

Abstract
Steady progress has been made in defining both the viral and cellular determinants of retroviral assembly and release. Although it is widely accepted that targeting of the Gag polypeptide to the plasma membrane is critical for proper assembly of HIV-1, the intracellular interactions and trafficking of Gag to its assembly sites in the infected cell are poorly understood. HIV-1 Gag was shown to interact and co-localize with calmodulin (CaM), a ubiquitous and highly conserved Ca2+-binding protein expressed in all eukaryotic cells, and is implicated in a variety of cellular functions. Binding of HIV-1 Gag to CaM is dependent on calcium and is mediated by the N-terminally myristoylated matrix (myr(+)MA) domain. Herein, we demonstrate that CaM binds to myr(+)MA with a dissociation constant (Kd) of ∼2 μm and 1:1 stoichiometry. Strikingly, our data revealed that CaM binding to MA induces the extrusion of the myr group. However, in contrast to all known examples of CaM-binding myristoylated proteins, our data show that the myr group is exposed to solvent and not involved in CaM binding. The interactions between CaM and myr(+)MA are endothermic and entropically driven, suggesting that hydrophobic contacts are critical for binding. As revealed by NMR data, both CaM and MA appear to engage substantial regions and/or undergo significant conformational changes upon binding. We believe that our findings will provide new insights on how Gag may interact with CaM during the HIV replication cycle.
Keywords: Calmodulin, HIV, NMR, Protein Structure, Virus Assembly, Gag, Matrix, Myristyl
Introduction
Gag is the major structural protein encoded by HIV-1 and contains all of the viral elements required to drive virus assembly (1–3). HIV-1 Gag targeting to the plasma membrane (PM)2 is critical for proper and efficient assembly to produce progeny virions (1, 3–9). During virus maturation, Gag is cleaved into myristoylated matrix (myr(+)MA), capsid, and nucleocapsid proteins, inducing major morphological reorganization of the virus (1, 2, 4, 5, 10). In many cell types, HIV-1 Gag budding and assembly has been shown to occur predominantly on the PM (4–9, 11–18). Gag binding to the PM is mediated by the MA domain and enhanced by multimerization. Proper assembly and efficient binding of Gag to the PM requires a myristyl (myr) group as a membrane anchor and a cluster of basic residues localized within the N-terminal domain to facilitate interactions with acidic phospholipids (1, 2, 19, 20).
Steady progress has been made in defining both the viral and cellular determinants of HIV-1 assembly and release (6). However, the trafficking pathway used by Gag to reach assembly sites in the infected cell is poorly understood. Studies by Freed, Ono, and co-workers (21–23) demonstrated that the ultimate localization of HIV-1 Gag at virus assembly sites is dependent on phosphatidylinositol-(4,5)-bisphosphate (PI(4,5)P2), a cellular factor localized at the inner leaflet of the PM (24–26). Our structural studies revealed that PI(4,5)P2 binds directly to HIV-1 MA, inducing a conformational change that triggers myr exposure (27). In addition to PI(4,5)P2, there is mounting evidence that HIV-1 Gag interacts with several cellular proteins during the virus replication cycle and that these interactions are mediated by the MA domain. These factors include calmodulin (CaM) (28), the human adaptor protein-3 complex (AP-3) (11), the tail-interacting protein (TIP47) (29), and the suppressor of cytokine signaling 1 (SOCS1) (30, 31). However, evidence for direct interactions between Gag and any of these cellular proteins is either absent or very limited.
CaM is a ubiquitous and highly conserved calcium-binding protein expressed in all eukaryotic cells and is implicated in a variety of cellular functions (32–37). It can be localized in various subcellular locations, including the cytoplasm, within organelles, or associated with the plasma or organelle membranes (32–37). CaM and its interactions with cellular proteins have been extensively studied (32, 38–40). Structurally, binding of calcium ions (Ca2+) to CaM triggers a conformational change, enabling it to bind to specific proteins for a specific response (33, 36, 37). CaM has been shown to specifically interact with and activate over 100 distinct target proteins (32, 41, 42).
It is well established that binding of Ca2+ to CaM triggers dramatic helical rearrangements in the N- and C-terminal lobes, resulting in the opening of large binding pockets on the surface of each domain consisting of hydrophobic residues that are essentially buried in the apoprotein (Ca2+-free) (33, 36, 37). As a result, each domain moves from a closed conformation in the apo form to an open conformation when Ca2+ is bound, allowing cellular targets to dock. The CaM protein is highly acidic and described as having “dumbbell-like” architecture with the N- and C-terminal lobes connected by a flexible helix called the central linker. The N- and C-terminal lobes each possess two helix-loop-helix motifs called “EF hands” (43).
A potential role of CaM in the HIV replication cycle has been proposed by several research groups. A combination of in vivo and in vitro studies revealed that, in addition to Gag, CaM interacts with HIV-1 Nef and the envelope glycoprotein (Env) (44–47). Interaction between Gag and CaM is Ca2+-dependent (28). Gag and CaM were also found to co-localize in a diffuse pattern spread throughout the cytoplasm. Recent studies have shown that myr(−)MA and short peptides derived from the MA protein interact directly with CaM (48, 49). However, it is well established that several myristoylated proteins like the myristoylated alanine-rich C kinase substrate (MARCKS), brain-specific protein kinase C substrate (CAP-23/NAP-22), and HIV-1 Nef interact with CaM via the myr group to facilitate their intracellular localization and membrane targeting (44, 45, 50, 51). Thus, we seek to determine whether the myr group of MA is important for CaM binding and/or whether binding of CaM to MA alters the myr switch mechanism in vitro. Membrane association and dissociation of myristoylated proteins are often regulated by intracellular Ca2+ signaling through a “Ca2+-myr switch.” Some examples include the Ca2+ sensor proteins recoverin (52), neurocalcin (53), and S-modulin (54), which alter their intracellular localization from the cytosol to the membrane through Ca2+ binding (55). In this report, we present structural, biochemical, and biophysical data on the interactions between myr(+)MA and CaM. Our data revealed that CaM binds directly to myr(+)MA in a calcium-dependent manner and 1:1 stoichiometry. We also demonstrate that CaM binding to myr(+)MA induces the extrusion of the myr group, which may suggest a potential role in the trafficking and/or targeting of Gag during the late stage of HIV replication.
EXPERIMENTAL PROCEDURES
Sample Preparation
A co-expression vector encoding the HIV-1 MA gene and yeast N-myristyl transferase was kindly provided by Michael Summers (Howard Hughes Medical Institute and University of Maryland, Baltimore County). Uniformly 15N-labeled samples for myr(−)MA and myr(+)MA proteins were prepared as described (56–58). The same protocols have been followed for preparation of recombinant myr(+)MA protein with a 13C-labeled myr group except that 5 mg/liter of [13C]myristic acid (Sigma) was added to cells at ∼40 min (A600 = ∼0.2) before induction with isopropyl β-d-thiogalactopyranoside. A plasmid encoding full-length (amino acids 1–148) Norvegicus rattus calmodulin was a kind gift from Madeline Shea (University of Iowa). CaM expression and purification were conducted according to a previously published protocol (59, 60). CaM samples were stored in a buffer containing 50 mm Tris (pH 7.0), 100 mm NaCl, and 10 mm CaCl2. Molecular masses for all of the protein samples were confirmed by electrospray ionization mass spectrometry.
NMR Spectroscopy
NMR data were collected at 35 °C (unless noted otherwise) on a Bruker Avance II (700 MHz 1H) spectrometer equipped with a cryogenic triple-resonance probe, processed with NMRPIPE (61), and analyzed with NMRVIEW (62). All of the NMR samples were prepared in a buffer containing 50 mm Tris-d11 (pH 6.3) and 10 mm CaCl2. Signal assignments of myr(−) and myr(+)MA were described elsewhere (27, 56–58). 1H-1H NOE cross-peaks between the 13C-labeled myr group and unlabeled protein residues were assigned from three-dimensional (13C-edited and 13C-edited/12C-double-half-filtered) HMQC-NOESY data (see Refs. 63–65 and citations therein). 1H, 13C, and 15N NMR chemical shifts for CaM have been reported (66). Because the CaM construct used here is different by four residues from that used by Ikura et al. (66) (Phe-99, Asn-129, Thr-143, and Ser-147 substituted with Tyr, Asp, Gln, and Ser, respectively), a complete signal assignment was achieved by a combination of two-, three-, and four-dimensional NOESY data collected on 15N- and 15N,13C-labeled protein samples. Protein backbone signals were assigned using standard triple resonance methods (HNCA, HNCO, HNCOCA, HNCACB, and HNCOCACB), and side chain signals were assigned from three- and four-dimensional 15N-, 13C-, and 15N/13C-edited NOESY data (see Refs. 64 and 65 and citations therein).
Gel Filtration Assay
The mobility of myr(+)MA, CaM, and their complex was analyzed with or without calcium. Briefly, 500 μl of CaM, MA, or their complex at ∼300 μm were run through a HiLoad 16/60 Superdex 75 column or HiPrep 16/60 Sephacryl S-200 HR (GE Healthcare) columns in a buffer containing 50 mm Tris (pH 7) and 10 mm CaCl2 (or 2 mm EDTA). Protein fractions were analyzed by SDS-PAGE. Molecular mass calibration kits (GE Healthcare and Sigma) were used to determine the approximate molecular masses of loaded proteins.
Analytical Ultracentrifugation
Sedimentation velocity measurements were collected on a Beckman XL-I Optima system equipped with a four-hole An-60 rotor (Beckman Coulter). Protein samples were prepared in 50 mm Tris, 100 mm NaCl, and 10 mm CaCl2. Loading concentrations were at 110, 250, and 100 μm for myr(+)MA, CaM, and myr(+)MA·CaM, respectively. Rotor speed was set at 40,000 rpm (20 °C), and scans were acquired at 295 nm. Partial specific volumes (v-bar) and molar extinction coefficients were calculated by using the program SEDNTERP, and buffer densities were measured pycnometrically. Sedimentation velocity data analysis were performed by using SEDFIT (67–70).
Isothermal Titration Calorimetry (ITC)
ITC experiments were conducted on protein samples in a buffer containing 50 mm Tris (pH 7.0), 100 mm NaCl, and 10 mm CaCl2. Dissociation equilibrium constants for CaM binding to myr(−) and myr(+)MA proteins were determined using a VP-isothermal titration microcalorimeter (MicroCal Corp., Northampton, MA) (71). CaM (∼400 μm) was titrated into the cell sample containing ∼20 μm of myr(+) or myr(−)MA. Endothermic heats of reaction were measured at 37 °C for 30 injections. Heats of dilution were measured by titrating CaM into a buffer under identical conditions. Base-line corrections were performed by subtracting heats of dilution from the raw MA·CaM titration data. Binding curves were analyzed, and dissociation constants were determined by nonlinear least square fitting of the base line-corrected data.
Fluorescence Spectroscopy
All of the fluorescence measurements were collected on a Fluorolog-3 spectrofluorimeter (Horiba Jobin Yvon Inc.). The following parameters have been used: λexcitation = 295 nm; λemission = 300–500 nm; data interval = 1 nm; slit width = 5 nm. CaM (∼500 μm) and MA (∼5.8–6.5 μm) samples were prepared at identical buffer conditions containing 50 mm HEPES (pH 7.0) and 10 mm CaCl2. To study the effect of salt on myr(+)MA·CaM interactions, fluorescent spectra were obtained for myr(+)MA as a function of added CaM at various NaCl concentrations (0, 100, 200, 300, and 500 mm). As a control, we examined the effect of salt on fluorescence signal by collecting spectra of myr(+)MA at the same salt concentrations described above. We found no detectable effect of salt on the fluorescence signal. Emission spectra of buffers were subtracted from the MA spectra to correct for the solvent effect. Additional control experiments were obtained by titrating CaM into buffer to correct for the Tyr fluorescence signal from CaM. These spectra have been subtracted from the Trp fluorescence spectra. Dissociation constants were calculated using ORIGIN 8.1 software (OriginLab, Northampton, MA) by plotting relative fluorescent enhancement (ΔF = Fn − Fo, where Fn and Fo are fluorescence intensities for MA in the CaM-bound and free forms) versus CaM concentration.
RESULTS
In vivo and in vitro studies have shown that Gag interactions with CaM are mediated by the MA domain and dependent on calcium (28). More recent studies have shown that myr(−)MA and short peptides derived from the MA protein interact directly with CaM (48, 49). However, because it is not yet known how CaM interacts with the native myr(+)MA protein and whether binding affects the myr switch mechanism in vitro, we have conducted structural, biochemical, and biophysical studies on the interactions between CaM and myr(+)MA.
Gel Mobility Assay
The solution properties of unbound CaM, MA, and their complex were initially analyzed with size exclusion chromatography in the presence or absence of calcium. Three samples were run separately on gel filtration columns (Superdex 75 and Sephacryl S-200 HR) under identical experimental conditions. As shown in Fig. 1 (top panel), the elution volumes of unbound CaM and myr(+)MA were at 66 and 77 ml, respectively. A sample of the myr(+)MA·CaM complex made with a 1:1 stoichiometry eluted at 57 ml. The chromatogram for the complex shows an additional small peak at 77 ml, indicating a slight excess of myr(+)MA. A gel filtration mobility assay with known protein standards (Fig. 1, bottom panel) revealed that the apparent molecular mass of CaM is significantly high (27 kDa) compared with MA despite the fact that both proteins have similar molecular masses (∼17 kDa). The migration behavior of CaM has been attributed to its elongated dumbbell shape (72). The myr(+)MA·CaM complex migrates as a ∼45–50-kDa species, significantly higher than a 1:1 complex (Fig. 1B and supplemental Fig. S1).
FIGURE 1.

Characterization of the MA·CaM complex by gel filtration chromatography. Top, chromatographic separation of myr(+)MA, CaM, and myr(+)MA·CaM in the presence of calcium. Bottom, gel filtration calibration assay showing mobility of CaM, MA, and the CaM·MA complex.
To determine whether myr(+)MA is able to bind to apo CaM, the myr(+)MA·CaM complex was made in the absence of Ca2+. Two separate peaks for CaM and myr(+)MA were eluted at 67 and 74 ml, respectively (supplemental Fig. S2). No peak was observed at ∼57 ml to indicate the formation of complex. The elution volume of apo CaM is very similar to that observed in the presence of Ca2+, indicating that the migration behavior is Ca2+-independent. Consistent with earlier studies, our results demonstrate that CaM interaction with myr(+)MA is Ca+2-dependent.
Sedimentation Studies of myr(+)MA·CaM Interactions
Although the results described above provide insights on the mobility of the myr(+)MA and CaM proteins as well as their complex, the oligomerization and equilibrium properties and stoichiometry have yet to be determined. To do so, we conducted sedimentation velocity experiments on samples prepared at loading concentrations 100–250 μm for myr(+)MA, CaM, and their complex. The myr(+)MA·CaM sample used for these experiments was first subjected to a gel filtration chromatography column to ensure sample homogeneity. As shown in Fig. 2, sedimentation velocity profiles of myr(+)MA, CaM, and the myr(+)MA·CaM complex exhibit a single sedimentation boundary. Analysis of the data using SEDFIT (67–70) yielded peaks of ∼1.5, 1.7, and 2.9 S for myr(+)MA, CaM, and myr(+)MA·CaM, respectively. Further analysis of the sedimentation velocity data using molecular mass distribution revealed approximate molecular masses of 16 and 17 kDa for myr(+)MA and CaM, respectively, whereas the apparent molecular mass for the myr(+)MA·CaM complex was calculated to be ∼35 kDa. The monomeric character of myr(+)MA at the current buffer conditions (pH 7.2) is consistent with our recent studies, which show that oligomerization of MA is pH-dependent (73). Taken together, the sedimentation velocity results indicate the formation of a 1:1 myr(+)MA·CaM complex.
FIGURE 2.
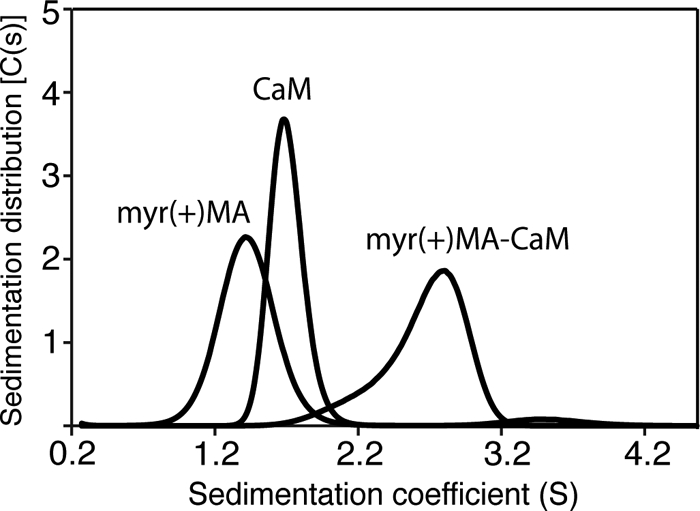
Sedimentation coefficient distributions (C(s)) obtained from the sedimentation profiles for CaM, myr(+)MA, and the myr(+)MA·CaM complex.
Characterization of CaM Binding to myr(+)MA by ITC
As discussed above, central to the cellular activity of Ca2+-bound CaM in binding to target proteins is the presence of two hydrophobic batches localized on the N- and C-terminal lobes (36). Additional electrostatic interactions were also shown to stabilize CaM-protein interactions (44, 45, 50, 51). However, the importance and relative contribution of hydrophobic versus electrostatic factors in myr(+)MA·CaM interactions are not known, especially that the hydrophobic myr group can potentially play a role as was observed in other CaM-protein interactions (44, 45, 50, 51). ITC has been used to determine the affinity and thermodynamic parameters of CaM binding to myr(+)MA. ITC measures heat absorbed or generated upon binding and provides values for the dissociation constant (Kd), the stoichiometry (n), and the enthalpy change (ΔH°). The Kd value is then used to calculate the change in Gibbs energy (ΔG°), which together with ΔH° allows the calculation of the entropic term TΔS°. The separation of the free energy of binding into entropic and enthalpic components reveals the nature of the forces that drive the binding reaction.
As shown in Fig. 3, one-site fitting of the ITC data yielded the following thermodynamic parameters: n = 1.08, Kd = 1.9 ± 0.1 μm, ΔH° = 11.3 ± 0.1 kcal/mol, and ΔS° = 62.6 cal/mol/deg. As indicated by the sign of the heat of enthalpy, CaM binding to myr(+)MA is strongly endothermic, which confirms the entropically driven nature of interactions. However, we do not rule out the presence of electrostatic interactions to further stabilize binding. Of particular note, CaM binding to myr(−)MA has been shown to be salt-dependent, suggesting a contribution of electrostatic interactions (48). To examine the effect of salt on myr(+)MA·CaM interactions, tryptophan fluorescence spectroscopy experiments were conducted at salt concentrations of 0–500 mm. The MA protein contains two Trp residues at positions 16 and 36, whereas CaM lacks any Trp residues. A change in the intensity of the Trp emission spectrum occurs upon binding of CaM. Our data revealed that binding affinity is reduced only by ∼6-fold upon increasing salt concentration from 0 to 500 mm (supplemental Table S1 and Fig. S3), indicating that the electrostatic factor in myr(+)MA·CaM binding is inconsequential.
FIGURE 3.
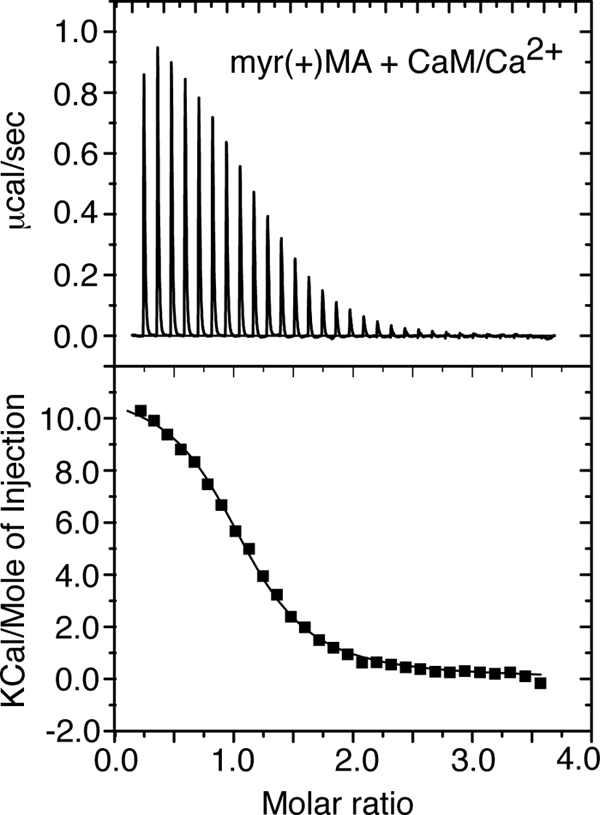
Upper panel, ITC data obtained upon titration of CaM (402 μm) into myr(+)MA (20 μm). The data best fit one-site binding mode and afforded a Kd of 1.9 μm (lower panel).
Myristate Group of MA Is Not Required for CaM Binding
Several myristoylated proteins including MARCKS, CAP-23/NAP-22, and HIV-1 Nef were found to bind CaM to facilitate their intracellular localization and membrane targeting (44, 45, 50, 51). Our ITC data described above show that CaM binding to myr(+)MA is entropically driven, indicating a major contribution of hydrophobic contacts. To examine whether the myr group is involved in CaM binding, ITC experiments were carried on the myr(−)MA protein as titrated with CaM (supplemental Fig. S4). Interestingly, thermodynamic parameters (Kd = 2.1 ± 0.2 μm, ΔH° = 12.90 ± 0.02 kcal/mol, and ΔS° = 67.5 cal/mol/deg) are almost identical to those obtained for the myr(+)MA·CaM complex. Taken together, these results indicate that the myr group is not involved in CaM binding.
CaM Binding to myr(+)MA as Detected by NMR
NMR methods have been utilized to determine how CaM interacts with myr(+)MA and to identify the interacting regions for both proteins. Chemical shift perturbations not only provide insights on the interacting regions but can also be used to identify allosteric conformational changes distant from the binding site. All NMR studies were obtained in the presence of calcium. We first conducted NMR binding studies on myr(+)MA as a function of added CaM. The two-dimensional 1H-15N HSQC spectrum collected for a uniformly 15N-labeled myr(+)MA at 100 μm mainly indicates a myr-sequestered conformation (57, 73). The addition of substoichiometric amounts of unlabeled CaM led to significant chemical shift changes and signal broadening/loss (Fig. 4). Interestingly, at 0.25:1 myr(+)MA:CaM, the 1H-15N signals corresponding to residues in the N-terminal domain (Gly-2 to Arg-20) became broad beyond detection, whereas residues Leu-21 to Glu-45 either disappeared or exhibited a significant loss in intensity (Fig. 4). These results suggest that the N-terminal region of MA is perturbed by CaM binding. A steady decrease in intensity for most of the 1H-15N signals in the HSQC spectra was clearly observed as a function of added CaM. At 1:1 myr(+)MA:CaM, the vast majority of 1H-15N signals were severely broadened, but many new others have appeared (Fig. 4).
FIGURE 4.

Top panel, overlay of two-dimensional 1H-15N HSQC spectra obtained for a uniformly 15N-labeled myr(+)MA upon titration with unlabeled CaM. Black, myr(+)MA·CaM ratio of 0:1; blue, myr(+)MA·CaM ratio of 0.25:1; red, myr(+)MA·CaM ratio of 1:1. Bottom panel, selected 1H-15N HSQC signals from the top panel are shown to emphasize the significant loss of signal intensity in the N-terminal domain of MA upon CaM binding at 0.25:1 stoichiometry.
Severe broadening or loss of signals can be explained by one or more of the following events. First, binding of CaM induces a large conformational change, leading to alteration of tertiary and/or secondary structure of MA. This possibility has been recently suggested by Chow et al. (48) upon studying the interactions of CaM and myr(−)MA. Second, a slow exchange on the NMR scale between free and bound forms at 1:1 stoichiometry. This is ruled out because no changes in spectra were detected by further addition of CaM (up to 3:1 CaM:MA). Third, an increase in the line widths of signals as a result of increasing the molecular size of the myr(+)MA·CaM complex. This possibility has been tested by collecting two-dimensional 1H-15N transverse relaxation optimized spectroscopy data for 15N-labeled myr(+)MA complexed with CaM (data not shown). Although a few 1H-15N signals were sharper, the transverse relaxation optimized spectroscopy and HSQC spectra were essentially similar. Fourth, binding kinetics are in intermediate exchange on the NMR time scale. To determine whether this was the case, we collected a set of two-dimensional HSQC data on an 15N-labeled myr(+)MA complexed with CaM as a function of temperature (15–35 °C; supplemental Fig. S5). Changing the temperature has not resulted in any major improvement of signal intensity. However, in the spectrum collected at 25 °C, two new signals have appeared at 10–11 ppm (1H). These two signals, which likely correspond to the side chains Trp-16 and Trp-36, are substantially shifted from their original positions, suggesting that these residues are probably involved in CaM binding or undergo a conformational change upon binding of CaM.
CaM Binding to myr(+)MA Triggers myr Exposure
Previous studies have shown that 1H-15N resonances corresponding to residues Gly-2 to Arg-20 are usually sensitive to the positioning of the myr group (27, 57). The finding that these signals completely disappeared at a substoichiometric addition of CaM (Fig. 4) may suggest a perturbation of the myr switch mechanism. To examine this hypothesis, we devised three approaches. First, we collected two-dimensional HSQC spectra for myr(−)MA as a function of added CaM. Interestingly, the 1H-15N signals for residues Gly-2 to Arg-20 did not exhibit significant intensity loss or broadening at substoichiometric CaM (supplemental Fig. S6). However, as was observed for myr(+)MA when complexed with CaM at an equimolar ratio, the HSQC spectrum of myr(−)MA became very broad, and several new peaks have appeared. Thus, the loss of signals for residues Gly-2 to Arg-20 in the HSQC spectrum of myr(+)MA (Fig. 4) suggests a slow exchange on the NMR scale between two conformations, likely to be myr-sequestered and exposed states.
In the second approach, we collected two-dimensional 1H-13C HMQC data for myr(+)MA containing a 13C-labeled myr group. Only methylene and methyl resonances for the myr group are expected to appear in the spectra. As shown in Fig. 5, the 1H-13C signals of the myr group in the free myr(+)MA protein (black) are identical to those reported previously for the myr-sequestered form (57) and are very different from those observed for free myristic acid (green) (74). Interestingly, for the myr(+)MA·CaM complex at 1:1 stoichiometry, the 1H-13C signals of the myr group (red) shift toward values of the free ligand, suggesting extrusion of the myr group upon CaM binding. At a substoichiometric amount of CaM (0.25:1 myr(+)MA:CaM), the HMQC spectrum (supplemental Fig. S7) shows two separate sets of 1H-13C signals for free and bound forms, confirming a slow exchange between myr-sequestered and exposed forms that is induced by CaM binding.
FIGURE 5.

Overlay of two-dimensional 1H-13C HMQC spectra obtained for myr(+)MA containing a 13C-labeled myr group in the unbound (black) and CaM-bound (red) states. For comparison, a spectrum of free myristic acid is shown (green). An asterisk indicates a breakthrough of 1H-13C signals from CaM.
In the third approach and to confirm that binding of CaM triggers myr exposure, three-dimensional 13C-edited and 13C-edited/12C-double half-filtered HMQC-NOESY spectra were obtained for myr(+)MA containing a 13C-labeled myr group in the free or CaM-bound states. Previous studies have shown that the myr group occupies a hydrophobic cavity and makes contacts with a number of hydrophobic and aromatic residues including Val-7, Leu-8, Leu-16, Ile-34, Leu-51, and Ile-85 (27, 57, 58, 73). For free myr(+)MA, several NOE cross-peaks are observed between the side chains of these residues and the methylene (H5–H11) and terminal methyl (H14) signals of the myr group, confirming sequestration of the myr group (supplemental Fig. S8). However, no NOE cross-peaks are observed between the myr group and any of these residues in the CaM-bound spectrum. These results were confirmed by three-dimensional 13C-edited HMQC-NOESY spectra (supplemental Fig. S8), demonstrating that CaM binding to myr(+)MA induces the extrusion of the myr group (supplemental Fig. S8). In addition, no new NOE cross-peaks are observed between the myr group and CaM, indicating that the myr group does not interact with CaM but is rather exposed to solvent.
NMR Studies of CaM upon Binding to MA
All of the NMR data described above were obtained for 15N- or 13C-labeled MA proteins as titrated with unlabeled CaM. To map out the interaction region(s) on the CaM protein as a function of MA binding, reciprocal NMR experiments were conducted on uniformly 15N- or 13C-labeled samples of CaM as a function of added MA.
Complete assignments of 1H, 15N, and 13C signals for CaM were achieved by using a combination of two-, three-, and four-dimensional NMR methods (see details under “Experimental Procedures”). Representative 1H-15N HSQC spectra obtained for 15N-labeled CaM in the unbound form and in complex with myr(+)MA are shown in Fig. 6A. At 1.4:1 MA:CaM, the majority of signals exhibited significant chemical shift changes. No changes in the spectrum were observed upon further addition of myr(+)MA. Interestingly, residues that exhibited substantial chemical shift changes are not localized in a well defined region of the CaM protein but rather spread throughout the N- and C-terminal lobes as well as the central helix. A surface representation of the CaM protein (Protein Data Base code 3CLN) shown in Fig. 6B highlights residues that exhibited chemical shift changes upon binding of myr(+)MA. Residues that exhibited substantial chemical shift changes (ΔδHN ((Δδ1H)2 + (Δδ15N)2)½) > 0.1 ppm) are colored in red, whereas those with modest changes are colored in orange (ΔδHN < 0.1). Perturbations of the vast majority of amide signals may suggest an engagement of a wide interacting interface or induction of significant conformational changes in CaM upon binding to MA.
FIGURE 6.

A, overlay of two-dimensional 1H-15N HSQC spectra obtained for 15N-labeled CaM in the unbound state (black) and in complex with unlabeled myr(+)MA at 1. 4:1 (MA:CaM) (red). B, surface representation of the CaM structure of (Protein Data Base code 3CLN). Residues that exhibited substantial chemical shift changes (> 0.1 ppm) or signal loss are colored in red, whereas those modestly perturbed (< 0.1 ppm) are colored in orange. Calcium ions are colored in green.
Because of partial assignment (∼70%) of the 1H-15N signals in the HSQC spectrum of CaM, Chow et al. (48) have recently proposed that CaM undergoes only modest structural changes upon binding with myr(−)MA. To assess whether the myr group is responsible for the significant changes observed in the HSQC spectrum shown in Fig. 6, two-dimensional HSQC data were also collected for 15N-labeled CaM as titrated with myr(−)MA (supplemental Fig. S9). The spectral changes in the HSQC spectrum of CaM upon binding to myr(−)MA are almost identical to those observed in the corresponding spectrum when myr(+)MA is bound (Fig. 6), which demonstrates that the substantial chemical shift changes are not caused by the myr group but rather by protein-protein contacts.
Hydrophobic Regions in CaM Are Important for MA Binding
A prominent feature that contributes to the flexibility of CaM is the presence of two hydrophobic surfaces on the N- and C-terminal lobes. These regions are only formed upon binding of Ca2+ (37). Central to the formation of these hydrophobic batches is exposure of eight Met residues that contribute ∼46% to the solvent-accessible surface area of these two regions (75, 76). Methionine residues are considered essential for the unique promiscuous binding behavior of CaM to target proteins (76). This novel structural feature renders the Met methyl groups excellent “NMR reporters.” Interactions of target proteins or CaM antagonists often led to significant perturbations in the 1H-13C chemical shifts of Met side chains (77). To assess whether the two hydrophobic batches are involved in the binding of MA, we collected two-dimensional 1H-13C HMQC data on a uniformly 13C-labeled CaM sample as titrated with myr(+)MA. A selected region showing the 1H-13C signals of the methionine methyl groups (Cϵ) is shown in Fig. 7. In this spectrum, the 1H-13C signals of all nine methionine residues are observed. The 1H-13C signals of Met-51, Met-71, Met-72, Met-144, and Met-145 exhibited significant chemical shift changes upon titration of myr(+)MA, whereas those corresponding to Met-36, Met-76, Met-109, and Met-124 were less pronounced. Methionine residues at positions 36, 51, 71, and 72 are located in the N-terminal hydrophobic batch, whereas those at positions 109, 124, 144, and 145 are located within the hydrophobic patch in the C-terminal lobe (Fig. 7). Our two-dimensional HMQC data suggest that both the N- and C-terminal lobes of CaM are probably involved in myr(+)MA binding because methionine groups from both motifs exhibited significant chemical shift changes. Similar two-dimensional HMQC experiments have also been conducted on CaM upon titration with the myr(−)MA protein (data not shown). Chemical shift changes observed for the 1H-13C signals of Met residues are very similar to those shown in Fig. 7. The absence of any additional chemical shift perturbations in the 1H-13C signals when the myr group is present confirms that the myr group is not involved in CaM binding.
FIGURE 7.

A, overlay of two-dimensional 1H-13C HMQC spectra obtained for 13C-labeled CaM as a function of added unlabeled myr(+)MA. Black, myr(+)MA·CaM ratio of 0:1; blue, myr(+)MA·CaM ratio of 0.25:1; magenta, myr(+)MA·CaM ratio of 0.5:1; green, myr(+)MA·CaM ratio of 1:1. B, surface representation of the CaM structure (Protein Data Base code 3CLN) showing the nine Met residues on the N- and C-terminal lobes (red). Calcium ions are colored in green.
DISCUSSION
CaM is a ubiquitous protein expressed in all eukaryotic cells and is implicated in a variety of cellular functions (32–37). It can be localized in various subcellular locations, including the cytoplasm, within organelles, or associated with the plasma or organelle membranes (32–37). Earlier studies have attempted to identify specific roles of CaM in viral replication and infectivity. CaM has been shown to possess a functional role in budding of Ebola virus-like particles through interactions with the viral matrix protein VP40 (78). Others have proposed a potential role of CaM in the HIV and simian immunodeficiency virus replication cycles (28, 44–47, 79). A combination of in vivo and in vitro studies revealed that CaM interacts with HIV-1 Gag, Nef, and Env proteins (28, 44–47). It has also been shown that HIV-infected cells express elevated levels of CaM (80). HIV-1 Gag was shown to co-localize with CaM in a diffuse pattern spread throughout the cytoplasm (28). These findings may suggest a link between the CaM cell signaling pathway and HIV replication and infectivity.
The interactions between Gag and CaM are mediated by the myr(+)MA protein and are Ca2+-dependent (28). Although recent studies have provided evidence for direct interactions between CaM and myr(−)MA (48) or short peptides derived from the MA protein (49), the molecular mechanism of CaM binding to myr(+)MA and the subsequent effect on the myr switch mechanism are not known. In this report, we have established that: (i) CaM binds to myr(+)MA with 1:1 stoichiometry in a Ca2+-dependent manner, (ii) CaM binding to myr(+)MA induces a structural change that triggers myr exposure, (iii) the myr group is not involved in CaM binding, and (iv) CaM-MA interaction is hydrophobic and entropically driven, whereas electrostatic interactions appear to be inconsequential.
Several myristoylated proteins including MARCKS, CAP-23/NAP-22, and HIV-1 Nef are known to bind CaM to facilitate their intracellular localization and membrane targeting (44, 45, 50, 51). Structural, biochemical, and biophysical characterization of CaM complexed with these proteins or short peptides derived from within revealed that the myr group is involved in CaM binding, suggesting that the myr group is not only important for membrane targeting but also for mediating protein-protein interactions. Membrane association and dissociation of myristoylated proteins are often regulated by intracellular Ca2+ signaling through a Ca2+-myr switch (52–54), which alters their intracellular localization from the cytosol to the membrane through Ca2+ binding (55). However, intracellular targeting of Ca2+ nonsensing proteins like MARCKS, CAP-23/NAP-22, and Nef is regulated by CaM binding. Although the MA protein belongs to the Ca2+ nonsensing protein, the finding that the myr group is not “grabbed” by CaM may suggest a novel functional role of CaM in the trafficking and/or targeting of Gag to the PM for assembly.
CaM-binding proteins typically contain a region that is characterized by a basic amphiphilic helix consisting of ∼20 residues. In many classical CaM-binding targets, hydrophobic residues usually occupy conserved positions at 1-5-10 or 1-8-14, which point to one face of the helix (34). Additional basic residues are responsible for stabilization of the complex via electrostatic interactions with CaM acidic residues. Although these patterns are typically found in many CaM-binding proteins, numerous unclassified examples were also identified (34). A web-based tool has been developed by the Ikura laboratory (Ontario Cancer Institute) to identify sequences with potential CaM-binding sites (41). Upon analysis of the protein sequence, it provides scores from 0 to 9 based on several criteria including hydropathy, α-helical propensity, residue weight, residue charge, hydrophobic residue content, helical class, and occurrence of particular residues. The most likely binding site in a given sequence is highlighted by repeated 9s. This method has been used to analyze the MA protein sequence and yielded the highest score for residues Lys-32 to Glu-40. These residues make up the majority of helix II. Interestingly, this region contains several hydrophobic residues (Ile-34, Val-35, Trp-36, and Ala-38) that do not match any of the known patterns for CaM-binding sites. Although sequence analysis using the web-based data base tool did not reveal a potential binding site within helix I (score 0), solution x-ray scattering data (49) and fluorescence studies on short MA peptides (28) revealed that both helices I and II of MA (residues 11–47) are important for CaM binding. Although it appears that a synthetic peptide spanning region Gly-11 to Gln-28 (helix I and β-hairpin) binds CaM with 2:1 stoichiometry, Gly-26 to Asn-47 (helix II) and Gly-11 to Asn-47 (helix I-II) bind with 1:1 stoichiometry. Taken together, our results combined with the previous findings may suggest a novel CaM-binding mode that requires engagement of hydrophobic residues from both helices I and II of MA.
During the preparation of this manuscript, Chow et al. (48) reported their findings on the interactions between CaM and full-length myr(−)MA. In this study, it was suggested that CaM disrupts the MA structure to interact with helices I and II. Based on CD data, it was suggested that the MA protein undergoes a 20% loss in helical character upon binding to CaM. Our current data combined with previous studies (28, 48, 49) indicate that the CaM-binding site in MA is localized within residues 11–45. Although it is not yet clear which specific residues are critical for binding, data suggest that several hydrophobic residues within helices I and/or II probably interact with CaM. The N-terminal region of MA (residues 2–47) is critical for various MA functions. Among these functions is its role in mediating Gag-membrane interactions and regulation of the myr switch mechanism (4, 5, 22, 23, 27, 57, 58, 73, 81, 82). Helix I of MA is implicated in Gag binding to several cellular constituents including the AP-3 complex (11) and TIP47 (29). Several residues in helix I (Leu-8, Ser-9, Leu-13, Trp-16, Glu-17, and Lys-18) have been reported as being critical for Env incorporation (83–85), which led to the suggestion that MA protein interacts directly with the Env protein via helix I (83, 86). Indeed, our NMR data presented here suggest that helix I is likely to undergo a structural change or readjustment of position in response to CaM binding, which in turn leads to extrusion of the myr group.
Structural studies revealed that myr exposure is increased by several factors including PI(4,5)P2 binding, pH, increasing protein concentration, and inclusion of the capsid domain (27, 57, 73). Equilibrium data revealed that although myr(+)MA resides in monomer-trimer equilibrium, the myr(−)MA protein maintains the monomeric character in solution under all conditions (57). In addition, exposure of the myr group is coupled with protein trimerization (57). Despite the evidence that MA can form trimers and assemble on membrane as hexamers (87, 88), the role of MA oligomerization in stabilizing Gag-Gag interactions is still controversial (57, 88–93). Our data presented here show that exposure of the myr group upon binding to CaM does not induce the formation of MA trimer. It has yet to be determined whether binding of CaM to Gag or Gag-like construct (containing the capsid domain) enhances oligomerization.
Induction of a transient rise in cytoplasmic Ca2+ has been shown to increase the amounts of HIV-1 Gag, Gag assembly intermediates, and virus-like particles in multivesicular bodies, and resulted in a dramatic increase in virus-like particle release (94). It was suggested that Ca2+-mediated fusion of vacuolar/multivesicular body-like compartments promotes virus-like particle production. However, it is not known whether manipulating the cytoplasmic Ca2+ level plays a role in stabilizing Gag-CaM interactions and facilitates Gag trafficking and assembly. In summary, although it is now established that CaM binds directly with the MA protein leading to extrusion of the myr group, the precise functional role of CaM in the HIV replication cycle has yet to be examined and warrants further investigation.
Supplementary Material
Acknowledgments
Thanks to Dr. Madeline Shea (University of Iowa) for providing the CaM molecular clone, Michael Summers (University of Maryland, Baltimore County) for the MA co-expression clone, and David Graves and Aaron Lucius (Department of Chemistry, University of Alabama at Birmingham) for help with the ITC and fluorescence experiments.
This work was supported, in whole or in part, by National Institutes of Health Grant 1R01AI087101. This work was also supported by intramural funding from the University of Alabama at Birmingham Center for AIDS Research and Comprehensive Cancer Center Grant NCI CA13148-35.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Table S1 and Figs. S1–S9.
- PM
- plasma membrane
- MA
- matrix protein
- CaM
- calmodulin
- myr
- myristyl
- myr(−)
- unmyristoylated protein
- myr(+)
- myristoylated protein
- HSQC
- heteronuclear single quantum coherence
- HMQC
- heteronuclear multiple quantum coherence
- PI(4,5)P2
- phosphatidylinositol-(4,5)-bisphosphate
- MARCKS
- myristoylated alanine-rich C kinase substrate
- NOESY
- nuclear Overhauser effect spectroscopy
- ITC
- isothermal titration calorimetry.
REFERENCES
- 1.Adamson C. S., Freed E. O. (2007) Adv. Pharmacol. 55, 347–387 [DOI] [PubMed] [Google Scholar]
- 2.Ganser-Pornillos B. K., Yeager M., Sundquist W. I. (2008) Curr. Opin. Struct. Biol. 18, 203–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bieniasz P. D. (2009) Cell Host Microbe 5, 550–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ono A. (2010) Biol. Cell 102, 335–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ono A. (2010) Vaccine 28, Suppl. 2, B55–B59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chu H., Wang J. J., Spearman P. (2009) Curr. Top. Microbiol. Immunol. 339, 67–84 [DOI] [PubMed] [Google Scholar]
- 7.Jouvenet N., Neil S. J., Bess C., Johnson M. C., Virgen C. A., Simon S. M., Bieniasz P. D. (2006) PLoS Biol. 4, e435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jouvenet N., Simon S. M., Bieniasz P. D. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 19114–19119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Finzi A., Orthwein A., Mercier J., Cohen E. A. (2007) J. Virol. 81, 7476–7490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turner B. G., Summers M. F. (1999) J. Mol. Biol. 285, 1–32 [DOI] [PubMed] [Google Scholar]
- 11.Dong X., Li H., Derdowski A., Ding L., Burnett A., Chen X., Peters T. R., Dermody T. S., Woodruff E., Wang J. J., Spearman P. (2005) Cell 120, 663–674 [DOI] [PubMed] [Google Scholar]
- 12.Gousset K., Ablan S. D., Coren L. V., Ono A., Soheilian F., Nagashima K., Ott D. E., Freed E. O. (2008) PLoS Pathog. 4, e1000015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hermida-Matsumoto L., Resh M. D. (2000) J. Virol. 74, 8670–8679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Joshi A., Ablan S. D., Soheilian F., Nagashima K., Freed E. O. (2009) J. Virol. 83, 53755387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jouvenet N., Bieniasz P. D., Simon S. M. (2008) Nature 454, 236–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li H., Dou J., Ding L., Spearman P. (2007) J. Virol. 81, 12899–12910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Waheed A. A., Freed E. O. (2009) Virus Res. 143, 162–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Welsch S., Keppler O. T., Habermann A., Allespach I., Krijnse-Locker J., Kräusslich H. G. (2007) PLoS Pathog. 3, e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ono A. (2009) Future Virol. 4, 241–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reil H., Bukovsky A. A., Gelderblom H. R., Göttlinger H. G. (1998) EMBO J. 17, 2699–2708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ono A., Ablan S. D., Lockett S. J., Nagashima K., Freed E. O. (2004) Proc. Natl. Acad. Sci. 101, 14889–14894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chukkapalli V., Hogue I. B., Boyko V., Hu W. S., Ono A. (2008) J. Virol. 82, 2405–2417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chukkapalli V., Oh S. J., Ono A. (2010) Proc. Natl. Acad. Sci. U.S.A. 107, 1600–1605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martin T. F. (2001) Curr. Opin. Cell Biol. 13, 493–499 [DOI] [PubMed] [Google Scholar]
- 25.Behnia R., Munro S. (2005) Nature 438, 597–604 [DOI] [PubMed] [Google Scholar]
- 26.McLaughlin S., Murray D. (2005) Nature 438, 605–611 [DOI] [PubMed] [Google Scholar]
- 27.Saad J. S., Miller J., Tai J., Kim A., Ghanam R. H., Summers M. F. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 11364–11369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Radding W., Williams J. P., McKenna M. A., Tummala R., Hunter E., Tytler E. M., McDonald J. M. (2000) AIDS Res. Hum. Retroviruses 16, 1519–1525 [DOI] [PubMed] [Google Scholar]
- 29.Lopez-Vergès S., Camus G., Blot G., Beauvoir R., Benarous R., Berlioz-Torrent C. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 14947–14952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ryo A., Tsurutani N., Ohba K., Kimura R., Komano J., Nishi M., Soeda H., Hattori S., Perrem K., Yamamoto M., Chiba J., Mimaya J., Yoshimura K., Matsushita S., Honda M., Yoshimura A., Sawasaki T., Aoki I., Morikawa Y., Yamamoto N. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 294–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nishi M., Ryo A., Tsurutani N., Ohba K., Sawasaki T., Morishita R., Perrem K., Aoki I., Morikawa Y., Yamamoto N. (2009) FEBS Lett. 583, 1243–1250 [DOI] [PubMed] [Google Scholar]
- 32.Chin D., Means A. R. (2000) Trends Cell Biol. 10, 322–328 [DOI] [PubMed] [Google Scholar]
- 33.Hoeflich K. P., Ikura M. (2002) Cell 108, 739–742 [DOI] [PubMed] [Google Scholar]
- 34.Ishida H., Vogel H. J. (2006) Protein Pept. Lett. 13, 455–465 [DOI] [PubMed] [Google Scholar]
- 35.Osawa M., Tokumitsu H., Swindells M. B., Kurihara H., Orita M., Shibanuma T., Furuya T., Ikura M. (1999) Nat. Struct. Biol. 6, 819–824 [DOI] [PubMed] [Google Scholar]
- 36.Vetter S. W., Leclerc E. (2003) Eur. J. Biochem. 270, 404–414 [DOI] [PubMed] [Google Scholar]
- 37.Yamniuk A. P., Vogel H. J. (2004) Mol. Biotechnol. 27, 33–57 [DOI] [PubMed] [Google Scholar]
- 38.Chattopadhyaya R., Meador W. E., Means A. R., Quiocho F. A. (1992) J. Mol. Biol. 228, 1177–1192 [DOI] [PubMed] [Google Scholar]
- 39.Fallon J. L., Quiocho F. A. (2003) Structure 11, 1303–1307 [DOI] [PubMed] [Google Scholar]
- 40.Finn B. E., Evenäs J., Drakenberg T., Waltho J. P., Thulin E., Forsén S. (1995) Nat. Struct. Biol. 2, 777–783 [DOI] [PubMed] [Google Scholar]
- 41.Yap K. L., Kim J., Truong K., Sherman M., Yuan T., Ikura M. (2000) J. Struct. Funct. Genomics 1, 8–14 [DOI] [PubMed] [Google Scholar]
- 42.Vogel H. J. (1994) Biochem. Cell Biol. 72, 357–376 [PubMed] [Google Scholar]
- 43.Kretsinger R. H. (1996) Nat. Struct. Biol. 3, 12–15 [DOI] [PubMed] [Google Scholar]
- 44.Hayashi N., Matsubara M., Jinbo Y., Titani K., Izumi Y., Matsushima N. (2002) Protein Sci. 11, 529–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Matsubara M., Jing T., Kawamura K., Shimojo N., Titani K., Hashimoto K., Hayashi N. (2005) Protein Sci. 14, 494–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Srinivas S. K., Srinivas R. V., Anantharamaiah G. M., Compans R. W., Segrest J. P. (1993) J. Biol. Chem. 268, 22895–22899 [PubMed] [Google Scholar]
- 47.Towler D. A., Adams S. P., Eubanks S. R., Towery D. S., Jackson-Machelski E., Glaser L., Gordon J. I. (1988) J. Biol. Chem. 263, 1784–1790 [PubMed] [Google Scholar]
- 48.Chow J. Y., Jeffries C. M., Kwan A. H., Guss J. M., Trewhella J. (2010) J. Mol. Biol. 400, 702–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Izumi Y., Watanabe H., Watanabe N., Aoyama A., Jinbo Y., Hayashi N. (2008) Biochemistry 47, 7158–7166 [DOI] [PubMed] [Google Scholar]
- 50.Matsubara M., Nakatsu T., Kato H., Taniguchi H. (2004) EMBO J. 23, 712–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Matsubara M., Titani K., Taniguchi H., Hayashi N. (2003) J. Biol. Chem. 278, 48898–48902 [DOI] [PubMed] [Google Scholar]
- 52.Tanaka T., Ames J. B., Harvey T. S., Stryer L., Ikura M. (1995) Nature 376, 444–447 [DOI] [PubMed] [Google Scholar]
- 53.Faurobert E., Chen C. K., Hurley J. B., Teng D. H. (1996) J. Biol. Chem. 271, 10256–10262 [DOI] [PubMed] [Google Scholar]
- 54.Matsuda S., Hisatomi O., Ishino T., Kobayashi Y., Tokunaga F. (1998) J. Biol. Chem. 273, 20223–20227 [DOI] [PubMed] [Google Scholar]
- 55.Braunewell K. H., Gundelfinger E. D. (1999) Cell Tissue Res. 295, 1–12 [DOI] [PubMed] [Google Scholar]
- 56.Massiah M. A., Starich M. R., Paschall C., Summers M. F., Christensen A. M., Sundquist W. I. (1994) J. Mol. Biol. 244, 198–223 [DOI] [PubMed] [Google Scholar]
- 57.Tang C., Loeliger E., Luncsford P., Kinde I., Beckett D., Summers M. F. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 517–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Saad J. S., Loeliger E., Luncsford P., Liriano M., Tai J., Kim A., Miller J., Joshi A., Freed E. O., Summers M. F. (2007) J. Mol. Biol. 366, 574–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Putkey J. A., Slaughter G. R., Means A. R. (1985) J. Biol. Chem. 260, 4704–4712 [PubMed] [Google Scholar]
- 60.Sorensen B. R., Shea M. A. (1998) Biochemistry 37, 4244–4253 [DOI] [PubMed] [Google Scholar]
- 61.Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 62.Johnson B. A., Blevins R. A. (1994) J. Biomol. NMR 4, 603–614 [DOI] [PubMed] [Google Scholar]
- 63.Folkers P. J., Folmer R. H., Konings R. N., Hilbers C. W. (1993) J. Am. Chem. Soc. 115, 3798–3799 [Google Scholar]
- 64.Kay L. E., Clore G. M., Bax A., Gronenborn A. M. (1990) Science 249, 411–414 [DOI] [PubMed] [Google Scholar]
- 65.Wüthrich K. (1986) NMR of Proteins and Nucleic Acids, John Wiley & Sons, New York [Google Scholar]
- 66.Ikura M., Kay L. E., Bax A. (1990) Biochemistry 29, 4659–4667 [DOI] [PubMed] [Google Scholar]
- 67.Lebowitz J., Lewis M. S., Schuck P. (2002) Protein Sci. 11, 2067–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schuck P. (2000) Biophys. J. 78, 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schuck P. (2003) Anal. Biochem. 320, 104–124 [DOI] [PubMed] [Google Scholar]
- 70.Schuck P., Perugini M. A., Gonzales N. R., Howlett G. J., Schubert D. (2002) Biophys. J. 82, 1096–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wiseman T., Williston S., Brandts J. F., Lin L. N. (1989) Anal. Biochem. 179, 131–137 [DOI] [PubMed] [Google Scholar]
- 72.Majava V., Kursula P. (2009) PLoS One 4, e5402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fledderman E. L., Fujii K., Ghanam R. H., Waki K., Prevelige P. E., Freed E. O., Saad J. S. (2010) Biochemistry 49, 9551–9562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ames J. B., Tanaka T., Ikura M., Stryer L. (1995) J. Biol. Chem. 270, 30909–30913 [DOI] [PubMed] [Google Scholar]
- 75.O'Neil K. T., DeGrado W. F. (1990) Trends Biochem. Sci. 15, 59–64 [DOI] [PubMed] [Google Scholar]
- 76.Yuan T., Ouyang H., Vogel H. J. (1999) J. Biol. Chem. 274, 8411–8420 [DOI] [PubMed] [Google Scholar]
- 77.Elshorst B., Hennig M., Försterling H., Diener A., Maurer M., Schulte P., Schwalbe H., Griesinger C., Krebs J., Schmid H., Vorherr T., Carafoli E. (1999) Biochemistry 38, 12320–12332 [DOI] [PubMed] [Google Scholar]
- 78.Han Z., Harty R. N. (2007) Virus Genes 35, 511–520 [DOI] [PubMed] [Google Scholar]
- 79.Miller M. A., Mietzner T. A., Cloyd M. W., Robey W. G., Montelaro R. C. (1993) AIDS Res. Hum. Retroviruses 9, 1057–1066 [DOI] [PubMed] [Google Scholar]
- 80.Radding W., Pan Z. Q., Hunter E., Johnston P., Williams J. P., McDonald J. M. (1996) Biochem. Biophys. Res. Commun. 218, 192–197 [DOI] [PubMed] [Google Scholar]
- 81.Bryant M., Ratner L. (1990) Proc. Natl. Acad. Sci. U.S.A. 87, 523–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhou W., Parent L. J., Wills J. W., Resh M. D. (1994) J. Virol. 68, 2556–2569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Davis M. R., Jiang J., Zhou J., Freed E. O., Aiken C. (2006) J. Virol. 80, 2405–2417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dorfman T., Bukovsky A., Ohagen A., Höglund S., Göttlinger H. G. (1994) J. Virol. 68, 8180–8187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Freed E. O., Martin M. A. (1996) J. Virol. 70, 341–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dorfman T., Mammano F., Haseltine W. A., Göttlinger H. G. (1994) J. Virol. 68, 1689–1696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Alfadhli A., Barklis R. L., Barklis E. (2009) Virology 387, 466–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Alfadhli A., Huseby D., Kapit E., Colman D., Barklis E. (2007) J. Virol. 81, 1472–1478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dou J., Wang J. J., Chen X., Li H., Ding L., Spearman P. (2009) Virology 387, 341–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Morikawa Y., Hockley D. J., Nermut M. V., Jones I. M. (2000) J. Virol. 74, 16–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Morikawa Y., Zhang W. H., Hockley D. J., Nermut M. V., Jones I. M. (1998) J. Virol. 72, 7659–7663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Franke E. K., Yuan H. E., Bossolt K. L., Goff S. P., Luban J. (1994) J. Virol. 68, 5300–5305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Datta S. A., Zhao Z., Clark P. K., Tarasov S., Alexandratos J. N., Campbell S. J., Kvaratskhelia M., Lebowitz J., Rein A. (2007) J. Mol. Biol. 365, 799–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Perlman M., Resh M. D. (2006) Traffic 7, 731–745 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.