functionally relevant supercomplexes involved in pathways for H2 oxidation and O2 reduction and for H2S oxidation and O2 reduction (5). However, the precise protein composition of these complexes has not been established. Based on the probable interactions between certain of the respiratory components involved in O2 reduction in A. aeolicus (5), we have begun a biochemical, functional, and physico-chemical characterization of the putative protein assemblies present in the membranes of this bacterium. Here, we describe the purification and characterization of an enzymatically functional and thermostable supercomplex containing all the protein components required for the electron transfer from H2S to O2. It includes the sulfide quinone reductase (Sqr),3 the bc1 complex, and the cytochrome c oxidase enzymes, making this supercomplex the first of its kind to be recognized and analyzed. This work implies that A. aeolicus is the sole organism known to date that has two complementary energetic pathways (sulfur compounds reduction and oxidation) organized into supramolecular structures. Moreover, a modulation of properties of Sqr, depending on its environment, is suggested. A global view of the energetic sulfur pathway in A. aeolicus can be proposed on the basis of our results.
EXPERIMENTAL PROCEDURES
Growth of Bacteria
A. aeolicus VF5 was routinely grown at 85 °C in 2-liter bottles with 400 ml of medium containing only inorganic compounds, in the presence of elemental sulfur (7.5 g/liter) as described previously (4). Cells were harvested and stored at −80 °C.
Isolation of the A. aeolicus Supercomplex
Membranes (from 45 g of cells) were isolated as described previously (5). They were washed in 75 ml of 50 mm Tris-HCl, pH 7.6, 5% (v/v) glycerol, and 1.5 m NaBr for 30 min at room temperature with gentle shaking and ultracentrifuged at 45,000 rpm (Ti 45 rotor, Beckman) for 1 h at 4 °C to pellet them. They were resuspended in solubilization buffer (50 mm Tris-HCl, pH 7.6, 5% (v/v) glycerol, 750 mm aminocaproic acid) to give a protein concentration of 20 mg/ml and then solubilized with n-dodecyl β-d-maltoside (DDM) at 1% (w/v) final concentration. The solubilization was performed for 1 h, at 37 °C, with gentle shaking, and the solubilized membranes were ultracentrifuged as above. The supernatant was applied to a hydroxyapatite column (80 ml) equilibrated with 50 mm Tris-HCl, pH 7.6, 5% glycerol, 0.5% (w/v) aminocaproic acid, 0.02% (w/v) DDM (buffer A) at room temperature. The supercomplex, carrying cytochrome c oxidase activity, was followed during purification by detecting this activity directly in gel as described below. It was eluted from the first column with 300 mm potassium phosphate in buffer A. The eluate was concentrated ∼10-fold to give a volume of about 10 ml with concentrators (Vivaspin 100,000 molecular weight cutoff membranes, Sartorius Stedim Biotech) and applied to a Superdex 200 gel filtration column (FPLC, 120 ml, GE Healthcare) equilibrated with buffer A at a flow rate of 0.6 ml/min. Fractions (1 ml) with cytochrome c oxidase activity were pooled and concentrated as described above. The fraction was then applied to a Q-Sepharose column (FPLC, 20 ml, Hiload, GE Healthcare) equilibrated with buffer A. Proteins were eluted by a gradient of 0–500 mm NaCl in buffer A at a flow rate of 1 ml/min. Fractions with cytochrome c oxidase activity (200 mm NaCl) were pooled, concentrated, and washed with buffer A as described above.
Isolation of A. aeolicus Sqr
Membranes were prepared as described above. Sqr was solubilized from membranes with 2% (w/v) DDM in 50 mm Tris-HCl, pH 7.6, 5% (v/v) glycerol, 750 mm aminocaproic acid (in the same conditions as for the supercomplex). The supernatant of ultracentrifugation as described above was applied on a DEAE-cellulose equilibrated with buffer A. Sqr was eluted at 150 mm NaCl in buffer A. The eluate, concentrated and washed in buffer A, was applied to a hydroxyapatite column in buffer A and eluted at 150 mm potassium phosphate in buffer A. The fraction was then applied to a Mono Q column (FPLC, 1 ml, Hiload, GE Healthcare) equilibrated with buffer A, at a flow rate of 0.5 ml/min. Fractions with Sqr activity (175 mm NaCl) were pooled, concentrated, and washed with buffer A as described above. This Sqr fraction is called Sqrfree in the remainder of the paper.
Analytical Procedures
Electrophoresis
Proteins were separated by blue native gel by the method of Schägger et al. (18), as described previously (5). Proteins were also loaded on denaturing or regular native gels (Tris-glycine gels), using a mini-Protean III cell electrophoresis apparatus (Bio-Rad) in all cases (5). After migration, gels were either stained with Coomassie Blue R-250, or enzymatic activities were revealed, or proteins were detected with antibodies, or c-type cytochromes were specifically revealed with 3,3′,5,5′-tetramethylbenzidine (19). Protein bands were cut out from gels and stored at −20 °C before analysis by mass spectrometry.
In-gel Enzyme Activities
Cytochrome c oxidase activity was revealed on the gels as described previously (5). Cytochrome bc1 complex was detected at 37 °C as described previously (20). Sulfide quinone reductase activity was anaerobically revealed on the blue native gel, at high temperature, using sulfide as electron donor and quinone as electron acceptor. The assay mixture contained 50 mm BisTris, pH 7, 30 μm decyl-ubiquinone (DB), and 100 μm Na2S. Anoxic conditions were established by flushing the assay mixture with argon gas. Gels were incubated at 85 °C, and 1 mm 2,3,5-triphenyltetrazolium chloride was added to fix the red coloration of the bands corresponding to the Sqr.
Western Blotting
Proteins were subjected to native gels and transferred to nitrocellulose membranes (Electran) with the fast blot apparatus (Biometra) at 5 mA/cm2 for 30 min. The blots were then processed and developed as described previously (21). Primary antibodies used were directed against Sqr from A. aeolicus or against subunit 2 (CoxB) of cytochrome c oxidase from the bacterium Acidithiobacillus ferrooxidans, which we have shown here to cross-react with the terminal oxidase of Aquifex.
Activity Measurements
Individual Enzyme Activity Assays
All reactions were recorded on a Cary 50 Bio (Varian) spectrophotometer equipped with a Peltier-type temperature control system (single cell peltier accessory, Varian) using the purified supercomplex or the Sqrfree prepared as detailed above. All activities were measured at least three times to ensure reproducibility.
Sulfide quinone reductase activity was determined as described previously (5) using sodium sulfide as the electron donor and DB as the electron acceptor. This activity has been determined at various temperatures. When used, antimycin A was added at a final concentration of 50 μm. One enzyme unit is defined as the amount of Sqr that catalyzes the reduction of 1 μmol of DB/min.
bc1 complex activity was measured anaerobically at 289–300 nm, at 40 °C, after the reoxidation of reduced decyl-ubiquinone (DBH2) using horse heart cytochrome c as electron acceptor. DB was reduced with NaBH4, and a freshly prepared solution was used. The reaction mixture contained 50 mm Tris-HCl, pH 7, 50 μm ferric cytochrome c, 50 μm DBH2, and 0.6 mm KCN final concentration. When used, antimycin A was added at a final concentration of 0.2 mm. One enzyme unit is defined as the amount of bc1 complex that catalyzes the oxidation of 1 μm DBH2/min. The bc1 complex was also assayed, at 550 nm, after reduction of the ferric cytochrome c.
Cytochrome c oxidase activity was measured at 550 nm at 40 °C, by oxidation of horse heart ferrous cytochrome c as described previously (5). When used, KCN was added at a final concentration of 0.6 mm. One enzyme unit is defined as the amount of cytochrome c oxidase that catalyzes the oxidation of 1 μmol of cytochrome c/min.
The stability of the Sqr was measured following the enzyme activity after preincubation of the Sqrfree and the supercomplex at 80 °C. The activity was measured at 25 °C in the spectrophotometer, as described above.
Measurement of Oxygen Uptake
Respiratory activities using sulfide as electron donor were determined with a Clark-type oxygen electrode (Rank Brothers oxygraph). Oxygen consumption was measured in a 3-ml reaction system at 60 °C in 50 mm Tris-HCl, pH 7, with 3–15 μg of supercomplex prepared as detailed above or solubilized membranes, supplemented (when specified) with 4 μm DB and 1 μm horse heart cytochrome c. Sulfide oxidase activity was started by the addition of 100 μm Na2S. When used, KCN and antimycin A were added at final concentrations of 0.6 and 0.2 mm respectively. The oxygen uptake rate was negligible in the absence of sample. All reactions (enzymatic and nonenzymatic) were measured at least in triplicate. To avoid damage to the apparatus, oxygen uptake was not determined at higher temperatures. One enzyme unit corresponds to the amount of enzyme catalyzing the consumption of 1 nmol of O2/min.
Spectroscopy
UV-visible Spectroscopy
Spectra were recorded on a Cary 50 Bio (Varian) spectrophotometer at room temperature. Heme contents were calculated from the reduced spectra using the following adsorption coefficients, Δϵ560–571 = 18 mm−1·cm−1 for heme B and Δϵ552–540 = 19 mm−1·cm−1 for heme C (21).
Electron Paramagnetic Resonance (EPR)
Spectra were recorded at liquid helium temperatures with a Bruker ESP 300e X-band spectrometer equipped with an Oxford Instruments cryostat and temperature control system. Spectra were recorded on untreated (“as prepared”) and reduced A. aeolicus supercomplex. For that purpose, freshly prepared solution of 1 m sodium dithionite in 20 mm Tris-HCl, pH 7.6, was added to the sample to obtain a 5 mm final concentration of the reductant. The sample was frozen in liquid nitrogen.
Fluorescence Spectroscopy
The presence of FAD was determined using a FluoroMax-3 spectrofluorometer (Horiba Jobin Yvon). Emission spectra were obtained using an excitation wavelength of 375 nm with a slit of 5 mm. The sample (purified supercomplex) was diluted in 50 mm Tris-HCl, pH 7.6, 0.02% DDM at a concentration of 0.5 mg/ml.
Mass Spectrometry
Tryptic digestion of excised gel plugs, ion trap, and MALDI-TOF mass spectrometry protein identification were performed as described previously (5).
Analytical Ultracentrifugation
Sedimentation velocity experiments were performed using a Beckman XL-I analytical ultracentrifuge and an AN-60 TI rotor (Beckman Coulter). The purified supercomplex at 3.5 mg/ml was dialyzed against buffer A at 4 °C and diluted in the same buffer for sedimentation velocity experiments, which were carried out at 1.75, 0.35, and 0.10 mg/ml. A volume of about 100 μl, for the most concentrated sample, or 400 μl was loaded into 0.3- or 1.2-cm optical path two-channel cells, with buffer A but without DDM in the reference channel. Scans were recorded at 12 °C and 42,000 rpm every 15 min, overnight, at 278 and 413 nm, and by interference.
The solvent density, ρ = 1.017 g/ml, and viscosity, η = 1.58 centipoise at 12 °C, were estimated from measurements with the density meter DMA 5000 and viscosity meter AMVn (Anton PAAR) at 20 °C and extrapolation to 12 °C on the basis of the ratio of the values for water. We estimate the partial specific volume of the polypeptide chains with the Sednterp software (free and available on line) and considered the mean value, v̄P = 0.75 ml/g at 12 °C and v̄Det = 0.82 ml/g for DDM (22). The sedimentation profiles were analyzed with the size distribution analysis function (23) of the program Sedfit (free and available on line).
The value of the sedimentation coefficient depends on the complex shape (described by the hydrodynamic radius RS), partial specific volume (inverse of the particle density), v̄, and molar mass, M, according to the Svedberg Equation 1,
The term M(1 − ρv̄), for the supercomplex, solubilized in DDM and in a solvent containing glycerol, with a density significantly different from water, depends on supercomplex protein molar mass, MP, detergent binding BDet (g/g), and hydration Bw (g/g), with v̄P, v̄Det, and v̄w the corresponding partial specific volumes (24, 25) as shown in Equation 2,
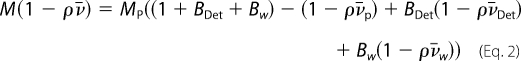 |
We took a typical value of Bw to be 0.3 g/g. The frictional ratio f/fmin relates RS to the minimum radius from the anhydrous volume. f/fmin = 1.25 for a globular compact macromolecule. From the amino acid composition, we calculate that the putative supercomplex of 310 kDa, including one dimeric bc1 complex, one cytochrome c oxidase, and one Sqr, has an extinction coefficient at 280 nm ϵ280 = 1.63 cm−1·mg−1·ml. For relating the fringe displacement, J, measured using interference optics, to concentration, we assumed refractive index increments of 0.187 and 0.143 ml/g for protein and DDM.
Analytical Size-exclusion Chromatography with Light Scattering Detection
The molecular mass of the supercomplex was also determined by multiangle laser light scattering and refractometry (refractive index) combined with size-exclusion chromatography. The supercomplex (20 μl) at 3.5 mg/ml in buffer A was injected onto a KW 804 column preceded by a guard column (Shodex), equilibrated in 20 mm Tris-HCl, pH 7.6, 100 mm NaCl, 5% glycerol, 0.02% DDM, 0.5% aminocaproic acid, at 20 °C with a flow rate of 0.5 ml/min. The elution profiles were measured on line by absorbance and refractive index using a multiwavelength detector (SPDM 20A from Shimazu) and an Optilab rEX (Wyatt Technology Corp.). On-line multiangle laser light scattering detection was performed with a miniDAWN TREOS detector (Wyatt Technology Corp.) using a laser emitting at 658 nm. Data were analyzed, and weight-averaged molar masses were calculated using the ASTRA version 5.3.4.18 software (Wyatt Technology Corp.), with the values of ∂n/∂c and ϵ280 given above. The molecular mass distribution was determined from combined multiangle laser light scattering and refractive index data and the protein content, taking account of the signal at 280 nm.
RESULTS
Purification of a Supercomplex Containing a Cytochrome c Oxidase
Based on previous studies, we decided to purify a superstructure that reduces O2, starting from cells cultured in H2/S0/O2 medium. The fractions displaying cytochrome c oxidase activity on blue native gel were combined, to give the final preparation of the purified complex. A supercomplex with a functional cytochrome c oxidase was then purified as described under “Experimental Procedures.”
Blue native gel electrophoresis of the purified fraction shows one major band of molecular mass around 350 kDa stained with Coomassie Blue (Fig. 1A), associated with cytochrome c oxidase activity (Fig. 1B), indicating that the complex is homogeneous. Two other very faint bands could be detected in some preparations (around 50 and 100 kDa), but these were not associated with the cytochrome c oxidase activity.
FIGURE 1.
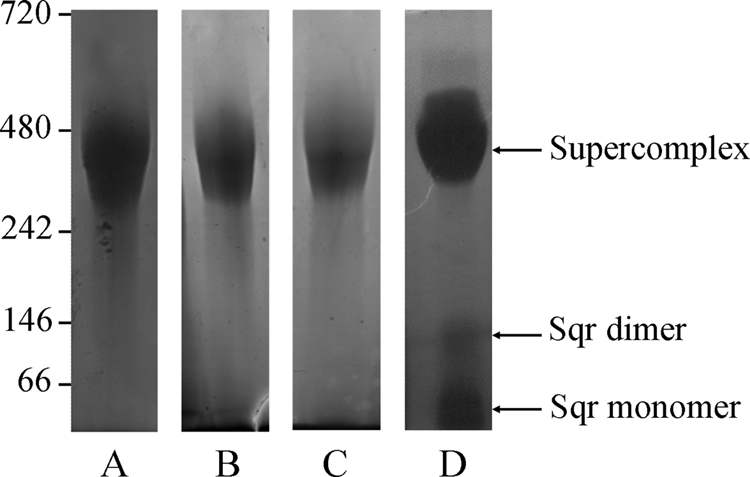
Migration of the purified supercomplex on blue native gel and in-gel detection of supercomplex components. 25 μg of supercomplex were loaded on 4–12% gels in all cases. A, detection by Coomassie Blue staining. B, detection of cytochrome c oxidase activity by in-gel assay. C, detection of bc1 complex. D, detection of Sqr activity. Molecular mass markers are indicated in kDa.
Identification of the Supercomplex Components
Mass Spectrometry
To precisely define the composition of the purified supercomplex, protein bands were directly cut out from blue native gels, digested with trypsin, and analyzed by ion trap mass spectrometry. The analysis of the 350-kDa band (Fig. 1A) confirms that the purified supercomplex contains a cytochrome c oxidase with identification of the two subunits Cox A2 and Cox B2 (Table 1). This enzyme, which we have named cytochrome c oxidase II, is the same as the one we previously detected in Aquifex membranes as part of potential supercomplexes (5). In addition, the fully assembled bc1 complex, containing the three subunits cytochrome c1 (cytochrome c in the table), cytochrome b, and the Rieske iron-sulfur protein, is present in the supercomplex as well as the Sqr, an enzyme of 47 kDa known to be involved in sulfur metabolism. Sqr transfers the electrons from H2S to a quinone pool. A protein of unknown function was also detected (Aq_814), which seems to have sequence similarities with serine dehydrogenase proteinases and the crotonase-like superfamily as found by a BLAST search. Analysis of the genetic environment of the Aq_814 gene showed that it is preceded by a gene encoding a possible DNA/pantothenate metabolism flavoprotein and followed by a gene encoding a holo-acyl carrier protein synthase. This protein is probably therefore just a contaminant present in the same protein gel band, although we cannot exclude the possibility that it is a true component of the supercomplex. No other proteins were identified by this approach.
TABLE 1.
Proteins composition of the purified supercomplex
Proteins were identified by ion trap mass spectrometry from the band cut out from the blue native gel shown in Fig. 1A.
| Protein namea | NCBI entryb | Gene | Coveragec | Peptidesd | Masse |
|---|---|---|---|---|---|
| Cytochrome c | 2982798 | cyc | 36.0 | 16 | 27.6 |
| Rieske-I iron-sulfur protein | 2982799 | petA | 18.2 | 6 | 19.4 |
| Cytochrome c oxidase subunit II | 2984392 | coxB2 | 23.3 | 5 | 9.5 |
| Sulfide quinone reductase | 2984385 | sqr | 18.4 | 5 | 47.4 |
| Hypothetical protein aq_814 | 2983372 | aq_814 | 9.7 | 6 | 32.5 |
| Cytochrome b | 2982800 | petB | 2.7 | 1 | 46.9 |
| Cytochrome c oxidase subunit I | 2984382 | coxA2 | 2.2 | 1 | 66.5 |
a Protein name is name as given in the NCBI database.
b NCBI entry is the accession number.
c Coverage indicates protein sequence coverage by the matching peptides (in %).
d Peptides indicate the number of different peptides matching the protein sequence.
e Mass indicates the theoretical molecular mass in kDa of the identified protein calculated from the amino acid sequence without possible processing or modifications.
The subunits of the supercomplex were visualized by gel electrophoreses on an SDS-PAGE (Fig. 2A). The presence of seven major protein bands was revealed by Coomassie Blue staining. Analysis by MALDI-TOF mass spectrometry of all of them confirmed the composition of the supercomplex (Fig. 2B). Three of these proteins (cytochromes c1 and b and Rieske protein) are components of the bc1 complex. Sqr was detected, as well as the small subunit of the cytochrome c oxidase (Cox B2). Cox A2 was absent from the gel, probably due to its high hydrophobicity (12 predicted transmembrane segments (5)). This result confirms the results obtained by ion trap mass spectrometry. Moreover, staining of cytochromes c with 3,3′,5,5′-tetramethylbenzidine in SDS gel revealed the presence of one band only with a molecular mass of about 25 kDa, corresponding to cytochrome c1 (data not shown).
FIGURE 2.

Protein composition of the purified supercomplex. A, separation of the subunits of the supercomplex (25 μg) on a 4–20% SDS gel and staining with Coomassie Blue. Arrowheads indicate subunits identified by mass spectrometry (B). The upper band (>100 kDa), not marked with an arrowhead, was not identified. B, identification of subunits by MALDI-TOF mass spectrometry. C, migration of the supercomplex (30 μg) on a 5–15% Tris-glycine native gel and detection with Coomassie Blue (lane 1), A. ferrooxidans Cox B antibodies (lane 2) for detecting the Aquifex cytochrome c oxidase, and A. aeolicus Sqr antibodies (lane 3).
Western Blotting
We used antibodies for detection of the components after separation of the supercomplex on a native PAGE, which confirms the presence of the Sqr and the cytochrome c oxidase in the same band (Fig. 2C). Sqr was also detected with antibodies, after migration on a blue native gel as well as on a denaturing gel (data not shown).
All the results indicate that the 350-kDa purified supercomplex contains three membrane-attached constituents, the Sqr, the bc1 complex, and the cytochrome c oxidase II. This is consistent with the proposition of the existence of an H2S oxidation/O2 reduction pathway in A. aeolicus (5, 26).
Homogeneity of the Supercomplex
The homogeneity and association state of the purified supercomplex were investigated by analytical ultracentrifugation. Fig. 3 shows the experimental and fitted sedimentation velocity profiles obtained at 413 nm for the supercomplex at 1.75 mg/ml (Fig. 3, A and B), the superposition for this sample of the distributions, c(s), of sedimentation coefficients obtained at 278 and 413 nm and by interference (Fig. 3D), and the superposition of the c(s) at 413 nm for the supercomplex at 1.75, 0.35, and 0.10 mg/ml (Fig. 3C). The three samples show qualitatively the same behavior, with limited nonideal effects, leading to smaller s values at larger protein concentration. The proportion of the largest major species increases slightly with increased concentration (from 67% at 0.10 mg/ml to 75% at 1.75 mg/ml of the total absorbance at 278 nm). This may indicate slow association-dissociation equilibrium but is not easily distinguishable from sample heterogeneity. The largest main species sediments at s = 6.8 S (s20,w = 11.4 S), corresponding to the protein-detergent supercomplex. The s value allows an estimation of the protein part within the complex, considering an amount of bound DDM/g of protein (BDet) in the range 0–1 g/g, for frictional ratios of 1.25 to 1.5 (i.e. for a globular to a moderately extended shape), of 240 ± 50 to 320 ± 70 kDa (complexes of 340 to 450 ± 50 kDa). The number of fringes per absorbance unit at 278 nm is 2.5. It corresponds, considering ϵ280 = 1.63 cm−1(mg/ml)−1 for the putative supercomplex and ϵ280 = 0 for the detergent, to BDet = 0.6 g/g. This species has rather a large absorbance at 413 nm, in agreement with the presence of cytochromes in the supercomplex (A413/A278 = 0.9). Two ill-resolved minor species are identified, from the superposition of the c(s) at different concentrations with the various optics, at s = 3.8 and 5.05 ± 0.5 S (s20,w = 6.4 and 8.4 ± 0.8 S), accounting for 10 and 5%, respectively, of the signal at 278 nm, with a low signal at 413 nm. The s20,w = 6.4 S species absorbs less at 413 nm compared with that of 8.4 S (A413/A278 ≈0.2 and 1.1, respectively). These s20,w values correspond to globular proteins of about 120 and 180 kDa, respectively. The species at 120 kDa might correspond to a dimer of Sqr (visualized on blue native gel in some supercomplex preparations, see below and Fig. 1D) for which A413/A278 between 0.1 and 0.2 is expected. The species at 180 kDa, not detected by blue native gel (Fig. 1), may correspond to a small amount of a dissociated species of the supercomplex. In addition, a contribution at s = 2.5 S (s20,w = 4.3 S assuming hydrated micelles of DDM) is characterized by a large interference signal with a number of fringes per absorbance unit at 278 nm between 34 for the most concentrated samples and 54 for the most diluted one. It corresponds reasonably well to the detergent micelles, with possibly dissolved absorbing impurities (22).
FIGURE 3.
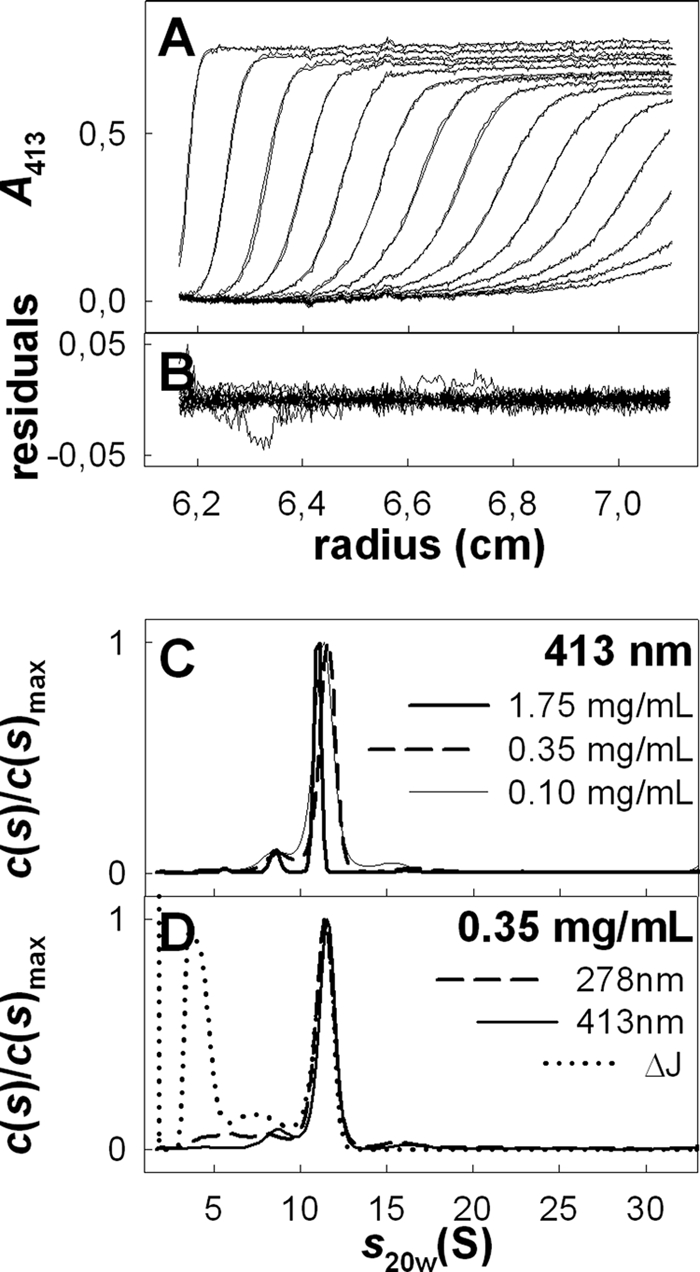
Sedimentation velocity experiments of A. aeolicus purified supercomplex. A, superimposition, for the supercomplex at 1.75 mg/ml in buffer A, of selected experimental sedimentation profiles obtained at 413 nm during 6 h at 42,000 rpm, at 12 °C, in 3-mm optical path length cell and of their modeled profiles with the c(s) analysis. B, corresponding residuals. C, superposition of the c(s) analysis for the supercomplex at 1.75, 0.35, and 0.10 mg/ml followed at 413 nm. D, result of the c(s) analysis for the supercomplex at 0.35 mg/ml followed at 278 and 413 nm and using interference optics. s20,w values were calculated using v̄ = 0.75 ml/g, corresponding to the protein part. The errors in s20,w related to the choice of s20,w are less than 3%.
When the purified supercomplex was subjected to a size exclusion chromatography with light scattering detection, the elution profile showed two resolved peaks (data not shown). The first is characterized by A413/A280 = 0.95 and corresponds to the supercomplex with a molecular mass of 375 ± 50 kDa. Combining the A280 and the refractive index signals allows estimation of an amount of bound DDM of 0.5 g/g (based on the extinction coefficient of the putative supercomplex) and, with the light scattering signal, a protein molecular mass of 235 kDa. The second peak corresponds at least in part to the detergent micelles with a low absorbance and a molecular mass of about 75 kDa. This result is consistent with the sedimentation velocity and blue native gels experiments and confirms that the purified complex is compact.
Spectroscopic Analysis
Optical Spectroscopy
In agreement with supercomplex composition, UV-visible absorption spectra of the purified 350-kDa complex, recorded at ambient temperature, showed the presence of cytochrome c, with peaks at 424 and 554 nm, cytochrome b, with peaks at 424 and 561 nm, and cytochrome c oxidase, with peaks at 448 and around 600 nm in the reduced state (Fig. 4A). These signals arise from the dihemic cytochrome b and monohemic cytochrome c1 from the bc1 complex and the cytochrome c oxidase II identified above (27, 28). Addition of sodium ascorbate to the “as prepared” sample leads to reduction of about 40% of total hemes C and 30% of total hemes B. Reduction of hemes C is in line with the redox midpoint potential previously determined for heme C1 (+160 mV) from the purified A. aeolicus bc1 complex (28). However, reduction of hemes B with ascorbate implies that the cytochrome c oxidase included in the supercomplex is a ba3-type enzyme with a positive redox potential because those from heme BL and heme BH from the cytochrome b of bc1 complex are negative (−190 and −60 mV, respectively (28)), and thus reducible by sodium dithionite only. In line with this result, the ratio hemes C/hemes B is around 1:2.6 confirming the presence of an extra heme B in the supercomplex, in addition to those of the dihemic cytochrome b. Signals from cytochrome c oxidase are similar to the ones obtained for the ba3 oxidase from Thermus thermophilus (29, 30).
FIGURE 4.

Spectral properties of the supercomplex. A, optical spectra. Proteins (3.5 mg/ml in buffer A) were analyzed as prepared (dotted line), reduced by sodium ascorbate (dashed line), and reduced by sodium dithionite (solid line). The inset shows the difference spectrum (dithionite-reduced minus as prepared). B and C, EPR spectra. The purified supercomplex (3.5 mg/ml in buffer A) was analyzed without any addition (as prep, solid line) or reduced with 5 mm sodium dithionite (dithionite-reduced, red, dashed line), in the region of the low field component of the low spin ferric heme resonances (B) and in the region of the Rieske iron-sulfur cluster (C). Instrument settings were as following: temperature, 15 K; microwave frequency, 9.406 GHz; microwave power, 6.4 milliwatts; modulation amplitude, 3 millitesla. Nine scans were averaged for each spectrum.
EPR Spectroscopy
EPR spectra of the purified A. aeolicus supercomplex confirmed the presence of cofactors of the bc1 complex and the ba3 terminal cytochrome c oxidase and in addition showed that the membrane-bound cytochrome c555 is present in very small amounts. Spectra, shown in Fig. 4, B and C, were recorded in a sample without any addition (Fig. 4B, as prepared sample, as prep, solid line) or treated with 5 mm sodium dithionite (Fig. 4, B and C, red, dashed lines). The untreated sample gave signals in the region of g = 3.7 to g = 2.9 due to oxidized low-spin heme species.
The signals at g = 3.71 and g = 3.36 arise from hemes from the bc1 complex. The position of these peaks and the line shape are essentially identical with those of isolated cytochrome bc1 complexes from bacteria or mitochondria (31–34). The peak at g = 3.71 corresponds to cytochrome bL, whereas peaks of cytochromes bH and c1 overlap with a peak at g = 3.36. In the EPR characterization of the cytochrome bc1 complex from A. aeolicus by Schütz et al. (28), the assignment of signals arising from hemes was different but also differs from that encountered in the bc1 complex of proteobacteria or mitochondria. In their study, many paramagnetic species overlapped to give a basically unresolved spectrum in which only few lines could be clearly distinguished (28).
The untreated sample showed also a relatively narrow peak at g = 3.00 arising from the low spin heme B of the cytochrome ba3 oxidase, as similarly found in mitochondrial or bacterial terminal oxidases (35–38). The g = 2.92 shoulder is due to the previously characterized membrane cytochrome c555 (39). This shoulder shows that it is present in a substoichiometric amount in the supercomplex and explains why it was not detected by mass spectrometry and 3,3′,5,5′-tetramethylbenzidine staining.
Reduction of the sample by sodium dithionite resulted in the complete disappearance of the signals in the gz region of cytochromes (Fig. 4B, red, dashed line) and in the appearance of the typical spectrum of the Rieske [2Fe-2S] cluster characterized by a gx trough at g = 1.80 and a derivative shaped gy line at g = 1.88 (28).
FAD, the cofactor of the Sqr embedded in the supercomplex, is not detectable by EPR spectroscopy, and its optical signals are masked by the ones from cytochromes. The presence of FAD was detected by a fluorescence emission around 510 nm, which is similar to the one we obtained for free Sqr (data not shown) (40).
These experiments demonstrate that the bc1 complex, the ba3 cytochrome c oxidase, and the Sqr in the supercomplex possess their redox cofactors suggesting a native and active state for all of them. We propose a stoichiometry of the supercomplex components on the basis of the spectroscopic analyses. The relative amount of hemes C and B in the supercomplex determined by optical spectroscopy suggests that it contains one cytochrome c oxidase for one homodimeric bc1 complex. The semi-empirical formula developed by de Vries and Albracht (41) allows calculation of the concentration of low spin hemes by EPR and thus determination that the two hemes at gz = 3.00 and gz = 3.71 are present in an approximate 1:2 stoichiometry, confirming the stoichiometry of one oxidase for one dimeric bc1 complex in the supercomplex.
Functional Role of the Supercomplex
To determine whether the three enzyme species associated in the supercomplex (bc1 complex, cytochrome c oxidase, and Sqr) are active, activities were determined in-gel and quantified by spectrophotometry.
Detection on Blue Native Gels
In addition to the cytochrome c oxidase activity, the bc1 complex was revealed at 350 kDa (Fig. 1C). To demonstrate the presence of active Sqr in the native gel, we developed the assay for specific detection of Sqr activity, as described under “Experimental Procedures.” Sqr activity was detected at 350 kDa, corresponding to the supercomplex, but also in two bands at about 50 and 100 kDa on the blue native gel with a faint intensity (Fig. 1D). They correspond to the two faint bands sometimes seen on the Coomassie Blue-stained gel and could also correspond to the minor species identified by analytical ultracentrifugation (at least for the 100-kDa species). When these two bands were cut out from the gel and analyzed by ion trap mass spectrometry, only the Sqr was identified (data not shown). These bands probably result from an equilibrium dissociation of the complex rather than an artifact of purification. Taken with the knowledge that Aquifex Sqr is a protein of 47 kDa, this experiment shows that it may exist in monomeric as well as in dimeric forms and that both are catalytically active on blue native gel. The fact that only Sqr appears to dissociate from the supercomplex in these conditions suggests that this component is associated less tightly than the rest.
Individual Enzyme Activities Measurements
Specific activities of the three enzymes in the supercomplex were determined spectrophotometrically at 40 °C (Table 2). Sqr, bc1 complex, and cytochrome c oxidase are active at this temperature. Sqr and bc1 complex activities are significantly inhibited by antimycin A, a quinone analog. Cyanide, an inhibitor of cytochrome c oxidases, inhibits the terminal enzyme demonstrating the specificity of the measured reactions (Table 2).
TABLE 2.
Specific activities of the supercomplex enzymatic constituents
The activity of the three enzymes (embedded in the protein supercomplex) has been determined at 40 °C with spectrophotometric assays. Inhibitions were measured with antimycin A and KCN as described under “Experimental Procedures.”
| Specific activity | Inhibited specific activity | Inhibitor used | |
|---|---|---|---|
| μmol·min−1·mg−1 | |||
| Sulfide quinone reductase | 20 ± 3.2 | 5.00 ± 0.98 | Antimycin A |
| bc1 complex | 1.85 ± 0.36 | 0.49 ± 0.1 | Antimycin A |
| Cytochrome c oxidase | 1.34 ± 0.29 | 0 | KCN |
Specific activities of the bc1 complex and cytochrome c oxidase are in the same range as those described in the literature (17), despite the fact that they were determined at a temperature far from the optimum (80 °C) and that the enzymes were not isolated but in association with proteins inside the supercomplex. Interestingly, the specific activity of the Sqr embedded in the supercomplex is very high compared with the specific activities of other purified Sqr described previously, except for the one of A. ferrooxidans (26, 42–44). These results show that the three enzymes are functional in the purified protein association.
Electron Transfer from Sulfide to Oxygen
To investigate whether the supercomplex is able to oxidize H2S (via Sqr) and reduce O2 (via the cytochrome c oxidase), we measured O2 consumption activities with a Clark-type electrode using Na2S as electron donor at 60 °C. The O2 was consumed by the supercomplex without any addition in the reactional medium (Fig. 5, −cyt −DB condition). Such an activity has also been detected with solubilized membranes (data not shown). The activity measurement was proportional to the concentration of the supercomplex in the reaction medium. This suggests that no dissociation or aggregation process of the enzymatic species occurs during the catalysis. O2 consumption was inhibited by cyanide (100% inhibition) and antimycin A (68% inhibition), demonstrating that O2 is reduced by the oxidase, and the electrons are provided by the Sqr and by the bc1 complex. Using quinol (DBH2) as substrate, a quinol oxidase-oxygen reductase activity was detected in the same conditions not only with the isolated supercomplex but also with the membranes (data not shown).
FIGURE 5.

H2S oxidase-O2 reductase activity of the supercomplex. Oxygen consumption was measured at 60 °C using Na2S as electron donor. The reactions were made in the presence (+) or absence (−) of horse heart cytochrome c (cyt) at 1 μm and quinone (DB) at 4 μm. Rates were corrected for the nonenzymatic reactions. The 100% activity corresponds to 3230 ± 610 units/mg.
It follows that the purified protein superstructure transfers electrons from H2S to O2 and that it is an active sulfide oxidase-oxygen reductase supercomplex. Quinone molecules are apparently contained in this supercomplex, and also cytochrome c555 (shown by EPR), both being electronic shuttles between the enzymes. This new protein assembly can be considered as a true respirasome.
Addition of exogenous quinone (DB) does not enhance the activity, suggesting that the amount of endogenous bound quinones in the isolated supercomplex allows a maximal electron transfer rate in our conditions (Fig. 5, −cyt +DB condition) (Sqr and bc1 complex are active with DB, although the physiological quinone is a demethylmenaquinone (28, 45)). On the other hand, a 2-fold higher activity could be obtained by addition of 1 μm of horse cytochrome c in the medium (Fig. 5, +cyt −DB and +cyt +DB conditions). In these conditions, an inhibition of the electron transfer from sulfide to oxygen of 71 and 100% is measured with antimycin A and KCN, respectively.
Functional Consequences of Association into a Supercomplex on the Sqr
To understand the impact on the Sqr enzyme of the physical association with its physiological partners (bc1 complex and cytochrome c oxidase), we studied the isolated Sqr enzyme purified from the membranes (Sqrfree). Its enzymatic and biochemical characteristics are essentially the same as those reported while this manuscript was under consideration (46), except for the value of the specific activity, which was lower than what we found. The N-terminal sequence confirms that the N-terminal part of the enzyme is not cleaved for translocation across the membrane, despite the fact that it is proposed to be periplasmic (data not shown). The signal required for translocation may be at the C-terminal part of the protein as in the Sqr of Rhodobacter capsulatus (47).
In addition to the pure Sqrfree, we could also obtain a pure Sqr fraction released from the supercomplex (called Sqrreleased). In contrast to what we and others (46) obtained for Sqrfree, migration on blue native gel shows beyond a doubt the absence of the trimeric state for Sqrreleased (Fig. 6). The nature of the protein in each band has been confirmed by ion trap mass spectrometry and Western blotting (data not shown). This suggests that the oligomeric state of the Sqr is different according to whether it is free or embedded in the supercomplex.
FIGURE 6.

Migration of the purified Sqr on blue native gel. 4–15% gels were used, and 10 μg of Sqr were loaded in all cases and detected by Coomassie Blue staining. A, Sqrreleased. B, Sqrfree. Protein bands are indicated by arrowheads and correspond to the monomer (58 kDa), dimer (106 kDa), trimer (175 kDa), and an higher oligomeric state (240 kDa) of the Sqr. Molecular mass markers are indicated in kDa.
The thermal stability (half-life determined at 80 °C) and the kinetic parameters of the Sqr have been compared in both states (Sqrfree versus associated with their redox partners within the supercomplex), as reported in Table 3; the properties of this enzyme are not significantly modified, except for the Michaelis constant (Km) for the quinone and the catalytic efficiency. The data suggest a higher affinity for this substrate when the Sqr is integrated in the multienzymatic complex.
TABLE 3.
Comparison of the properties of the isolated Sqr and the Sqr integrated in the supercomplex
| State of the Sqr | Isolated (Sqrfree, this study) | Integrated in the supercomplex (this study) | Isolated (from Ref. 46) |
|---|---|---|---|
| Oligomeric state on blue native gel | 4 states at 58, 106, 175 and 240 kDa | 2 states at 58 and 106 kDa | 4 states at 50, 90, 150, 280 kDa |
| t½ at 80 °C | 18 h | 15 h | 32 h |
| kcat (s−1)a | 75b | 60b | 59c |
| Km (DB) | 1.66 ± 0.68 μmb | 0.71 ± 0.4 μmb | 2.16 ± 0.19 μmd |
| kcat/Kma | 45.2 s−1·μm−1b | 84.5 s−1·μm−1b |
a Kinetic parameters were calculated using the monomeric concentration of isolated Sqr and on the basis of one monomeric Sqr in the supercomplex.
b Data were determined at 25 °C.
c Data were determined at 40 °C.
d Temperature was not specified.
DISCUSSION
Stable Association of All Components Involved in H2S Oxidation-O2 Reduction Pathway
We have described a new functional respiratory supercomplex containing the Sqr enzyme, the bc1 complex, and a ba3 cytochrome c oxidase, which are properly arranged into a functional structure. Thus, it contains all the molecular components required for the electron transfer from H2S as electron donor to O2 as an acceptor. It has a sulfide oxidase-oxygen reductase activity and is thus a complete respirasome. To our knowledge, this is the first time that Sqr is found as part of a complex.
In a variety of phototrophic and chemolithoautotrophic bacteria, Sqr feeds electrons into the photosynthetic or respiratory electron transfer chain via a bc-type complex (43). The implication of Sqr in the oxygen reduction pathway in A. aeolicus was proposed a few years ago by Nübel et al. (26) and recently by our group (5). However, this work is the first evidence for the association of all enzymes involved in this pathway into a membrane-bound supercomplex. We have demonstrated that it is very stable, as it is not destabilized by relatively harsh solubilization conditions and three purification steps, unlike other mitochondrial or bacterial respiratory supercomplexes that are stabilized by relatively weak interactions (15, 48). This superstructure could not have been predicted from the organization of the genes in the genome.
Organization of the Supercomplex
This new multienzyme protein assembly is shown in the scheme in Fig. 7 with the bc1 complex interacting with the cytochrome c oxidase, as we have previously shown that they both associate in the membrane of A. aeolicus (5). Because Sqr is considered to be an integral monotopic membrane protein (44, 49) and uses quinone as substrate, we propose that the Sqr interacts with the bc1 complex inside the supercomplex, which includes one dimeric bc1 complex and one cytochrome c oxidase complex (Fig. 7). This is consistent with the fact that dimers of bc1 complexes constitute the minimal structural and functional unit (50, 51). Supercomplexes containing a dimeric bc1 complex and one or two copies of cytochrome c oxidase have been purified from yeast mitochondria. Structural models showed that the terminal enzyme is peripherally located in these supercomplexes and that one copy of this enzyme is lacking in a substantial number of particles (52).
FIGURE 7.

Model of the A. aeolicus energetic sulfur metabolism. The H2/S0 and H2S/O2 respiratory pathways are structurally organized in supercomplexes. Cytochrome oxidase, ba3 cytochrome c oxidase (composed of Cox A2 and Cox B2 subunits); bc, monomeric bc1 complex (composed of cytochrome b, cytochrome c1, and the Rieske protein); c, monoheme cytochrome c555; Sqr, sulfide quinone reductase; SOR, sulfur oxygenase reductase; S(n), elemental sulfur (the true substrate and the number of sulfur atoms in the molecule in vivo are not known); S2O3, thiosulfate; SO3, sulfite; H2S, hydrogen sulfide. Evidence for cytoplasmic sulfur globules was reported previously (4). The membrane-bound protein symbolized by dotted lines is a putative H2S transporter.
The A. aeolicus bc1 complex is typical (28), but the isolated Sqr enzyme, characterized very recently, has been proposed to be a trimer (46, 49), which differs from other organisms. We detected this trimer on blue native gel with Sqrfree. We have shown that the Sqrreleased appears to be a monomer as well as a homodimer on the blue native gel and that this Sqr fraction seems to be less stable than Sqrfree or Sqr inserted in the supercomplex (data not shown). Moreover, we have shown that Sqr is active in the oligomeric as well as in the monomeric state. The precise stoichiometry of the Sqr in the supercomplex remains unknown. However, the experimental molecular mass of the purified superstructure (around 350 kDa) and the proposed stoichiometry of components based on spectroscopy exclude the possibility of a trimeric state of the Sqr in the supercomplex. We propose that Sqr in the supercomplex is a monomer or a dimer. Sqr from A. aeolicus, whether in the supercomplex or free, is much more active than purified Sqr from other organisms, even though the specific activity may well be substantially underestimated, because it was determined at 25 and 40 °C, much lower temperatures than those optimal for growth. These data call for further investigation.
Presence of Electronic Intermediates in the Supercomplex
The supramolecular assembly contains one of the two cytochromes c555 known in A. aeolicus, which is probably the membrane-tethered one (called c555m) (39, 53). This cytochrome is present in substoichiometric amounts, as was also the case for the purified NADH oxidase respirasome from P. denitrificans (0.4 cytochrome c552/1 cytochrome c oxidase) (17). In A. aeolicus membranes, however, the gz peak corresponding to this cytochrome in EPR spectrum has a very high intensity compared with that of the cytochrome c oxidase. This suggests that this electron carrier could be present in the supercomplex in a large amount and that part of it could even be free in membranes (data not shown). This would indicate that cytochrome c555 is partially lost during supercomplex purification steps as the c552 from P. denitrificans (40–80% loss). Such a loss would be consistent with the idea that this cytochrome is a “shuttle” between the two partners (bc1 complex and cytochrome c oxidase) and might thus be loosely bound to be able to interact with both of them. As the sulfide oxidase-oxygen reductase activity is significantly enhanced by the addition of horse heart cytochrome c, the electron transport between bc1 complex and cytochrome c oxidase was impeded by loss of cytochrome c555. This indicates that the bc1 complex and the cytochrome c oxidase can tightly interact in the absence of this electron carrier. In addition, quinones are retained in the supercomplex after isolation, as with the NADH oxidase purified from P. denitrificans, where the ubiquinone is specifically enriched (17). We propose that electrons delivered from Sqr into the quinone pool pass through the cytochrome bc1 complex and finally reduce the cytochrome c oxidase via the cytochrome c, as shown in the scheme of Fig. 7.
Supramolecular Organization of the Respiratory Chains in A. aeolicus: Functional Significance
The sulfide oxidase/oxygen reductase supercomplex is the second respirasome that we have characterized from A. aeolicus. A few years ago, we demonstrated that enzymes that couple periplasmic oxidation of hydrogen to cytoplasmic sulfur reduction form a membrane-bound supercomplex containing a hydrogenase and a sulfur reductase connected via quinones (4). It thus appears that the two respiratory pathways of Aquifex, sulfur reduction and sulfur oxidation, which are probably complementary or antagonistic in vivo, are organized into supramolecular structures (Fig. 7), which is a unique feature among prokaryotic as well as eukaryotic organisms. Indeed, all the supercomplexes identified in the mitochondrial membranes belong to the same energetic pathway.
Several studies of mitochondrial as well as bacterial supercomplexes have highlighted that the isolated respiratory complexes could be structurally stabilized when associated with their physiological partners (17, 54). Another main functional advantage that may originate from the interaction of consecutive enzymes is substrate channeling. The enzymes embedded in multiprotein complexes can also have different kinetic behavior compared with free enzymes (55), particularly when the subunit composition is different. Our present results suggest that interaction of the H2S/O2 pathway components into a supercomplex modifies the oligomerization state of the Sqr but does not stabilize the Sqr. However, the kinetic behavior of the Sqr is slightly altered, as the catalytic efficiency (kcat/Km) of this enzyme is increased when it is present in the supercomplex. The fact that the solubilized supercomplex is stable with time (there appears to be no dissociation) suggests that the Sqrfree that we find could be part of a second pool of enzyme in the membrane that is not associated. The observation that the Sqrreleased appears to have a different quaternary structure from the Sqrfree (Fig. 6) supports the idea of the existence in vivo of two pools, and these two pools possibly have different physiological roles. The fact that the kinetic parameters of Sqr inserted in the supercomplex and Sqrfree appear to be different is compatible with this possibility.
Energetic Sulfur Metabolism in A. aeolicus
A. aeolicus requires a sulfur compound for growth, in addition to molecular hydrogen and molecular oxygen, which is puzzling. Why are these three components? Genes encoding enzymes involved in the oxidation as well as in the reduction of sulfur compounds are present in its genome. We have previously characterized several enzymes involved in sulfur compound utilization (Fig. 7) as follows: a cytoplasmic sulfur oxygenase reductase catalyzing the disproportionation of elemental sulfur in the presence of O2 (56), the membrane-bound sulfur reductase (4), and the Sqr (this work and see Refs. 26, 46, 49). All these enzymes are present under our growth conditions.
It is known that A. aeolicus can grow in the presence of elemental sulfur or thiosulfate if H2 and O2 are also present (4). We have also found that A. aeolicus can grow in the presence of sodium sulfide, but as in the previous case, H2 and O2 are required (data not shown). No growth was observed with H2S and O2 only, which raises some questions. This suggests that this bacterium can use oxidized or reduced sulfur compounds to grow and has a very versatile metabolism.
Brito et al. (42) have proposed that in Acidianus ambivalens, an acidophilic and hyperthermophilic archae, Sqr oxidizes sulfide produced by sulfur oxygenase reductase and thus participates into an energetic spiral allowing the maximum of energy to be obtained from sulfur compounds. We propose that an analogous process occurs in Aquifex with an energetic coupling between the sulfur reduction and oxidation pathways. The sulfur that might be stored in cytoplasmic inclusions by the cell (4) could be used by the sulfur oxygenase reductase and the sulfur reductase producing H2S (Fig. 7). H2S would in turn be used by Sqr to reduce oxygen and produce ATP. H2S transport across the membrane could occur by simple diffusion without facilitation by membrane channels as recently demonstrated (57). We have characterized most of the enzymes involved in the intricate sulfur metabolism in A. aeolicus, and we will now concentrate our efforts in understanding how the various energetic pathways are connected, as a way to gain insight of why A. aeolicus requires H2, O2, and a source of sulfur for growth.
Acknowledgments
We gratefully acknowledge the contribution of Aline Le Roy (Analytical Ultracentrifugation Platform of the IBS and PBS, Grenoble, France) for technical assistance and helpful discussions; Sabrina Lignon and Régine Lebrun (Proteomic Analysis Center, IFR88, CNRS, Marseilles) for proteomic analysis; Marielle Bauzan (Fermentation Plant Unit, IFR88, CNRS, Marseilles, France) for growing the bacteria; Violaine Bonnefoy (LCB, IFR88, CNRS, Marseilles) for kindly supplying the anti-CoxB antibodies; Bruno Guigliarelli (BIP, IFR88, Marseilles) for access to the EPR facilities, and Rainer Hienerwadel (Plant Genetic and Biophysic Laboratory, Marseilles) for access to the oxygraph apparatus. We also thank Maria Luz Cardenas, Athel Cornish-Bowden, and Wolfgang Nitschke (BIP, Marseilles) for helpful discussions and Athel Cornish-Bowden for correcting the English. The Proteomic Analysis Center of IFR88 is part of MaP (Marseille Protéomique, IBiSA).
This work was supported in part by CNRS, Région-Provence-Alpes Côte d'Azur, and Marseille Metropole.
- Sqr
- sulfide quinone reductase
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- DB
- decyl-ubiquinone
- DDM
- n-dodecyl β-d-maltoside.
REFERENCES
- 1.Huber R., Wilharm T., Huber D., Trincone A., Burggraf S., König H., Rachel R., Rockinger I., Fricke H., Stetter K. O. (1992) Syst. Appl. Microbiol. 15, 340–351 [Google Scholar]
- 2.Huber R., Stetter K. O. (1999) Aquificales. in Embryonic Encyclopedia of Life Sciences, Nature Publishing Group, London [Google Scholar]
- 3.Deckert G., Warren P. V., Gaasterland T., Young W. G., Lenox A. L., Graham D. E., Overbeek R., Snead M. A., Keller M., Aujay M., Huber R., Feldman R. A., Short J. M., Olsen G. J., Swanson R. V. (1998) Nature 392, 353–358 [DOI] [PubMed] [Google Scholar]
- 4.Guiral M., Tron P., Aubert C., Gloter A., Iobbi-Nivol C., Giudici-Orticoni M. T. (2005) J. Biol. Chem. 280, 42004–42015 [DOI] [PubMed] [Google Scholar]
- 5.Guiral M., Prunetti L., Lignon S., Lebrun R., Moinier D., Giudici-Orticonit M. T. (2009) J. Proteome Res. 8, 1717–1730 [DOI] [PubMed] [Google Scholar]
- 6.Brugna-Guiral M., Tron P., Nitschke W., Stetter K. O., Burlat B., Guigliarelli B., Bruschi M., Giudici-Orticoni M. T. (2003) Extremophiles 7, 145–157 [DOI] [PubMed] [Google Scholar]
- 7.Srere P. A. (1987) Annu. Rev. Biochem. 56, 89–124 [DOI] [PubMed] [Google Scholar]
- 8.Schägger H., Pfeiffer K. (2000) EMBO J. 19, 1777–1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eubel H., Heinemeyer J., Sunderhaus S., Braun H. P. (2004) Plant Physiol. Biochem. 42, 937–942 [DOI] [PubMed] [Google Scholar]
- 10.Genova M. L., Baracca A., Biondi A., Casalena G., Faccioli M., Falasca A. I., Formiggini G., Sgarbi G., Solaini G., Lenaz G. (2008) Biochim. Biophys. Acta 1777, 740–746 [DOI] [PubMed] [Google Scholar]
- 11.Berry E. A., Trumpower B. L. (1985) J. Biol. Chem. 260, 2458–2467 [PubMed] [Google Scholar]
- 12.Sone N., Sekimachi M., Kutoh E. (1987) J. Biol. Chem. 262, 15386–15391 [PubMed] [Google Scholar]
- 13.Keefe R. G., Maier R. J. (1993) Biochim. Biophys. Acta 1183, 91–104 [DOI] [PubMed] [Google Scholar]
- 14.Verméglio A., Joliot P. (2002) Biochim. Biophys. Acta 1555, 60–64 [DOI] [PubMed] [Google Scholar]
- 15.Niebisch A., Bott M. (2003) J. Biol. Chem. 278, 4339–4346 [DOI] [PubMed] [Google Scholar]
- 16.Megehee J. A., Hosler J. P., Lundrigan M. D. (2006) Microbiology 152, 823–829 [DOI] [PubMed] [Google Scholar]
- 17.Stroh A., Anderka O., Pfeiffer K., Yagi T., Finel M., Ludwig B., Schägger H. (2004) J. Biol. Chem. 279, 5000–5007 [DOI] [PubMed] [Google Scholar]
- 18.Schägger H., Cramer W. A., von Jagow G. (1994) Anal. Biochem. 217, 220–230 [DOI] [PubMed] [Google Scholar]
- 19.Thomas P. E., Ryan D., Levin W. (1976) Anal. Biochem. 75, 168–176 [DOI] [PubMed] [Google Scholar]
- 20.Wittig I., Karas M., Schägger H. (2007) Mol. Cell. Proteomics 6, 1215–1225 [DOI] [PubMed] [Google Scholar]
- 21.Castelle C., Guiral M., Malarte G., Ledgham F., Leroy G., Brugna M., Giudici-Orticoni M. T. (2008) J. Biol. Chem. 283, 25803–25811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Salvay A. G., Ebel C. (2006) Prog. Colloid. Polym. Sci. 131, 74–82 [Google Scholar]
- 23.Schuck P. (2000) Biophys. J. 78, 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ebel C. (2007) in Protein Structures: Methods in Protein Structure and Stability Analysis (Uversky V., Permyakov E. A. eds) pp. 229–260, Nova Science Publishers, Inc. New York [Google Scholar]
- 25.le Maire M., Arnou B., Olesen C., Georgin D., Ebel C., Møller J. V. (2008) Nat. Protoc. 3, 1782–1795 [DOI] [PubMed] [Google Scholar]
- 26.Nübel T., Klughammer C., Huber R., Hauska G., Schütz M. (2000) Arch. Microbiol. 173, 233–244 [DOI] [PubMed] [Google Scholar]
- 27.Scharf B., Wittenberg R., Engelhard M. (1997) Biochemistry 36, 4471–4479 [DOI] [PubMed] [Google Scholar]
- 28.Schütz M., Schoepp-Cothenet B., Lojou E., Woodstra M., Lexa D., Tron P., Dolla A., Durand M. C., Stetter K. O., Baymann F. (2003) Biochemistry 42, 10800–10808 [DOI] [PubMed] [Google Scholar]
- 29.Farver O., Chen Y., Fee J. A., Pecht I. (2006) FEBS Lett. 580, 3417–3421 [DOI] [PubMed] [Google Scholar]
- 30.Sousa F. L., Veríssimo A. F., Baptista A. M., Soulimane T., Teixeira M., Pereira M. M. (2008) Biophys. J. 94, 2434–2441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McCurley J. P., Miki T., Yu L., Yu C. A. (1990) Biochim. Biophys. Acta 1020, 176–186 [DOI] [PubMed] [Google Scholar]
- 32.Montoya G., te Kaat K., Rodgers S., Nitschke W., Sinning I. (1999) Eur. J. Biochem. 259, 709–718 [DOI] [PubMed] [Google Scholar]
- 33.Salerno J. C. (1984) J. Biol. Chem. 259, 2331–2336 [PubMed] [Google Scholar]
- 34.Zhu J., Egawa T., Yeh S. R., Yu L., Yu C. A. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 4864–4869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Van Gelder B. F., Beinert H. (1969) Biochim. Biophys. Acta 189, 1–24 [DOI] [PubMed] [Google Scholar]
- 36.Fee J. A., Choc M. G., Findling K. L., Lorence R., Yoshida T. (1980) Proc. Natl. Acad. Sci. U.S.A. 77, 147–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sone N., Naqui A., Kumar C., Chance B. (1984) Biochem. J. 223, 809–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dispirito A. A., Lipscomb J. D., Hooper A. B. (1986) J. Biol. Chem. 261, 17048–17056 [PubMed] [Google Scholar]
- 39.Baymann F., Tron P., Schoepp-Cothenet B., Aubert C., Bianco P., Stetter K. O., Nitschke W., Schütz M. (2001) Biochemistry 40, 13681–13689 [DOI] [PubMed] [Google Scholar]
- 40.Schütz M., Shahak Y., Padan E., Hauska G. (1997) J. Biol. Chem. 15, 9890–9894 [DOI] [PubMed] [Google Scholar]
- 41.de Vries S., Albracht S. P. (1979) Biochim. Biophys. Acta 546, 334–340 [DOI] [PubMed] [Google Scholar]
- 42.Brito J. A., Sousa F. L., Stelter M., Bandeiras T. M., Vonrhein C., Teixeira M., Pereira M. M., Archer M. (2009) Biochemistry 48, 5613–5622 [DOI] [PubMed] [Google Scholar]
- 43.Griesbeck C., Hauska G., Schütz M. (2000) in Recent Research Developments in Microbiology (Pandalai S. G. ed) Vol. 4, pp. 179–203, Research Signpost, Trivadrum, India [Google Scholar]
- 44.Cherney M. M., Zhang Y., Solomonson M., Weiner J. H., James M. N. (2010) J. Mol. Biol. 398, 292–305 [DOI] [PubMed] [Google Scholar]
- 45.Infossi P., Lojou E., Chauvin J. P., Herbette G., Brugna M., Giudici-Orticoni M. T. (2010) Int. J. Hydrogen Energy 35, 10778–10789 [Google Scholar]
- 46.Marcia M., Langer J. D., Parcej D., Vogel V., Peng G., Michel H. (2010) Biochim. Biophys. Acta 1798, 2114–2123 [DOI] [PubMed] [Google Scholar]
- 47.Schütz M., Maldener I., Griesbeck C., Hauska G. (1999) J. Bacteriol. 181, 6516–6523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stuart R. A. (2009) Methods Enzymol. 456, 191–208 [DOI] [PubMed] [Google Scholar]
- 49.Marcia M., Ermler U., Peng G., Michel H. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 9625–9630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Iwata S., Lee J. W., Okada K., Lee J. K., Iwata M., Rasmussen B., Link T. A., Ramaswamy S., Jap B. K. (1998) Science 281, 64–71 [DOI] [PubMed] [Google Scholar]
- 51.Esser L., Elberry M., Zhou F., Yu C. A., Yu L., Xia D. (2008) J. Biol. Chem. 283, 2846–2857 [DOI] [PubMed] [Google Scholar]
- 52.Heinemeyer J., Braun H. P., Boekema E. J., Kouril R. (2007) J. Biol. Chem. 282, 12240–12248 [DOI] [PubMed] [Google Scholar]
- 53.Aubert C., Guerlesquin F., Bianco P., Leroy G., Tron P., Stetter K. O., Bruschi M. (2001) Biochemistry 40, 13690–13698 [DOI] [PubMed] [Google Scholar]
- 54.Schägger H., de Coo R., Bauer M. F., Hofmann S., Godinot C., Brandt U. (2004) J. Biol. Chem. 279, 36349–36353 [DOI] [PubMed] [Google Scholar]
- 55.Lebreton S., Graciet E., Gontero B. (2003) J. Biol. Chem. 278, 12078–12084 [DOI] [PubMed] [Google Scholar]
- 56.Pelletier N., Leroy G., Guiral M., Giudici-Orticoni M. T., Aubert C. (2008) Extremophiles 12, 205–215 [DOI] [PubMed] [Google Scholar]
- 57.Mathai J. C., Missner A., Kügler P., Saparov S. M., Zeidel M. L., Lee J. K., Pohl P. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 16633–16638 [DOI] [PMC free article] [PubMed] [Google Scholar]