

Abstract
LIN28 (a homologue of the Caenorhabditis elegans lin-28 gene) is an evolutionarily conserved RNA-binding protein and a master regulator controlling the pluripotency of embryonic stem cells. Together with OCT4, SOX2, and NANOG, LIN28 can reprogram somatic cells, producing induced pluripotent stem cells. Expression of LIN28 is highly restricted to embryonic stem cells and developing tissues. In human tumors, LIN28 is up-regulated and functions as an oncogene promoting malignant transformation and tumor progression. However, the mechanisms of transcriptional and post-transcriptional regulation of LIN28 are still largely unknown. To examine microRNAs (miRNAs) that repress LIN28 expression, a combined in silico prediction and miRNA library screening approach was used in the present study. Four miRNAs directly regulating LIN28 (let-7, mir-125, mir-9, and mir-30) were initially identified by this approach and further validated by quantitative RT-PCR, Western blot analysis, and a LIN28 3′-UTR reporter assay. We found that expression levels of these four miRNAs were clustered together and inversely correlated with LIN28 expression during embryonic stem cell differentiation. In addition, the expression of these miRNAs was remarkably lower in LIN28-positive tumor cells compared with LIN28-negative tumor cells. Importantly, we demonstrated that these miRNAs were able to regulate the expression and activity of let-7, mediated by LIN28. Taken together, our studies demonstrate that miRNAs let-7, mir-125, mir-9, and mir-30 directly repress LIN28 expression in embryonic stem and cancer cells. Global down-regulation of these miRNAs may be one of the mechanisms of LIN28 reactivation in human cancers.
Keywords: Embryonic Stem Cell, MicroRNA, Oncogene, Tumor, Tumor Suppressor, LIN28, Post-transcriptional Regulation
Introduction
LIN28 is an evolutionarily conserved RNA-binding protein with two RNA-binding domains (a cold shock domain and retroviral type CCHC zinc finger motif), which was first characterized as a critical regulator of developmental timing in Caenorhabditis elegans (1, 2). The mammalian homologs of the C. elegans lin-28 gene (LIN28 and LIN28B) are important in processes such as embryogenesis (3), skeletal myogenesis (4), germ cell development (5, 6), and neurogliogenesis (7, 8). Genome-wide association studies have implicated the LIN28B locus in controlling both height and the timing of menarche in humans (9–13). This finding has been successfully phenocopied in a transgenic mouse model that expresses inducible LIN28 (14). Increasing evidence suggests that LIN28 may also be a master regulator controlling the pluripotency of ES cells (15–18). LIN28, together with OCT4, SOX2, and NANOG (the “reprogramming factors”), can reprogram somatic cells to induced pluripotent stem cells (19). Several reports have demonstrated that LIN28 binds to mRNAs, regulating their stability and translation (4, 16, 17). In addition, LIN28 can bind to the terminal loops of the precursor of the miRNA let-7, thereby blocking the processing of let-7 into its mature form (7, 8, 20–26). Importantly, expression of LIN28 is highly restricted to ES2 cells as well as developing tissues, and its expression dramatically decreases as differentiation proceeds (2, 4, 5, 8, 14, 27–29). In human tumors, LIN28/LIN28B is up-regulated/reactivated and functions as an oncogene promoting malignant transformation and tumor progression (30–37). However, the transcriptional and post-transcriptional regulation of mammalian LIN28 is still largely unknown.
miRNAs are endogenous ∼18–25-nucleotide non-coding small RNAs that regulate gene expression in a sequence-specific manner via degradation of target mRNAs or inhibition of protein translation (38–40). With the exception of miRNAs within the Alu repeats, which are transcribed by RNA polymerase III (41), most miRNAs are derived from primary miRNA transcripts transcribed by polymerase II and containing a 5′ cap and a poly(A) tail (42, 43). The primary miRNA transcript is cleaved within the nucleus into a ∼70-nucleotide hairpin precursor known as pre-miRNA by a multiprotein complex called Microprocessor, which is composed of the RNase III enzyme Drosha and the double-stranded RNA-binding domain protein DGCR8/Pasha (44–47). Next, the pre-miRNA is exported into the cytoplasm by Exportin-5 via a Ran-GTP-dependent mechanism (48–50). The pre-miRNA is further cleaved into the mature ∼22-nucleotide miRNA-miRNA* duplex by an RNase III enzyme, Dicer, in association with its partners TRBP/Loquacious and PACT in human cells (51, 52). Subsequently, an RNA-induced silencing complex called RISC is assembled with Argonaute 2 (53, 54). The miRNA strand is selectively incorporated into RISC (55, 56) and guides the complex specifically to its mRNA targets through base-pairing interactions.
EXPERIMENTAL PROCEDURES
Cell Culture
The cancer cell lines A2780, T47D, MCF7, and HeLa were cultured in RPMI1640 (Cellgro) supplemented with 10% fetal bovine serum (FBS; Invitrogen) and 1% penicillin/streptomycin (Invitrogen). Mouse embryonic fibroblasts were cultured in Dulbecco's Modified Eagle's medium (DMEM; Invitrogen) with 10% FBS and 1% penicillin/streptomycin. The mouse embryonic stem cell lines R1 (ATCC) and C57BL/6J-693 (The Jackson Laboratory) were maintained on gelatin-coated flasks with mitomycin C-treated mouse embryonic fibroblasts in DMEM containing 15% ES cell FBS (Invitrogen), 2 mm l-glutamine (Invitrogen), 1% MEM nonessential amino acids (Invitrogen), 1% penicillin/streptomycin, 0.1 mm 2-mercaptoethanol (Invitrogen), and 1000 units/ml mouse leukemia-inhibitory factor (Chemicon).
Plasmid Construction
A genome-wide miRNA expression library was generated in the laboratory of Dr. Qihong Huang (The Wistar Institute). The full-length sequence of the human LIN28 3′-untranslated region (3′-UTR) was cloned from human genomic DNA using the Expand High Fidelity PCR system (Roche Applied Science). The PCR product was ligated into the PCR2.1 TOPO cloning vector (Invitrogen) and then subcloned into the psiCHECK2 reporter vector (Promega). For mutagenesis of microRNA binding sites on reporter vectors, an overlap extension approach by PCR was used as described previously (57). Briefly, primers encompassing the site to be mutated were designed to have overlapping ends. The first round of PCR, yielding two fragments with mutated sequences at either end, was performed using the Expand High Fidelity PCR system with wild type reporter plasmid as template. The fragments were purified and annealed with each other for mutually primed extension. A full-length DNA fragment with the desired mutation was then produced via a second round of PCR. Purified DNA was digested and ligated with the reporter vector backbone.
Western Blot
Cells were lysed in mammalian protein extraction reagent (Pierce). After quantification using a BCA protein assay kit (Pierce), 15 μg of total protein was separated by 10% SDS-PAGE under denaturing conditions and transferred to PVDF membranes (Millipore). Membranes were blocked in 5% nonfat milk (Bio-Rad) and then incubated with an anti-LIN28 primary antibody (1:10,000; Abcam), followed by incubation with an anti-rabbit secondary antibody conjugated with HRP (1:10,000; Amersham Biosciences) together with an HRP-conjugated primary antibody for β-actin (1:10,000; Sigma). Immunoreactive proteins were visualized using the LumiGLO chemiluminescent substrate (Cell Signaling).
Quantitative Real-time RT-PCR
Total RNA was extracted using TRIzol reagent (Invitrogen) and reverse-transcribed using a high capacity RNA-to-cDNA kit (Applied Biosystems) under conditions provided by the supplier. cDNA was quantified by RT-PCR on an ABI Prism 7900 sequence detection system (Applied Biosystems) using the primers listed in Table 1. PCR was performed using SYBR Green PCR core reagents (Applied Biosystems) according to the manufacturer's instructions. PCR amplification of the housekeeping gene GAPDH was performed for each sample as a control for sample loading and to allow normalization across samples.
TABLE 1.
Primer sequences
| Gene | Accession number | Primer sequence |
|---|---|---|
| hLIN28 | NM_024674.4 | Forward: caaaaggaaagagcatgcagaag |
| Reverse: gcatgatgatctagacctccaca | ||
| mLin28 | NM_145833.1 | Forward: gttcggcttcctgtctatgacc |
| Reverse: cttccatgtgcagcttgctct | ||
| mOct-4 | NM_013633.2 | Forward: atggcatactgtggacctca |
| Reverse: agcagcttggcaaactgttc | ||
| mNanog | NM_028016.1 | Forward: ctcatcaatgcctgcagtttttca |
| Reverse: ctcctcagggcccttgtcagc | ||
| mGata6 | NM_010258.3 | Forward: acagcccacttctgtgttccc |
| Reverse: gtgggttggtcacgtggtacag | ||
| mGata4 | NM_008092.3 | Forward: cctggaagacaccccaatctc |
| Reverse: aggtagtgtcccgtcccatct | ||
| mBrachyury (T) | NM_009309.2 | Forward: ctctaatgtcctcccttgttgcc |
| Reverse: tgcagattgtctttggctactttg | ||
| mGSC | NM_010351.1 | Forward: ttcgggaggagaaggtgga |
| Reverse: cggcgaggcttttgagga | ||
| mFgf5 | NM_010203.3 | Forward: aaagtcaatggctcccacgaa |
| Reverse: ggcacttgcatggagttttcc | ||
| mNestin | NM_016701.3 | Forward: tgagggtcaggtggttctg |
| Reverse: agagcagggagggacattc |
TaqMan miRNA Profiling Fluidic Cards
Total RNA was extracted with TRIzol reagent (Invitrogen). 700 ng of total RNA was subjected to reverse transcription using the Megaplex RT primer pool (Applied Biosystems) and the Taqman microRNA reverse transcription kit (Applied Biosystems) according to the manufacturer's instructions. cDNA was loaded to the microfluidic card of Taqman miRNA panel A (Applied Biosystems), and quantitative PCR was performed following the manufacturer's instructions.
TaqMan miRNA Assay
Expression of mature miRNAs were analyzed using the TaqMan miRNA assay (Applied Biosystems) under conditions defined by the supplier. Briefly, single-stranded cDNA was synthesized from 5 ng of total RNA in a 15-μl reaction volume, using the TaqMan microRNA reverse transcription kit (Applied Biosystems). The reactions were incubated first at 16 °C for 30 min and then at 42 °C for 30 min and then inactivated by incubation at 85 °C for 5 min. Each cDNA generated was amplified by quantitative PCR using sequence-specific primers from the TaqMan microRNA assays on an Applied Biosystems 7900HT sequence detection system (Applied Biosystems). Each 20-μl PCR included 10 μl of 2× Universal PCR Master Mix (without AmpErase UNG), 1 μl of 20× TaqMan microRNA assay mix, and 2 μl of reverse transcription product. The reactions were incubated in a 384-well plate at 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min.
Cell Transfection
For transient transfections, cells were plated 24 h before transfection at 50% confluence. Plasmid and miRNA mimic transfections were performed with the FuGENE6 transfection reagent (Roche Applied Science) or Lipofectamine RNAiMAX (Invitrogen), respectively.
Lentiviral Transduction and Stable Cell Line Generation
Lentiviral vector and packaging vectors were transfected into the packaging cell line 293T (ATCC) using the FuGENE6 Transfection Reagent (Roche Applied Science). The medium was changed 8 h post-transfection, and the medium containing lentivirus was collected 48 h later. Tumor cells were infected with lentivirus in the presence of 8 μg/ml Polybrene (Sigma).
Reporter Assay
Cells were plated on a 24-well plate and transfected with 0.125 μg of reporter vector together with 0.25 μg of miRNA expression plasmid using the FuGENE6 transfection reagent. Alternatively, a sequential transfection method was used as follows. 30 nm miRNA mimics were transfected using Lipofectamine RNAiMAX; 24 h post-transfection, 0.125 μg of reporter vector was transfected using FuGENE6 transfection reagent. In both cases, 48 h after reporter vector transfection, cells were harvested, and reporter assays were performed using a dual luciferase reporter assay system (Promega) according to the manufacturer's instructions. Reporter activity was measured on the Fluoroskan Ascent FL fluorometer (Thermo Fisher Scientific).
Immunohistochemistry and Confocal Image
Sections were sequentially incubated in 5% normal serum, rabbit anti-LIN28 antibody diluted at 1:500 overnight at 4 °C, and Alexa Fluor 594 goat anti-rabbit IgG (Invitrogen) at 1:150 for 1.5 h at room temperature. Sections were counterstained with DAPI (Vector). The image was collected using an Axiovert 200 M inverted microscope (Zeiss).
Embryoid Body (EB) Formation Assay
Undifferentiated ES cells were trypsinized and resuspended in ES cell culture medium without mouse leukemia-inhibitory factor. ES cell suspensions were applied to gelatin-coated dishes and incubated at 37 °C for 40 min to remove mouse embryonic fibroblast cells. ES cells were collected and resuspended in ES cell culture medium without mouse leukemia-inhibitory factor. For suspension cultures, 1 × 106 cells were placed in 100-mm Petri dishes, and the medium was changed every 2 days.
let-7-responsive Sensor Construction and Transfection
An miRNA-responsive sensor, a technique for monitoring miRNA activity, has been used to detect let-7 expression in mouse embryos in vivo and in mammary epithelial progenitor cells in vitro. The sensor contains a constitutively expressed reporter bearing sequences complementary to let-7 in the downstream region of the 3′-UTR of a reporter gene. In cultured cells transfected with the sensor, expression of the reporter is decreased when let-7 is present. Thus, the let-7-responsive sensor is able to monitor let-7 activity in real time. The let-7 sensor was constructed by introducing two copies of let-7b perfect complement sequences into the 3′-UTR region of the Renilla luciferase gene in the psiCheck 2 vector (Promega). The let-7-complementary oligonucleotides (sense, TCG AGA ACC ACA CAA CCT ACT ACC TCA GGA TCC AAC CAC ACA ACC TAC TAC CTC AGC; antisense, GGC CGC TGA GGT AGT AGG TTG TGT GGT TGG ATC CTG AGG TAG TAG GTT GTG TGG TTC) were purchased from IDT and annealed in buffer containing 100 mm potassium acetate, 30 mm HEPES-KOH, pH 7.4, and 2 mm magnesium acetate. A2780 cells were seeded in 6-well plates and grown overnight to 40% confluence prior to transfection. To test let-7 activity, a total of 1 μg of sense vector was introduced to the cells using FuGENE6 transfection reagent (Roche Applied Science). All transfection experiments were done in triplicate and repeated at least twice. Luciferase assays were performed 48 h post-transfection with a Fluoroskan Ascent FL fluorometer (Thermo Fisher Scientific) using the dual luciferase reporter assay system (Promega) according to the manufacturer's instructions.
Statistics
Statistical analysis was performed using the SPSS statistics software package (SPSS, Chicago, IL). All results are expressed as mean ± S.D. and with significance at p < 0.05.
RESULTS
A Combined in Silico Prediction and miRNA Library Screening Approach Identified miRNAs Targeting LIN28
The expression of LIN28 is highly restricted to ES cells, somatic progenitor cells, and developing tissues but is not detectable in most adult organs (2, 4, 5, 8, 14, 27–29). However, LIN28 is dramatically up-regulated/reactivated in human cancers (30–35). There is a double negative regulation loop between LIN28 and the miRNA let-7 (7, 8, 20–26). To identify miRNAs regulating LIN28, our initial screening was performed on a platform containing four well characterized cancer cell lines, two of which (A2780 and T47D) highly expressed endogenous LIN28 and two of which (MCF7 and HeLa) were LIN28-negative (Fig. 1A). Consistent with previous reports (7, 8, 20–26), the levels of let-7 in the LIN28-positive cell lines were remarkably lower than the levels in LIN28-negative lines (Fig. 1B).
FIGURE 1.

A combined in silico prediction and miRNA library screening approach identified miRNAs targeting LIN28. A, Western blots were used to detect endogenous LIN28 expression in the four cancer cell lines selected for experimental screening. Two of these (A2780 and T47D) were LIN28-positive, and two (MCF7 and HeLa) were LIN28-negative. B, real-time RT-PCR was used to detect let-7b expression in these cell lines. As expected, the LIN28-negative lines expressed relatively higher levels of let-7b. C, illustration of the miRNA expression vector and reporter vector used in the screening assay. Pro., promoter; hRluc, Renilla luciferase; hluc, firefly luciferase. D, the known LIN28-regulatory miRNA let-7 was used for the pilot screening. Co-transfection with the let-7b expression vector significantly (*, p < 0.05) reduced the luciferase activity of the LIN28 3′-UTR reporter vector in the two LIN28-negative cell lines. E, schematic structure of LIN28 mRNA. The miRNA binding sites were predicted by TargetScan. UTR, untranslated region; ORF, open reading frame; E, exon. F, the summary heat map of the miRNA library screening in four cell lines. Here, miRNAs are listed from left to right according to their prediction scores (high to low). Nine miRNAs (marked in green) significantly reduced the reporter activity in all four cancer cell lines. G, stable cell lines overexpressing each of these nine miRNAs as well as five miRNA controls that did not reduce luciferase activity were generated by lentiviral infection. Three of the nine candidate miRNAs (mir-30, mir-125, and mir-9) markedly reduced both LIN28 mRNA and protein expression.
Genome-wide miRNA library screening has been used successfully to identify miRNAs that regulate certain protein-coding genes (58, 59). Because this method is time- and labor-intensive, we designed a bioinformatics-driven screening approach, in which a concentrated miRNA library was generated by miRNA binding site prediction (i.e. we used bioinformatics for miRNA target prediction as a filter to generate a restricted/selected miRNA library on which to perform the functional screening). This allowed us to perform the experimental screening in a co-transfection system in which a luciferase reporter was co-transfected with a pre-miRNA-expressing vector (Fig. 1C). The reporter vector contained the full-length human LIN28 3′-UTR sequence, which was cloned downstream of the reporter gene Renilla (hRluc), such that the reporter gene expression was regulated by the LIN28 3′-UTR. The firefly luciferase (hluc) reporter served as an internal control for transfection efficiency (Fig. 1C). To test our screening system, we first chose the miRNA let-7, one of the known miRNAs regulating LIN28. As expected, let-7 significantly reduced luciferase activity in LIN28-negative cell lines but not in LIN28-positive cell lines, in which the pre-miRNA maturation was blocked by endogenous LIN28 (Fig. 1D).
Next, we predicted miRNA binding sites in the 3′-UTR of the human LIN28 gene using the bioinformatics miRNA target prediction program TargetScan (available on the World Wide Web) (60). A total of 23 potential miRNA binding sites that were broadly conserved among vertebrates were identified (Fig. 1E). The individual miRNA expression vectors for the above miRNAs were selected from our genome-wide miRNA library. A pilot single transfection for each vector was performed in cancer cell lines, and enforced miRNA expression was confirmed by RT-PCR (data not shown). The miRNA vectors that were not efficiently expressed were excluded from the co-transfection screening. Finally, a total of 17 miRNAs were successfully used for the functional screen in all four cell lines. Briefly, nine of these miRNAs significantly reduced luciferase activity in all four cell lines (marked as green in Fig. 1F). To further test whether these 9 candidate miRNAs led to reduced expression of endogenous LIN28, we generated stable cell lines that overexpressed each of these nine miRNAs individually as well as stable cell lines expressing five miRNAs (as controls) that did not reduce luciferase activity. We found that three of the nine candidate miRNAs (mir-30, mir-125, and mir-9) markedly reduced both LIN28 mRNA and protein expression (Fig. 1G). The direct regulation of two miRNAs in the LIN28 3′-UTR (mir-9 and mir-30, which were first identified in our study) were confirmed by a mutant LIN28 3′-UTR reporter assay (Fig. 2). The seeding sequences of mir-9 or mir-30 in the LIN28 3′-UTR were mutated, and overexpression of mir-9 or mir-30 was shown to significantly reduce luciferase activity in the wild type but not the binding site mutant LIN28 3′-UTR reporters. Although mir-18 decreased LIN28 expression at a level comparable with that of mir-30 (Fig. 1G), overexpression of mir-18 was not able to reduce luciferase activity in the wild type and the binding site mutant LIN28 3′-UTR reporters in both A2780 and HeLa cells (data not shown). Taken together, four miRNAs, let-7, mir-30, mir-125, and mir-9, were identified in our initial screening. Importantly, two miRNAs known to regulate LIN28 in mammalian (let-7 and mir-125) (7, 61) were successfully identified using our approach.
FIGURE 2.

LIN28-regulatory function of mir-9 and mir-30 was validated by the 3′-UTR reporter assay. A, schematic diagram of the mir-125, let-7, mir-30, and mir-9 binding sites in the LIN28 3′-UTR. The seeding sequences (marked in gray) were broadly conserved among different species. Hsa, Human; Ptr, chimpanzee; Mml, Rhesus; Mmu, mouse; Rno, Rat; Cpo, Pig; Ocu, rabbit; Eeu, Hedgehog; Cfa, Dog; Eca, Horse; Bta, Cow; Dno, armadillo; Laf, elephant; Mdo, opossum. B and C, summary of the reporter assays on the wild type and mir-9 binding site mutant reporter (B) and mir-30 binding site mutant reporter (C) in A2780 (LIN28-positive) and HeLa (LIN28-negative) cells. WT, wild type LIN28 3′-UTR reporter; mut9, mir-9 binding site mutant LIN28 3′-UTR reporter; mut30, mir-30 binding site mutant LIN28 3′-UTR reporter. Overexpression of mir-9 or mir-30 was able to significantly (*, p < 0.05) reduce luciferase activity in the wild type but not the binding site mutant LIN28 3′-UTR reporters.
miRNAs Regulate Lin28 in Undifferentiated ES Cells
Next, we examined whether the above miRNAs identified in cancer cell lines were able to regulate Lin28 expression under physiological conditions, such as in undifferentiated ES cells. To address this question, we transiently transfected the mouse ES cell line R1 with the mimics of let-7, mir-125, mir-9, and mir-30 and a control mimic. At 24 and 48 h post-transfection, total RNA and protein were collected, and the endogenous Lin28 expression was examined by RT-PCR and Western blots. We found that the expression of both Lin28 protein and mRNA was significantly decreased in the cells transfected with miRNA mimics compared with the control transfections (Fig. 3, A and B). For the two miRNAs (mir-9 and mir-30) that were first identified as LIN28 regulators in this study, we also confirmed the protein expression and localization by immunohistochemical staining. Consistent with the Western blot results, both mir-9 and mir-30 led to a reduction of endogenous Lin28 in ES cells, but they had no effect on the cellular distribution of Lin28 in these cells (Fig. 3C).
FIGURE 3.

miRNAs regulate Lin28 in undifferentiated ES cells. The miRNA mimics (30 nm) of let-7, mir-125, mir-9, mir-30, and a control mimic were transiently transfected into the mouse ES cell line R1. At 24 and 48 h post-transfection, total RNA and protein were collected, and the endogenous Lin28 expression was examined by real-time RT-PCR (A) and Western blots (B). A, the Lin28 mRNA expression was significantly (*, p < 0.05) decreased in the cells transfected with miRNA mimics compared with the control transfections. B, Lin28 protein expression was markedly decreased in the cells transfected with miRNA mimics compared with the control transfections. C, immunohistochemical staining further confirmed that mir-9 and mir-30 decreased LIN28 expression in ES cells.
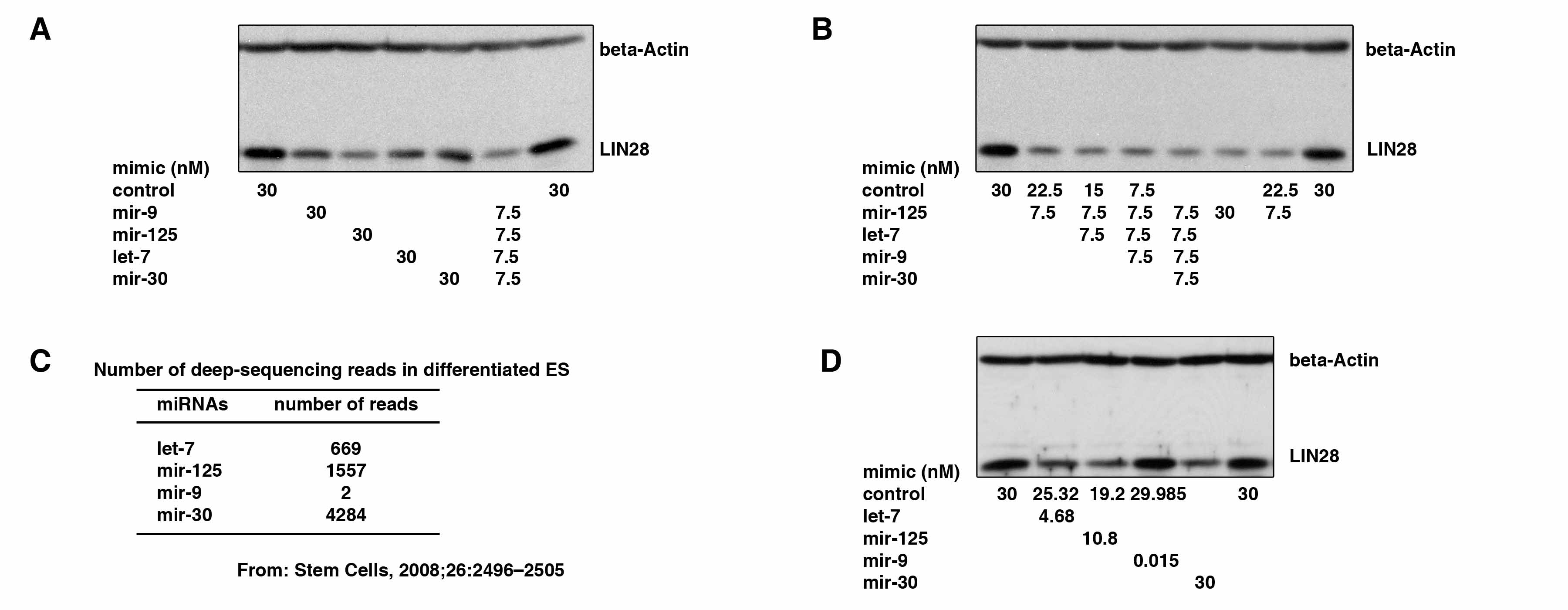
Finally, we asked whether the effects of the four miRNA families on LIN28 expression are cumulative or if the effect of one miRNA is dominant over the others during ES cell differentiation. We transfected undifferentiated ES cells with each individual miRNA as well as a combination of all four miRNAs. We found that mir-125 more efficiently suppressed LIN28 expression compared with the other three miRNAs (supplemental Fig. S1A) and that the transfection of all four miRNAs combined had a similar efficiency on the suppression of LIN28 expression as mir-125 alone (supplemental Fig. S1A). Serial combination transfections further demonstrated that mir-125 was indeed the major functional miRNA in the combination transfection and that the effect of mir-125 is dominant on LIN28 expression when these miRNA are present in equal concentrations in ES cells (supplemental Fig. S1B). However, the mature miRNA expression levels during ES cell differentiation will be another important factor affecting LIN28 expression. To address this question, we retrieved previously published miRNA profiling data that was obtained by deep sequencing of small RNA libraries during ES cell differentiation (62). In differentiated ES cells, mir-125a/b yielded 1,557 sequence reads, and let-7a/b/c/d/e/f/g/i, mir-9, and mir-30a/b/c/d/e yielded 669, 2, and 4,284 sequence reads, respectively (supplemental Fig. S1C). We transfected the four miRNAs in the above ratios into undifferentiated ES cells in which all four miRNAs were absent. Consistently, we found that mir-125 was most efficient at suppressing endogenous LIN28 expression (supplemental Fig. S1D). Based on the above results, we conclude that the mir-125 family is the most dominant miRNA regulating LIN28 expression during ES cell differentiation, and that the let-7 and mir-30 families may also play important roles in the suppression of LIN28 expression in the early stages of differentiation. Because the transfection of miRNA mimics only transiently affects miRNA expression, it is technically difficult to manipulate the levels of miRNAs throughout the entire process of ES cell differentiation. Therefore, to address the same question at later stages of ES cell differentiation, we retrieved three published miRNA profiling data sets from ES cell differentiation studies (63–65). We found that during ES cell differentiation, let-7 is ubiquitously up-regulated/expressed in most somatic progenitor cells, whereas the other three miRNAs are probably tissue type-specific. For example, during the differentiation of ES cells to neurons, let-7 is remarkably up-regulated, and mir-9 is also significantly increased (63–65). This suggests that during the late differentiation stages in ES cells, these four miRNAs may serve distinct functions in different tissue lineages and that one or two of them may be more dominant at suppressing LIN28 expression.
Expression Levels of LIN28 and Its Regulatory miRNAs Are Inversely Correlated during ES Cell Differentiation
Expression of LIN28 is highly restricted to ES cells as well as developing tissues, and its expression dramatically decreases as differentiation proceeds (2, 4, 5, 8, 14, 27–29). Therefore, we examined how the expression levels of LIN28 and its regulatory miRNAs dynamically changed during ES cell differentiation. ES cells are commonly differentiated in vitro by spontaneously self-assembling in suspension culture into three-dimensional cell aggregates called EBs, which model many of the hallmarks of early embryonic development. We chose two mouse ES cell lines (R1 and C57) and induced them to differentiate and form EBs (Fig. 4A). The germ layer maker expression patterns during EB formation (days 0–18) were monitored by real-time RT-PCR (Fig. 4B), and the expression of Lin28 was examined by real-time RT-PCR and Western blots. Lin28 mRNA levels were markedly decreased at day 6 of ES differentiation and EB formation (Fig. 4C), and LIN28 protein expression was reduced at days 6–8 of differentiation (Fig. 4, D and E). To examine the global miRNA expression changes during EB formation, a low density TaqMan assay containing 381 miRNA probes was used (Fig. 4F). Interestingly, unsupervised cluster analysis indicated that all of the above four miRNA families (let-7, mir-125, mir-9, and mir-30) identified by our screening were clustered in the same group and shared similar expression patterns (Fig. 4F, blue cluster). Importantly, their expression levels were nearly non-detectable in undifferentiated ES cells and remarkably increased from day 6 at the same time that Lin28 expression was decreased. Their expression levels then remained high in the differentiated EB cells (Fig. 4, F and G). This high throughput result was further validated by real-time RT-PCR assays in the two ES cell lines (Fig. 4H). In summary, during ES cell differentiation and EB formation, the expression level of Lin28 decreases, whereas its regulatory miRNAs (let-7, mir-125, mir-9, and mir-30) display an inversely correlated expression pattern. This result strongly suggests that these four miRNAs play a functional role in Lin28 regulation under physiological conditions such as early development.
FIGURE 4.

Expression levels of Lin28 and its regulatory miRNAs are inversely correlated during ES cell differentiation. A, ES cells were differentiated in vitro by spontaneously self-assembling in suspension culture into three-dimensional cell aggregates (EBs). Immunostaining demonstrated that Lin28 was highly expressed in the undifferentiated ES cells. B, pluripotency and differentiation markers during differentiation and EB formation were monitored by real-time RT-PCR. C, Lin28 mRNA expression during EB formation was analyzed by real-time RT-PCR. D and E, Lin28 protein expression during EB formation was analyzed using Western blots. F, the global miRNA expression profile during EB formation was analyzed using a TaqMan miRNA assay. An unsupervised cluster analysis indicated that all four Lin28-regulatory miRNAs grouped together. G, detailed miRNA expression changes during EB formation identified by the TaqMan miRNA assay. All four Lin28-regulatory miRNAs were markedly up-regulated from day 6 of EB formation. H, real-time RT-PCR validations of the TaqMan miRNA assay in two ES cell lines.
LIN28-regulatory miRNAs Are Globally Down-regulated in LIN28-positive Cancer Cell Lines
LIN28 has also been identified as a putative oncogene up-regulated/reactivated in 5–15% of human tumors (30–37). We examined whether the LIN28-regulatory miRNAs were also deregulated in these tumors. First, we reanalyzed a publicly available miRNA data set containing 218 specimens (normal control tissues, n = 46; tumors, n = 172) (66), and we successfully retrieved 16 miRNAs from the four LIN28-regulatory miRNA families. Interestingly, we found that 11 of these 16 miRNAs were markedly down-regulated (decreased more than 20%) in human tumors (Fig. 5, A and B). This indicates that the LIN28-regulatory miRNAs are deregulated in cancer. To provide further experimental evidence of this, we chose two cancer cell lines (A2780 and T47D) that highly express LIN28 as well as 17 cancer cell lines in which LIN28 protein was not detectable (Fig. 5C). The expression of LIN28-regulatory miRNAs was analyzed in these cell lines by real-time RT-PCR. As shown in Fig. 5C, all four miRNA families (let-7, mir-9, mir-30, and mir-125) displayed lower levels of expression in the LIN28-positive lines compared with the LIN28-negative lines. The negative correlation of LIN28 expression with its suppressing miRNAs suggests that the global deregulation of these miRNAs may be an important mechanism of oncogenic LIN28 up-regulation/reactivation in human cancer.
FIGURE 5.

LIN28-regulatory miRNAs are globally down-regulated in LIN28-positive cancer cell lines. A, a publicly available miRNA microarray data set was retrieved from the Broad Institute (66). Normalized expression levels of miRNAs regulating LIN28 were analyzed and are shown as a heat map. We found that 11 of 16 LIN28-regulatory miRNAs (marked in green) were markedly down-regulated (more than 20% reduction) in human tumors compared with normal control tissues. B, summary of the average expression levels of the miRNAs regulating LIN28 in normal and tumor specimens in the public miRNA microarray data set. C, LIN28 protein levels in 19 human cancer cell lines detected by Western blot. Two of these cell lines (10.5%) were LIN28-positive. The LIN28-regulatory miRNA expression levels were analyzed by real-time RT-PCR in these 19 cell lines. Shown is a summary of the average expression of each individual miRNA in the LIN28-positive lines (green) and LIN28-negative lines (white).
Next, we tested whether miRNA inhibitors were able to rescue/up-regulate endogenous LIN28 expression in tumor cells. We transfected each individual miRNA inhibitor and a combination of these inhibitors as well as control oligonucleotides to both LIN28-positive (T47D) and LIN28-negative (HeLa) cell lines. We found that in the LIN28-positive cell line T47D, all four inhibitors, both individually and in a combination, were able to increase LIN28 protein expression. However, in the LIN28-negative cell line HeLa, none of these activated LIN28 expression to detectable levels (supplemental Fig. S2). Blocking all four miRNAs did not activate endogenous LIN28 expression in LIN28-negative tumor cells. This suggests that the silencing of LIN28 in adult tissues may be controlled by multiple mechanisms and not only by miRNA suppression. In LIN28-positive tumors, there may be at least two LIN28-suppressing pathways that are not functioning (e.g. miRNA suppression and epigenetic silencing).
miRNAs Regulate let-7 Expression Mediated by LIN28
Our finding suggested a novel regulatory mechanism in which one miRNA may indirectly regulate another miRNA via a protein coding gene; for example, mir-125, mir-9, and mir-30 could regulate let-7 expression via repression of LIN28 expression. To test this hypothesis, we transfected mir-9 and mir-125 mimics into a LIN28-positive cell line (T47D). As expected, both mir-9 and mir-125 significantly reduced endogenous LIN28 expression as detected by real-time RT-PCR (Fig. 6A). We then examined endogenous let-7b expression by real-time RT-PCR. As shown in Fig. 6B, both mir-9 and mir-125 significantly increased let-7b expression in T47D cells. Importantly, when we transfected mir-9 or mir-125 into a LIN28-negative cell line (MCF7), the let-7b expression levels were not affected (Fig. 6B). Finally, in order to monitor let-7 activity, a let-7 sensor (Fig. 6C) was co-transfected with the mir-9 and mir-125 mimics. As shown in Fig. 6D, mir-9 and mir-125 significantly reduced the luciferase activity of the let-7 sensor in T47D cells, indicating that the endogenous let-7 activity was increased by the mir-9 and mir-125 mimics. In the LIN28-negative line MCF7, the mimic transfection consistently did not change the activity of the let-7 sensor. Taken together, our results demonstrate a novel regulatory mechanism where LIN28-regulatory miRNAs regulate let-7 expression and activity via the protein-coding gene LIN28.
FIGURE 6.

miRNAs regulate let-7 expression mediated by LIN28. A, the transfection of mir-9 and mir-125 mimics significantly (*, p < 0.05) reduced LIN28 mRNA expression in T47D cells. B, transfection of mir-9 and mir-125 mimics significantly (*, p < 0.05) increased let-7b expression in the LIN28-positive cell line T47D but not in the LIN28-negative cell line MCF7. C, a miRNA-responsive sensor, a technique for monitoring miRNA activity, bearing sequences complementary to let-7b in the downstream region of the 3′-UTR of a constitutively expressed reporter gene. D, transfection of mir-9 and mir-125 mimics significantly (*, p < 0.05) decreased the let-7b-responsive sensor in the LIN28-positive cell line T47D but not in the LIN28-negative cell line MCF7.
DISCUSSION
It is estimated that the human genome contains ∼1,000 miRNAs (67), more than 900 of which have already been identified, according to the latest version of miRBase. miRNAs are predicted to target up to one-third of human mRNAs (60). Each miRNA can target hundreds of transcripts (68–71) and proteins (70, 71) directly or indirectly, and more than one miRNA can converge on a single transcript target (72). Therefore, the potential regulatory circuitry afforded by miRNA is enormous, but identification of miRNAs regulating protein-coding genes still remains challenging. Both bioinformatic prediction methods and whole genome-wide genetic screening have been used to characterize miRNA targets (58, 59). Here, using a combinatorial approach, we successfully identified four miRNA families, including two newly identified families (mir-9 and mir-30), which regulate LIN28 expression in ES cells and cancer cells. However, similar to the results obtained when miRNA library screening has been performed on other protein coding genes (58, 59), many predicted LIN28-regulatory miRNAs showed no significant effect on endogenous LIN28 expression. For example, using a whole genome-wide miRNA library, Sage et al. (58) reported that only the mir-221/mir-222 family regulated p27Kip1, and Nagel et al. (59) also found that only the mir-135 family suppressed APC expression. This suggests that protein-coding genes may be regulated by a smaller number of miRNAs than has previously been thought. However, in this study, we only included miRNAs that were predicted to bind to the LIN28 3′-UTR in our screening. Therefore, it is possible that other miRNAs may target other regions of LIN28. All four miRNA families identified in our study contain multiple members (e.g. the human let-7 family contains 12 members; the mir-125 family and the mir-9 family each contain three members; and the mir-30 family contains six members) (Table 2). These families are located in distinct chromosomal locations, suggesting that their expression is regulated by different 5′-UTR regulatory sequences and transcription factors. In addition, several LIN28-regulatory miRNAs are located in the same genomic loci and clustered together (e.g. let-7e and mir-125a are located in the same genomic locus, as are mir-30b and mir-30d) (Table 2). This suggests that they may be processed from the same primary miRNA transcript, and could coordinately regulate LIN28 expression. Therefore, the potential regulatory circuitry afforded by these four miRNA families (at least 24 members) on LIN28 is extensive and complex.
TABLE 2.
Genomic location and organization of the LIN28 regulatory miRNAs in humans
| miRNA name | Cluster | Genomic location |
|---|---|---|
| mir-30e | mir-30e and mir-30c-1 | 1: 41220027–41220118 (+) |
| mir-30c-1 | mir-30c-1 and mir-30e | 1: 41222956–41223044 (+) |
| mir-30c-2 | 6: 72086663–72086734 (−) | |
| mir-30d | mir-30b and mir-30d | 8: 135817119–135817188 (−) |
| mir-30a | 6: 72113254–72113324 (−) | |
| mir-30b | mir-30b and mir-30d | 8: 135812763–135812850 (−) |
| let-7a-1 | let-7a-1, let-7f-1, and let-7d | 9: 96938239–96938318 (+) |
| let-7a-2 | mir-100 and let-7a-2 | 11: 122017230–122017301 (−) |
| let-7a-3 | let-7a-3 and let-7b | 22: 46508629–46508702 (+) |
| let-7b | let-7a-3 and let-7b | 22: 46509566–46509648 (+) |
| let-7c | mir-99a and let-7c | 21: 17912148–17912231 (+) |
| let-7d | let-7a-1, let-7f-1, and let-7d | 9: 96941116–96941202 (+) |
| let-7e | mir-99b, let-7e and mir-125a | 19: 52196039–52196117 (+) |
| let-7f-1 | let-7a-1, let-7f-1, and let-7d | 9: 96938629–96938715 (+) |
| let-7f-2 | let-7f-2 and mir-98 | X: 53584153–53584235 (−) |
| let-7g | 3: 52302294–52302377 (−) | |
| let-7i | 12: 62997466–62997549 (+) | |
| mir-98 | let-7f-2 and mir-98 | X: 53583184–53583302 (−) |
| mir-125a | mir-99b, let-7e, and mir-125a | 19: 52196507–52196592 (+) |
| mir-125b-1 | 11: 121970465–121970552 (−) | |
| mir-125b-2 | 21: 17962557–17962645 (+) | |
| mir-9-1 | 1: 156390133–156390221 (−) | |
| mir-9-2 | 5: 87962671–87962757 (−) | |
| mir-9-3 | 15: 89911248–89911337 (+) |
Expression of LIN28 is highly restricted to ES cells as well as developing tissues and dramatically decreases as differentiation proceeds (2, 4, 5, 8, 14, 27–29). We found that the expression of all four miRNA families that we identified in this study was nearly undetectable in undifferentiated ES cells as well as in the first 6 days of EB formation and differentiation. During this time window, LIN28 is expressed at a remarkably high level. From day 6 of EB formation, all four LIN28-regulatory miRNA families display a significantly increased expression, whereas both LIN28 mRNA and protein levels begin to dramatically decrease (Figs. 4 and 7). This inversely related expression pattern suggests that these miRNAs may play an important functional role in the suppression of LIN28 expression during development and differentiation. In most adult tissues, LIN28 is almost completely silenced; however, these four miRNAs are expressed at high levels (Figs. 5A and 7). Interestingly, some of these miRNAs (e.g. mir-9) appear to be tissue-specific, whereas some of them (e.g. mir-30) are expressed in nearly all adult tissues. This indicates that there is a complex temporal and spatial regulatory network of miRNAs affecting LIN28 expression under physiological conditions. Finally, given that LIN28 can act as a reprogramming factor together with OCT4, SOX2, and NANOG to reprogram somatic cells to induced pluripotent stem cells (19), the question arises as to whether co-transfection of LIN28-regulatory miRNA inhibitors with these reprogramming factors could increase the efficiency of reprogramming (Fig. 7). Supporting this hypothesis, Melton et al. (73) have recently reported that inhibition of let-7 promotes dedifferentiation of somatic cells to induced pluripotent stem cells.
FIGURE 7.

The regulatory circuitry afforded by LIN28-regulatory miRNAs in development and tumorigenesis.
In human cancer, LIN28 is up-regulated/reactivated in about 5–15% of patients and functions as an oncogene (30–37). Interestingly, we found that these four LIN28-regulatory miRNAs are globally down-regulated in tumors (Figs. 5 and 7), possibly due to genetic and/or genomic alterations in these miRNAs or deregulation of their biogenesis pathways (66). Notably, in the LIN28-positive tumor cell lines, these miRNAs were expressed at relatively lower levels compared with the LIN28-negative tumor cell lines. Therefore, down-regulation of LIN28-regulatory miRNAs may be an important mechanism of LIN28 reactivation in human cancer (Fig. 5C). Recent studies have demonstrated that LIN28 serves as an oncogene promoting malignant transformation and tumor growth (30–37). Importantly, LIN28 may contribute to the maintenance of cancer stem cells, a relatively rare subpopulation of tumor cells having the unique ability to initiate and perpetuate tumor growth (34). Rapidly accumulating evidence indicates that miRNAs are involved in the initiation and progression of cancer (66, 74–80). Interestingly, two of the LIN28-regulatory miRNAs (let-7 and mir-125) have been demonstrated as tumor suppressor genes. For example, the tumor suppressor role of let-7 in cancer was first demonstrated by the Slack laboratory (76). It was found that the let-7 family negatively regulates let-60/RAS in C. elegans by binding to multiple let-7-complementary sites in its 3′-UTR (76). Moreover, the finding that let-7 expression is lower in lung tumors than in normal lung tissue, whereas RAS protein is significantly higher in lung tumors, proposes let-7 as a tumor suppressor gene (76, 81–84). Therefore, our study could lead to new therapeutic strategies for cancer treatment (Fig. 7). For example, nanoparticle-delivered LIN28-regulatory miRNA mimics may be an attractive therapeutic method to target the LIN28-positive cancer stem cell population in tumors.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health, NCI, Grant R01CA142776 and Ovarian Cancer SPORE P50-CA83638-7951 Project 3. This work was also supported in part by grants from the Breast Cancer Alliance, the Ovarian Cancer Research Fund (Liz Tilberis Scholar), and the Mary Kay Ash Charitable Foundation and by United States Department of Defense Grant W81XWH-10-1-0082.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2.
- ES
- embryonic stem
- miRNA
- microRNA
- UTR
- untranslated region
- EB
- embryoid body.
REFERENCES
- 1.Ambros V., Horvitz H. R. (1984) Science 226, 409–416 [DOI] [PubMed] [Google Scholar]
- 2.Moss E. G., Lee R. C., Ambros V. (1997) Cell 88, 637–646 [DOI] [PubMed] [Google Scholar]
- 3.Yokoyama S., Hashimoto M., Shimizu H., Ueno-Kudoh H., Uchibe K., Kimura I., Asahara H. (2008) Gene Expr. Patterns 8, 155–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Polesskaya A., Cuvellier S., Naguibneva I., Duquet A., Moss E. G., Harel-Bellan A. (2007) Genes Dev. 21, 1125–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.West J. A., Viswanathan S. R., Yabuuchi A., Cunniff K., Takeuchi A., Park I. H., Sero J. E., Zhu H., Perez-Atayde A., Frazier A. L., Surani M. A., Daley G. Q. (2009) Nature 460, 909–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zheng K., Wu X., Kaestner K. H., Wang P. J. (2009) BMC Dev. Biol. 9, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rybak A., Fuchs H., Smirnova L., Brandt C., Pohl E. E., Nitsch R., Wulczyn F. G. (2008) Nat. Cell. Biol. 10, 987–993 [DOI] [PubMed] [Google Scholar]
- 8.Balzer E., Heine C., Jiang Q., Lee V. M., Moss E. G. (2010) Development 137, 891–900 [DOI] [PubMed] [Google Scholar]
- 9.Lettre G., Jackson A. U., Gieger C., Schumacher F. R., Berndt S. I., Sanna S., Eyheramendy S., Voight B. F., Butler J. L., Guiducci C., Illig T., Hackett R., Heid I. M., Jacobs K. B., Lyssenko V., Uda M., Boehnke M., Chanock S. J., Groop L. C., Hu F. B., Isomaa B., Kraft P., Peltonen L., Salomaa V., Schlessinger D., Hunter D. J., Hayes R. B., Abecasis G. R., Wichmann H. E., Mohlke K. L., Hirschhorn J. N. (2008) Nat. Genet. 40, 584–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ong K. K., Elks C. E., Li S., Zhao J. H., Luan J., Andersen L. B., Bingham S. A., Brage S., Smith G. D., Ekelund U., Gillson C. J., Glaser B., Golding J., Hardy R., Khaw K. T., Kuh D., Luben R., Marcus M., McGeehin M. A., Ness A. R., Northstone K., Ring S. M., Rubin C., Sims M. A., Song K., Strachan D. P., Vollenweider P., Waeber G., Waterworth D. M., Wong A., Deloukas P., Barroso I., Mooser V., Loos R. J., Wareham N. J. (2009) Nat. Genet. 41, 729–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sulem P., Gudbjartsson D. F., Rafnar T., Holm H., Olafsdottir E. J., Olafsdottir G. H., Jonsson T., Alexandersen P., Feenstra B., Boyd H. A., Aben K. K., Verbeek A. L., Roeleveld N., Jonasdottir A., Styrkarsdottir U., Steinthorsdottir V., Karason A., Stacey S. N., Gudmundsson J., Jakobsdottir M., Thorleifsson G., Hardarson G., Gulcher J., Kong A., Kiemeney L. A., Melbye M., Christiansen C., Tryggvadottir L., Thorsteinsdottir U., Stefansson K. (2009) Nat. Genet. 41, 734–738 [DOI] [PubMed] [Google Scholar]
- 12.He C., Kraft P., Chen C., Buring J. E., Paré G., Hankinson S. E., Chanock S. J., Ridker P. M., Hunter D. J., Chasman D. I. (2009) Nat. Genet. 41, 724–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perry J. R., Stolk L., Franceschini N., Lunetta K. L., Zhai G., McArdle P. F., Smith A. V., Aspelund T., Bandinelli S., Boerwinkle E., Cherkas L., Eiriksdottir G., Estrada K., Ferrucci L., Folsom A. R., Garcia M., Gudnason V., Hofman A., Karasik D., Kiel D. P., Launer L. J., van Meurs J., Nalls M. A., Rivadeneira F., Shuldiner A. R., Singleton A., Soranzo N., Tanaka T., Visser J. A., Weedon M. N., Wilson S. G., Zhuang V., Streeten E. A., Harris T. B., Murray A., Spector T. D., Demerath E. W., Uitterlinden A. G., Murabito J. M. (2009) Nat. Genet. 41, 648–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu H., Shah S., Shyh-Chang N., Shinoda G., Einhorn W. S., Viswanathan S. R., Takeuchi A., Grasemann C., Rinn J. L., Lopez M. F., Hirschhorn J. N., Palmert M. R., Daley G. Q. (2010) Nat. Genet. 42, 626–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Darr H., Benvenisty N. (2009) Stem Cells 27, 352–362 [DOI] [PubMed] [Google Scholar]
- 16.Xu B., Zhang K., Huang Y. (2009) RNA 15, 357–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qiu C., Ma Y., Wang J., Peng S., Huang Y. (2010) Nucleic Acids Res. 38, 1240–1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu B., Huang Y. (2009) Nucleic Acids Res. 37, 4256–4263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu J., Vodyanik M. A., Smuga-Otto K., Antosiewicz-Bourget J., Frane J. L., Tian S., Nie J., Jonsdottir G. A., Ruotti V., Stewart R., Slukvin I. I., Thomson J. A. (2007) Science 318, 1917–1920 [DOI] [PubMed] [Google Scholar]
- 20.Viswanathan S. R., Daley G. Q., Gregory R. I. (2008) Science 320, 97–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Newman M. A., Thomson J. M., Hammond S. M. (2008) RNA 14, 1539–1549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heo I., Joo C., Cho J., Ha M., Han J., Kim V. N. (2008) Mol. Cell. 32, 276–284 [DOI] [PubMed] [Google Scholar]
- 23.Piskounova E., Viswanathan S. R., Janas M., LaPierre R. J., Daley G. Q., Sliz P., Gregory R. I. (2008) J. Biol. Chem. 283, 21310–21314 [DOI] [PubMed] [Google Scholar]
- 24.Heo I., Joo C., Kim Y. K., Ha M., Yoon M. J., Cho J., Yeom K. H., Han J., Kim V. N. (2009) Cell 138, 696–708 [DOI] [PubMed] [Google Scholar]
- 25.Lehrbach N. J., Armisen J., Lightfoot H. L., Murfitt K. J., Bugaut A., Balasubramanian S., Miska E. A. (2009) Nat. Struct. Mol. Biol. 16, 1016–1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hagan J. P., Piskounova E., Gregory R. I. (2009) Nat. Struct. Mol. Biol. 16, 1021–1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Viswanathan S. R., Daley G. Q. (2010) Cell 140, 445–449 [DOI] [PubMed] [Google Scholar]
- 28.Moss E. G., Tang L. (2003) Dev. Biol. 258, 432–442 [DOI] [PubMed] [Google Scholar]
- 29.Yang D. H., Moss E. G. (2003) Gene Expr. Patterns 3, 719–726 [DOI] [PubMed] [Google Scholar]
- 30.Viswanathan S. R., Powers J. T., Einhorn W., Hoshida Y., Ng T. L., Toffanin S., O'Sullivan M., Lu J., Phillips L. A., Lockhart V. L., Shah S. P., Tanwar P. S., Mermel C. H., Beroukhim R., Azam M., Teixeira J., Meyerson M., Hughes T. P., Llovet J. M., Radich J., Mullighan C. G., Golub T. R., Sorensen P. H., Daley G. Q. (2009) Nat. Genet. 41, 843–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chang T. C., Zeitels L. R., Hwang H. W., Chivukula R. R., Wentzel E. A., Dews M., Jung J., Gao P., Dang C. V., Beer M. A., Thomas-Tikhonenko A., Mendell J. T. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 3384–3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iliopoulos D., Hirsch H. A., Struhl K. (2009) Cell 139, 693–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dangi-Garimella S., Yun J., Eves E. M., Newman M., Erkeland S. J., Hammond S. M., Minn A. J., Rosner M. R. (2009) EMBO J. 28, 347–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peng S., Maihle N. J., Huang Y. (2010) Oncogene 29, 2153–2159 [DOI] [PubMed] [Google Scholar]
- 35.Guo Y., Chen Y., Ito H., Watanabe A., Ge X., Kodama T., Aburatani H. (2006) Gene 384, 51–61 [DOI] [PubMed] [Google Scholar]
- 36.Lu L., Katsaros D., Shaverdashvili K., Qian B., Wu Y., de la Longrais I. A., Preti M., Menato G., Yu H. (2009) Eur. J. Cancer. 45, 2212–2218 [DOI] [PubMed] [Google Scholar]
- 37.Wang Y. C., Chen Y. L., Yuan R. H., Pan H. W., Yang W. C., Hsu H. C., Jeng Y. M. (2010) Carcinogenesis 31, 1516–1522 [DOI] [PubMed] [Google Scholar]
- 38.Lee R. C., Feinbaum R. L., Ambros V. (1993) Cell 75, 843–854 [DOI] [PubMed] [Google Scholar]
- 39.Lagos-Quintana M., Rauhut R., Lendeckel W., Tuschl T. (2001) Science 294, 853–858 [DOI] [PubMed] [Google Scholar]
- 40.Lau N. C., Lim L. P., Weinstein E. G., Bartel D. P. (2001) Science 294, 858–862 [DOI] [PubMed] [Google Scholar]
- 41.Borchert G. M., Lanier W., Davidson B. L. (2006) Nat. Struct. Mol. Biol. 13, 1097–1101 [DOI] [PubMed] [Google Scholar]
- 42.Lee Y., Kim M., Han J., Yeom K. H., Lee S., Baek S. H., Kim V. N. (2004) EMBO J. 23, 4051–4060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cai X., Hagedorn C. H., Cullen B. R. (2004) RNA 10, 1957–1966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee Y., Ahn C., Han J., Choi H., Kim J., Yim J., Lee J., Provost P., Rådmark O., Kim S., Kim V. N. (2003) Nature 425, 415–419 [DOI] [PubMed] [Google Scholar]
- 45.Denli A. M., Tops B. B., Plasterk R. H., Ketting R. F., Hannon G. J. (2004) Nature 432, 231–235 [DOI] [PubMed] [Google Scholar]
- 46.Gregory R. I., Yan K. P., Amuthan G., Chendrimada T., Doratotaj B., Cooch N., Shiekhattar R. (2004) Nature 432, 235–240 [DOI] [PubMed] [Google Scholar]
- 47.Landthaler M., Yalcin A., Tuschl T. (2004) Curr. Biol. 14, 2162–2167 [DOI] [PubMed] [Google Scholar]
- 48.Yi R., Qin Y., Macara I. G., Cullen B. R. (2003) Genes Dev. 17, 3011–3016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lund E., Güttinger S., Calado A., Dahlberg J. E., Kutay U. (2004) Science 303, 95–98 [DOI] [PubMed] [Google Scholar]
- 50.Bohnsack M. T., Czaplinski K., Gorlich D. (2004) RNA 10, 185–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hutvágner G., McLachlan J., Pasquinelli A. E., Bálint E., Tuschl T., Zamore P. D. (2001) Science 293, 834–838 [DOI] [PubMed] [Google Scholar]
- 52.Ketting R. F., Fischer S. E., Bernstein E., Sijen T., Hannon G. J., Plasterk R. H. (2001) Genes Dev. 15, 2654–2659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gregory R. I., Chendrimada T. P., Cooch N., Shiekhattar R. (2005) Cell 123, 631–640 [DOI] [PubMed] [Google Scholar]
- 54.Maniataki E., Mourelatos Z. (2005) Genes Dev. 19, 2979–2990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schwarz D. S., Hutvágner G., Du T., Xu Z., Aronin N., Zamore P. D. (2003) Cell 115, 199–208 [DOI] [PubMed] [Google Scholar]
- 56.Du T., Zamore P. D. (2005) Development 132, 4645–4652 [DOI] [PubMed] [Google Scholar]
- 57.Ho S. N., Hunt H. D., Horton R. M., Pullen J. K., Pease L. R. (1989) Gene 77, 51–59 [DOI] [PubMed] [Google Scholar]
- 58.le Sage C., Nagel R., Egan D. A., Schrier M., Mesman E., Mangiola A., Anile C., Maira G., Mercatelli N., Ciafrè S. A., Farace M. G., Agami R. (2007) EMBO J. 26, 3699–3708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nagel R., le Sage C., Diosdado B., van der Waal M., Oude Vrielink J. A., Bolijn A., Meijer G. A., Agami R. (2008) Cancer Res. 68, 5795–5802 [DOI] [PubMed] [Google Scholar]
- 60.Lewis B. P., Burge C. B., Bartel D. P. (2005) Cell 120, 15–20 [DOI] [PubMed] [Google Scholar]
- 61.Wu L., Belasco J. G. (2005) Mol. Cell. Biol. 25, 9198–9208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bar M., Wyman S. K., Fritz B. R., Qi J., Garg K. S., Parkin R. K., Kroh E. M., Bendoraite A., Mitchell P. S., Nelson A. M., Ruzzo W. L., Ware C., Radich J. P., Gentleman R., Ruohola-Baker H., Tewari M. (2008) Stem Cells 26, 2496–2505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Krichevsky A. M., Sonntag K. C., Isacson O., Kosik K. S. (2006) Stem Cells 24, 857–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wu H., Xu J., Pang Z. P., Ge W., Kim K. J., Blanchi B., Chen C., Südhof T. C., Sun Y. E. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 13821–13826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Marson A., Levine S. S., Cole M. F., Frampton G. M., Brambrink T., Johnstone S., Guenther M. G., Johnston W. K., Wernig M., Newman J., Calabrese J. M., Dennis L. M., Volkert T. L., Gupta S., Love J., Hannett N., Sharp P. A., Bartel D. P., Jaenisch R., Young R. A. (2008) Cell 134, 521–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lu J., Getz G., Miska E. A., Alvarez-Saavedra E., Lamb J., Peck D., Sweet-Cordero A., Ebert B. L., Mak R. H., Ferrando A. A., Downing J. R., Jacks T., Horvitz H. R., Golub T. R. (2005) Nature 435, 834–838 [DOI] [PubMed] [Google Scholar]
- 67.Zamore P. D., Haley B. (2005) Science 309, 1519–1524 [DOI] [PubMed] [Google Scholar]
- 68.Bartel D. P., Chen C. Z. (2004) Nat. Rev. Genet. 5, 396–400 [DOI] [PubMed] [Google Scholar]
- 69.Lim L. P., Lau N. C., Garrett-Engele P., Grimson A., Schelter J. M., Castle J., Bartel D. P., Linsley P. S., Johnson J. M. (2005) Nature 433, 769–773 [DOI] [PubMed] [Google Scholar]
- 70.Selbach M., Schwanhäusser B., Thierfelder N., Fang Z., Khanin R., Rajewsky N. (2008) Nature 455, 58–63 [DOI] [PubMed] [Google Scholar]
- 71.Baek D., Villén J., Shin C., Camargo F. D., Gygi S. P., Bartel D. P. (2008) Nature 455, 64–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Krek A., Grün D., Poy M. N., Wolf R., Rosenberg L., Epstein E. J., MacMenamin P., da Piedade I., Gunsalus K. C., Stoffel M., Rajewsky N. (2005) Nat. Genet. 37, 495–500 [DOI] [PubMed] [Google Scholar]
- 73.Melton C., Judson R. L., Blelloch R. (2010) Nature 463, 621–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Calin G. A., Dumitru C. D., Shimizu M., Bichi R., Zupo S., Noch E., Aldler H., Rattan S., Keating M., Rai K., Rassenti L., Kipps T., Negrini M., Bullrich F., Croce C. M. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 15524–15529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Calin G. A., Sevignani C., Dumitru C. D., Hyslop T., Noch E., Yendamuri S., Shimizu M., Rattan S., Bullrich F., Negrini M., Croce C. M. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 2999–3004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Johnson S. M., Grosshans H., Shingara J., Byrom M., Jarvis R., Cheng A., Labourier E., Reinert K. L., Brown D., Slack F. J. (2005) Cell 120, 635–647 [DOI] [PubMed] [Google Scholar]
- 77.O'Donnell K. A., Wentzel E. A., Zeller K. I., Dang C. V., Mendell J. T. (2005) Nature 435, 839–843 [DOI] [PubMed] [Google Scholar]
- 78.Voorhoeve P. M., le Sage C., Schrier M., Gillis A. J., Stoop H., Nagel R., Liu Y. P., van Duijse J., Drost J., Griekspoor A., Zlotorynski E., Yabuta N., De Vita G., Nojima H., Looijenga L. H., Agami R. (2006) Cell 124, 1169–1181 [DOI] [PubMed] [Google Scholar]
- 79.Calin G. A., Croce C. M. (2006) Nat. Rev. Cancer 6, 857–866 [DOI] [PubMed] [Google Scholar]
- 80.Esquela-Kerscher A., Slack F. J. (2006) Nat. Rev. Cancer. 6, 259–269 [DOI] [PubMed] [Google Scholar]
- 81.Kumar M. S., Erkeland S. J., Pester R. E., Chen C. Y., Ebert M. S., Sharp P. A., Jacks T. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 3903–3908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yu F., Yao H., Zhu P., Zhang X., Pan Q., Gong C., Huang Y., Hu X., Su F., Lieberman J., Song E. (2007) Cell 131, 1109–1123 [DOI] [PubMed] [Google Scholar]
- 83.Johnson C. D., Esquela-Kerscher A., Stefani G., Byrom M., Kelnar K., Ovcharenko D., Wilson M., Wang X., Shelton J., Shingara J., Chin L., Brown D., Slack F. J. (2007) Cancer Res. 67, 7713–7722 [DOI] [PubMed] [Google Scholar]
- 84.Trang P., Medina P. P., Wiggins J. F., Ruffino L., Kelnar K., Omotola M., Homer R., Brown D., Bader A. G., Weidhaas J. B., Slack F. J. (2010) Oncogene 29, 1580–1587 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}