

Abstract
Raman microspectroscopy (RMS) was used to detect and image molecular markers specific to cardiomyocytes (CMs) derived from human embryonic stem cells (hESCs). This technique is noninvasive and thus can be used to discriminate individual live CMs within highly heterogeneous cell populations. Principal component analysis (PCA) of the Raman spectra was used to build a classification model for identification of individual CMs. Retrospective immunostaining imaging was used as the gold standard for phenotypic identification of each cell. We were able to discriminate CMs from other phenotypes with >97% specificity and >96% sensitivity, as calculated with the use of cross-validation algorithms (target 100% specificity). A comparison between Raman spectral images corresponding to selected Raman bands identified by the PCA model and immunostaining of the same cells allowed assignment of the Raman spectral markers. We conclude that glycogen is responsible for the discrimination of CMs, whereas myofibril proteins have a lesser contribution. This study demonstrates the potential of RMS for allowing the noninvasive phenotypic identification of hESC progeny. With further development, such label-free optical techniques may enable the separation of high-purity cell populations with mature phenotypes, and provide repeated measurements to monitor time-dependent molecular changes in live hESCs during differentiation in vitro.
Introduction
Stem cell (SC) therapy is widely acknowledged as a key medical technology of the 21st century. SCs have enormous potential for use in cell-replacement therapies for age-related illnesses such as Alzheimer's disease and Parkinson's disease, as well as diabetes and cardiovascular disorders (1), and are a reliable source of cells for tissue engineering (2). Furthermore, SC-seeded scaffolds may provide an unlimited supply of grafts to replace and repair diseased tissues (3). Human embryonic SCs (hESCs) are derived from the inner cell mass of blastocyst-stage embryos (4). They are pluripotent by nature, and virtually all cell types can be derived after differentiation, including cardiomyocytes (CMs) (5). However, the ability to control the differentiation status and phenotypic purity of hESC-derived cell populations is a critical factor in addressing the efficiency and safety of SC therapies. It has been reported that excessive proliferation after transplantation of cells with unwanted phenotypes can cause tissue overgrowth and tumor formation (6).
However, the current conditions under which specific cell types are derived remain suboptimal, and generally result in low yields of the desired differentiated lineages within highly heterogeneous populations that are not suitable for clinical use due to the presence of mainly unwanted cell types. This current limitation in the delivery of validated hESCs suitable for clinical applications highlights the immediate need for noninvasive optical techniques that can phenotypically identify live cells within highly heterogeneous populations.
In this study, we developed a technique based on Raman microspectroscopy (RMS) to identify lineage-specific molecular markers for live cardiomyocytes (CMs) derived from hESCs in vitro. Our basic hypothesis was that RMS could be used to measure intrinsic chemical differences among different cell types without the use of labels or other invasive procedures. Indeed, chemical differences among cell types are expected, as differentiated cells are specialized to perform specific functions and consequently produce specific biochemicals. For example, CMs contain a large number of myofibrils, β-cells secrete insulin, and neurons produce neurotransmitters.
Compared with other techniques, Raman spectroscopy has several unique advantages. Conventional cell biology assays (e.g., polymerase chain reaction and Western blotting) are invasive and not suitable for characterizing heterogeneous cell populations because they require a large number of cells, and the results represent averages over entire cell populations. Fluorescence- and magnetic-activated cell sorting approaches rely on lineage-specific surface markers that are expressed on the cell membrane (7,8). However, there are numerous cases, including that of CMs, in which most of the lineage-specific markers used in these techniques are not expressed on the cell surface. Detection thus requires fixation and permeabilization, rendering the cell unusable in a clinical environment. Therefore, these techniques can only be used to identify a limited number of viable cell types (e.g., hematopoietic lineages). To circumvent the difficulties of antibody-based detection approaches, transgenic strategies can be used to express markers (e.g., green fluorescent protein) from lineage-specific promoters. However, recently developed strategies to enrich hESC-derived CMs to >90% purity (9,10) involve complex genetic modifications of cells, and thus are not ideal for clinical applications.
Recent studies have demonstrated the potential of RMS for measuring the chemical properties of live cells (11). This technique combines the high chemical specificity of Raman spectroscopy with the high spatial resolution of optical microscopy to provide detailed molecular information about complex biological samples, including cells (12). Because RMS has only a minimal background signal from water, it allows repeated observations of viable cells maintained under physiological sterile conditions, which is difficult to achieve by other molecular vibrational techniques (13,14).
The high spatial resolution of RMS has enabled the analysis of organelles (15) and the distribution of proteins (16), apoptotic bodies (17), lipid vesicles (18), and lipid deposits (19) in individual fixed cells. Other applications to live cells maintained in buffer solutions include analysis of subcellular components, such as the nucleus (12), heme moieties in erythrocytes (20), and cytotoxic granules in human killer T cells (21). RMS has also been used to detect biochemical changes related to various cellular processes, including differentiation (22) and interactions with drugs (23,24) and toxins (25). More recently, time-course RMS imaging has been used for short time periods (20 min) on cells in saline buffers (26) and on cells maintained under physiological conditions for extended periods of time (6 h) to monitor biochemical changes related to apoptosis (27).
In one study (28), the high chemical specificity of RMS enabled detection of a decrease in RNA abundance during in vitro differentiation of murine ESCs, and another (29) showed that the contribution of nucleic acids was higher in the Raman spectra of undifferentiated murine ESCs compared with differentiated cells. Similar results were also found in more recent studies using hESCs (30). A comparison between different age groups of rhesus monkey mesenchymal cells derived from bone marrow indicated a higher DNA and lower protein contribution in the Raman spectra of fetal cells compared with juvenile cells (31). In a recent study (32), investigators attempted to discriminate between undifferentiated hESC and hESC-derived CMs by RMS; however, although they were able to identify spectral differences, mainly related to nucleic acids, the reported discrimination accuracy for CMs against undifferentiated hESCs was only 66%.
In this study, we tackled for the first time (to our knowledge) the challenge of detecting noninvasive molecular markers specific to hESC-derived CMs. Such markers can be used to identify CMs from other differentiated cells within highly heterogeneous cell populations derived from hESCs. We analyzed individual live cells by RMS and carried out retrospective phenotypic identification of all cells by the gold-standard method of immunofluorescence imaging integrated with the Raman microscope. We evaluated the effects of laser irradiation on the cells by performing a beating frequency analysis on individual CMs. The Raman spectra of the cells were analyzed by means of multivariate statistical methods, with the aim of developing high-accuracy models for discriminating CMs from other unwanted cell types. The spectral markers identified in these models were then correlated with cellular biochemical changes related to differentiation toward the cardiac phenotype. The distribution of these spectral markers in individual live cells was mapped and compared with fluorescence images of the same cells to enable identification of the chemicals responsible for discrimination of CMs.
Materials and Methods
Materials and general cell culture
All tissue culture reagents were purchased from Invitrogen (Paisley, UK) and chemicals were obtained from Sigma-Aldrich (Poole, UK) unless otherwise stated. Mouse embryo fibroblasts and hESC cultures were maintained at 37°C, 5% CO2, in a humidified atmosphere. The medium was changed daily for hESC culture and every 3–4 days during differentiation. Purified chemicals used for comparison with Raman spectra of cells (i.e., myosin heavy chain from rabbit muscle (M7659-1MG), Troponin T from human heart (T0175-50UG), and glycogen from Mytilus edulis (G1767-1VL)) were purchased from Sigma-Aldrich. All chemicals were used without further purification.
hESC culture and differentiation
The hESC line HUES7 was cultured in feeder-free conditions in conditioned medium in a Matrigel-coated flask and cultured using trypsin passaging between passages 17–35. Differentiation was achieved by forced aggregation of defined numbers of hESCs as described previously (5) (see the Supporting Material). Beating clusters containing CMs and non-CMs were manually dissected, washed in phosphate-buffered saline (PBS), and then incubated for 30 min at room temperature in buffer 1 (120 mM NaCl, 5.4 mM KCl, 5 mM MgSO4, 5 mM sodium pyruvate, 20 mM taurine, 10 mM HEPES, 20 mM glucose, pH 6.9), for 45 min at 37°C in buffer 2 (120 mM NaCl, 5.4 mM KCl, 5 mM MgSO4, 5 mM sodium pyruvate, 20 mM taurine, 10 mM HEPES, 0.3 mM CaCl2, 20 mM glucose, 1 mg/mL collagenase B, pH 6.9), and for 1 h at room temperature in buffer 3 (85 mM KCl, 5 mM MgSO4, 5 mM sodium pyruvate, 20 mM taurine, 1 mM EGTA, 5 mM creatine, 30 mM K2HPO4, 20 mM glucose, 1 mg/mL Na2ATP, pH 7.2). Finally, cell clusters were dissociated by repeated pipetting through a P1000 tip, and the liberated cells were seeded in dialyzed fetal bovine serum in purpose-built cell chambers for the Raman measurements.
Immunostaining of cells
Immediately after the Raman measurements were obtained, the samples were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton-X100, and then incubated with mouse monoclonal anti-α-actinin (1:800; Sigma) for 1 h at room temperature. Cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI, 100 ng/mL) at 1:1000 dilution in PBS for 5 min at room temperature. For glycogen staining, a periodic acid-Schiff (PAS) kit (Sigma-395B) was used. After the cells were stained with α-actinin, the samples were rinsed with deionized water and oxidized in periodic acid solution for 5 min. The samples were rinsed three times with deionized water and treated with Schiff's reagent for 15 min. They were then washed with deionized water for 10 min and counterstained with hematoxylin for 90 s. The samples were rinsed again with deionized water for evaluation under a light microscope.
Analysis of beating frequency for individual CMs
A beating frequency analysis was carried out on individual beating CMs maintained on the Raman microscope under conditions similar to those used for the Raman measurements. Videos recorded under white-light illumination were analyzed using routines developed in MATLAB (The MathWorks, Natick, MA). First, all video frames were converted into grayscale and only pixels that contained the cell were selected for analysis (typically ∼400 pixels). To increase the accuracy of the analysis method, we increased the contrast between the lightest and darkest regions of a cell by digitizing the intensity shifts at each pixel using a threshold value. We obtained the beating frequency at each pixel of a cell by calculating the Fourier transform of the time-dependent intensity shift at each individual pixel. The beating frequency of the cell was then calculated as the mean of the frequency values obtained at all pixels. Assuming that beating was homogeneous within the cell, the digitizing threshold was selected as the value that provided the smallest standard deviation (SD) between frequencies measured at different pixels inside the cell. The SD was then used as a measure of the error for the beating frequency.
RMS measurements
Raman spectra were recorded with an in-house-built Raman microspectrometer optimized for live-cell studies. The instrument consisted of an inverted microscope with a water-immersion objective (60×/NA 0.90; Olympus, Essex, UK), a spectrometer equipped with an 830 lines/mm grating, a cooled deep-depletion back-illuminated CCD detector (Andor Technologies, Belfast, UK), and a high-precision automated step-motor stage (Prior, Cambridge, UK). The wave-number axis of the spectrometer was calibrated before each experiment with the use a standard Tylenol sample, and the spectral resolution was ∼1.5 cm−1 in the 600–1800 cm−1 region. The laser diode (785 nm wavelength) had ∼150 mW power (after objective), 2.2 mm beam diameter, and beam quality factor M2 = 1.7 (Toptica Photonics, Munich, Germany). The laser spot diameter was calculated to be 2.3 μm.
Purpose-designed titanium cell chambers were built that incorporated MgF2 coverslips (0.17 mm thick) at the bottom to enable acquisition of Raman spectra of the cells. The Raman microscope was equipped with an environmental enclosure (Solent, Segensworth, UK), which combined with the inverted optical configuration allowed the cells to be maintained under sterile physiological conditions (culture medium, 37°C temperature, 5% CO2).
We recorded the Raman spectra of CMs (50 cells) and non-CMs (40 cells) for several months using >20 cell culture flasks, including cells at various passages. For each cell population, both CMs and non-CMs were measured. The Raman spectrum of each individual cell represented the average of a total of 625 spectra that were measured at different positions inside the cell by raster-scanning the cell through the laser focus in 2 μm steps (equivalent to a grid of 25 × 25 points). The acquisition time at each position was 1 s. After the acquisition of Raman spectra was completed, the position coordinates of each cell were recorded and the cells were fixed and prepared for immunostaining (cardiac phenotype and cell nucleus). The cardiac phenotype of the cells was established with the use of a wide-field fluorescence staining system integrated on the Raman microscope. We retropositioned each cell by using the cell coordinates (accuracy ∼5 μm), which allowed us to assign each individual cell to the CM or non-CM group. We found that it was impractical to establish the exact phenotype of the non-CMs by the immunostaining method.
Data analysis and processing
Data preprocessing consisted of three steps: removal of spectra containing cosmic rays, background subtraction, and normalization. Because 625 Raman spectra were acquired for each individual cell, the small fraction of individual spectra that contained cosmic rays (typically <1%) could be eliminated without affecting the outcome of the data analysis. The average of the Raman spectra measured at points outside of the cell (automatically identified using a PCA routine) represented the background spectrum (contributions from the culture medium, MgF2 coverslip, and microscope objective). The Raman spectrum that was representative of each cell was obtained by algebraic subtraction of the background spectrum from the average of the Raman spectra at all positions inside the cell. All Raman spectra were then normalized using the standard normal variance method (zero mean and unity variance) (28).
The Raman spectra of the cells were analyzed by PCA using functions in MATLAB. Raman spectral images corresponding to selected Raman bands were obtained by calculating the area under the spectral bands after subtracting the estimated local linear baselines and representing the integrated intensity value at each measurement position in the cell.
Results and Discussion
Effect of laser irradiation on CMs: analysis of beating frequency
It is well known that biomolecules are sensitive to light, and exposure of cells to lasers can cause localized heating or induce toxic photochemical reactions. Although lasers in the visible range (400–600 nm) allow rapid measurement of high-quality Raman spectra of fixed cells (15), such lasers can induce cell damage even at low laser powers and short exposure times (33). In contrast, when cells are irradiated by lasers in the near-infrared region (700–1000 nm), they can remain viable even after longer exposures at much high laser power densities (34,35).
In a previous study on hESCs and hESC-derived CMs (32), it was noted that, visually, laser exposure at 70 mW for 5 min (785 nm laser) did not seem to affect the beating of the hESC-CM clusters, and trypan blue viability tests indicated that the cells remained viable. However, the experiments presented in that work used a fivefold higher laser power and fivefold longer acquisition time compared to previous studies, as required to sample the entire volume of the cell rather than to obtain single-point measurements. Therefore, this longer laser exposure required a new evaluation of the laser's effect on cells to ensure that the proposed RMS technique is noninvasive.
Because we observed no evident morphological changes indicating necrosis or apoptosis after laser irradiation (Fig. 1, A and B), we evaluated more subtle cellular changes by measuring the beating frequency of individual CMs before and 15 min after the Raman measurements. The beating frequency before laser irradiation was found to be in the 0.75–1.75 Hz range (Fig. 1 C), in agreement with recent reports for CMs (36). The mean SD values were ∼24%, indicating that the beating frequency can vary considerably within individual CMs. Fig. 1 C shows that the mean change in beating frequency of hESC-derived CMs after measurement of Raman spectra was only 18%, which is lower than the SD. Therefore, these results demonstrate that, apart from not inducing cell death, laser irradiation (as required for the acquisition of Raman spectra to phenotypically identify CMs) did not affect the beating frequency of the CMs.
Figure 1.
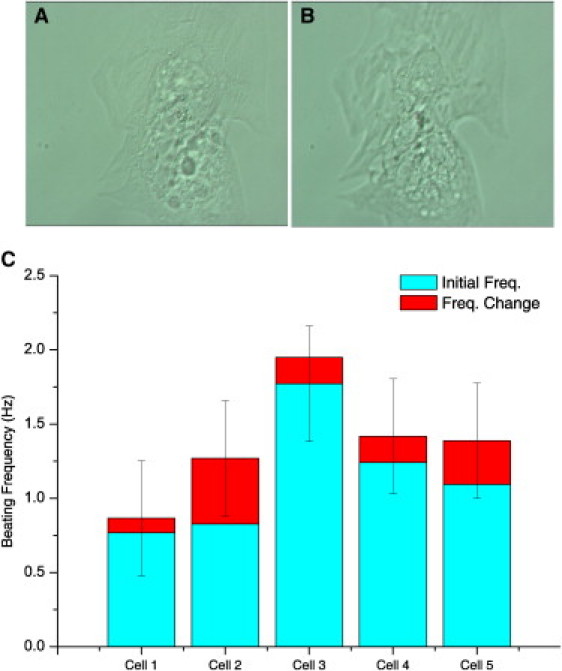
Analysis of beating frequency of CMs before and after laser irradiation, as required for collection of Raman spectra. (A and B) Phase-contrast image before (A) and after (B) irradiation. (C) Initial frequencies and frequency changes after laser irradiation. Error bars represent the variability in frequency measurement (SD) at various positions in each cell.
Molecular markers for phenotypic identification of CMs
Fig. 2 presents immunofluorescence images and Raman spectra of typical differentiated live cells derived from hESCs. The immunostaining images of the cardiac phenotype (α-actinin) and cell nuclei (DAPI) for the same cells highlight the high heterogeneity of the cell populations derived from hESCs (see Fig. S1 for additional low-magnification figures of cells derived from hESCs). Because the percentage of CMs in the cell population was typically <10%, retrospective phenotypic identification using the gold-standard immunostaining assay for each individual cell was essential. A recent RMS study that aimed to discriminate hESC-derived CMs from undifferentiated hESCs without using a gold-standard method for retrospective phenotypic identification of the CMs reported a modest accuracy of 66% (32). The authors acknowledged that the low accuracy might have been due to the high phenotypic heterogeneity of the cell populations derived from hESCs.
Figure 2.
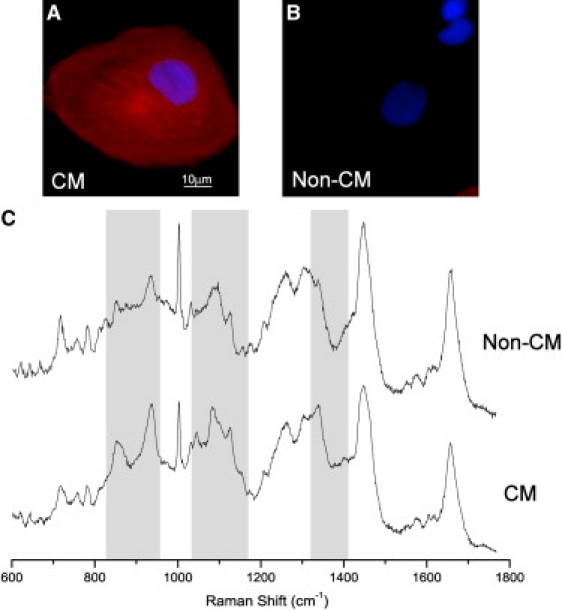
Typical immunostaining images of individual cardiomyocytes (CM) (A) and non-cardiomyocyte (non-CM) (B) derived from hESCs: cell nuclei DAPI (blue), cardiac α-actinin (red). (C) Raman spectra of the same cells.
Fig. 2 C shows that the sampling method used in this study led to high signal/noise ratio Raman spectra that are representative of the entire cell analyzed. Several spectroscopic features can be easily recognized in the Raman spectra of both CMs and non-CMs that correspond to the molecular vibrations of cellular biochemicals (i.e., nucleic acids, protein, lipids, and carbohydrates) and are consistent with previous reports on other cell types (11). To illustrate the spectral differences, the average Raman spectra of CMs (50 cells) and non-CMs (40 cells) derived from hESCs are shown in Fig. 3. The computed SD spectra were also included (gray lines) to show the intercellular variance within each group of cells. The computed difference spectrum (DS; CMs minus non-CMs) is also presented in Fig. 3, including the combined SD spectrum. Fig. 3 shows that spectral differences between CMs and non-CMs can be identified in several spectral regions. The first region is 825–950 cm−1 and is associated with the molecular vibrations of proteins, carbohydrates, and lipids. The second region is 1035–1170 cm−1 and is associated mainly with the vibrations of carbohydrates, lipids, and nucleic acids. The third region is the highly convoluted region between 1320 and 1420 cm−1, where contributions are dominated by proteins, carbohydrates, and nucleic acids. A more detailed discussion on the molecular assignments of these bands will be presented in the next section.
Figure 3.
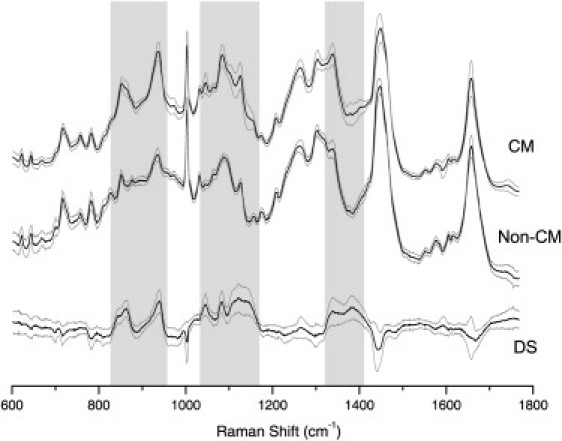
Average Raman spectra of CMs and non-CMs, and their computed DS. SD lines represent the SD calculated at each wave number. Shaded areas highlight the spectral regions that may be used for discrimination of CMs.
Because the spectral differences between CMs and non-CMs include multiple Raman bands, a multivariate statistical model has been developed for the discrimination of CMs. PCA decomposes multivariate data sets in uncorrelated PCs in such a way that the first few components will capture most of the variation present in the data set. PCA has been used extensively in the study of Raman spectra of cells, including ESCs (28,32).
Fig. 4 A shows that out of 90 PCs needed to identically reconstruct the original data set, only the first four PCs had a high signal/noise ratio and captured 86% of the variance between the Raman spectra of all cells, including CMs and non-CMs. After we inspected individual PCs and their contribution to the total variance, we concluded that using only the first PC (PC1), accounting for 65% of the variance, is adequate for class-discrimination purposes. It is also important to note that the main spectral bands in PC1 are also found in the computed DS between average Raman spectra of CMs and non-CMs. The inclusion of PC2, PC3, or PC4 (capturing 12.36%, 7.33%, and 2.15% of the variance, respectively) did not improve the discrimination between CMs and non-CMs.
Figure 4.
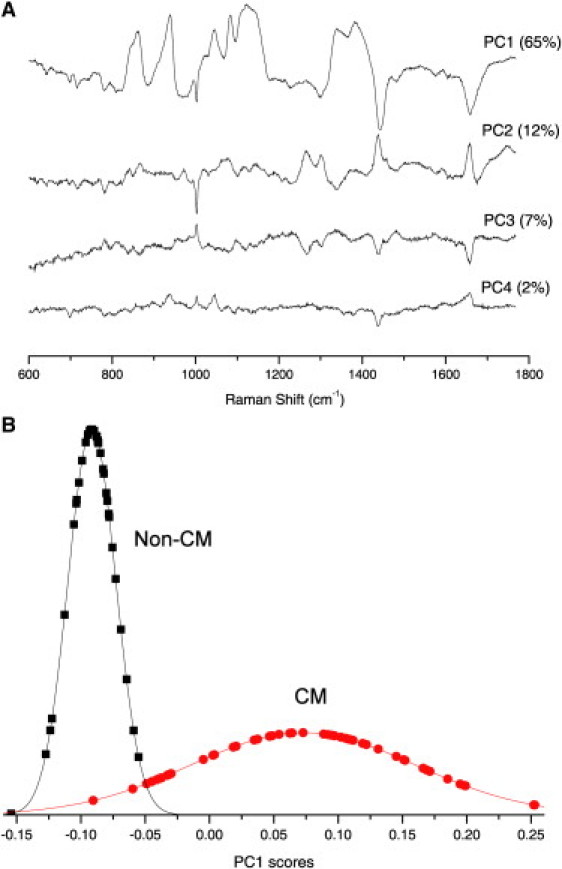
(A) First four PC loading spectra in the PCA of Raman spectra of CMs and non-CMs. The variance captured by each PC is shown in brackets. (B) Distribution of the scores corresponding to the first PC (PC1) for CMs (circles) and non-CMs (squares).
The probability distributions of the PC1 scores are presented in Fig. 4 B, which shows a clear distinction between CMs and non-CMs. By selecting the boundary between the two classes at the intersection of the two fitted probability functions (Fig. 4 B), we find that the classification accuracy of the PCA model has 96% sensitivity and 100% specificity. (In this case, the specificity is the ability of Raman spectroscopy to identify and exclude all non-CM cells, and the sensitivity parameter represents the ability to correctly identify CMs.)
Although PCA has been successfully used to identify Raman spectral bands that are able to discriminate CMs, the values for specificity and sensitivity give no indication about the ability of these spectral markers to enable phenotypic identification of new cells. To determine the true accuracy for the phenotypic identification of CMs, we used cross-validation (CV) to determine the sensitivity and specificity parameters for a certain target sensitivity or specificity. The different CV methods can also be labeled with the percentages corresponding to the data set splitting (where the first figure refers to the fraction of the data set used to build the classification model, and the second figure represents the fraction of the data set to which the model is applied). In leave-one-out CV (LOOCV), all spectra except one are used to build a model and then to classify the left-out spectrum. This method is repeated so that each spectrum is predicted once.
The PCA model gives us the flexibility to set the classification boundary between CMs and non-CMs to emulate a realistic scenario in which we can target a highly specific regime of 100% specificity, as required in regenerative medicine. This type of modeling is required to ensure that no unwanted cells are predicted as CM, thus leading to a lower-purity cell population. However, this regime may lead to more CMs being misclassified as non-CMs, thus decreasing the number of cells available for treatment. The CV specificity and sensitivity for 70/30%, 80/20%, and LOOCV calculated for target 100% specificity are shown in Table 1. For the LOOCV, all possible permutations were used to calculate the sensitivity and specificity because the number of possible permutations was relatively small. However, for the 70/30% and 80/20% methods, the number of possible permutations was high, and therefore only 3000 randomly chosen combinations were used. The CV results in Table 1 show that the Raman spectral bands determined by PC1 provide >97% specificity and >96% sensitivity. Table 1 also shows that the values for the predicted sensitivity and specificity are independent of the CV method used, demonstrating the robustness of the Raman spectral markers. It is worth noting that by adjusting the classifications boundary, one can further improve the prediction specificity by up to 100%, albeit at the expense of lower sensitivity.
Table 1.
Discrimination accuracy for CMs calculated by three CV methods
| CV method | Model target specificity | Model sensitivity | Prediction specificity | Prediction sensitivity |
|---|---|---|---|---|
| 70/30% | 100% | 96.25 ± 2% | 97.27 ± 4% | 96.67 ± 3% |
| 80/20% | 100% | 96.28 ± 1.45% | 96.79 ± 6.75 | 96.30 ± 5% |
| LOOCV | 100% | 96.08 ± 0.29% | 97.54 | 96.1 |
Imaging and assignment of the Raman spectral markers
CMs are specialized to perform specific functions and consequently express specific biochemicals to produce a large number of myofibrils. Beating CMs also require a higher amount of energy compared with other cell types, and therefore they store larger amounts of glycogen. In addition to the high classification and discrimination ability provided by assignment of the Raman bands identified by the PCA model, this approach can also provide insight into the chemical differences between CMs and non-CMs. A comparison between the computed DS and the Raman spectra of a few selected biochemicals known to be more abundant in CMs (i.e., myosin, troponin, and glycogen) is shown in Fig. 5. There is a striking similarity between the Raman spectra of glycogen and DS, suggesting that glycogen is one of the main contributors to the discriminant factor between CMs and non-CMs. Several Raman bands corresponding to glycogen (C-O-H ring vibrations at 860 cm−1, C-O-C glycosidic bond vibrations at 938 cm−1, and C-C and C-O stretchings at 1084 cm−1 and 1123 cm−1) can be identified, whereas C-H deformation vibrations contribute at 1340 cm−1,1381 cm−1, and 1450 cm−1 (37). The presence of glycogen inside the CMs has been well documented, as these cells have long been used as a valid model of glycogen metabolism (38). Fig. 5 also shows that myosin and troponin have several bands that overlap certain glycogen bands identified in the DS: 853 cm−1 assigned to C-C and CH3 stretching vibrations in lysine, aspartic acid, leucine, valine, and 936 cm−1 associated with C-C stretching of protein backbones (37). However, the contribution of myosin and troponin to the spectral marker is less evident than that for glycogen.
Figure 5.
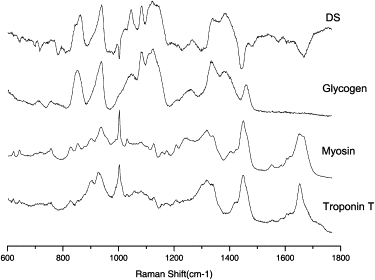
Comparison between the computed DS and selected chemicals that are more abundant in CMs compared with other phenotypes.
One of the main features of RMS is that it enables the collection of Raman spectra from micrometric regions of live cells. We used this feature to aid in the molecular assignment of Raman spectral markers of CMs by observing the spatial distributions of these biomolecules within individual cells and comparing them with immunostaining images. Fig. 6 compares the Raman spectral map of the 938 cm−1 Raman band for a typical CM (Fig. 6 C) with the immunostaining images for myofibrils (α-actinin; Fig. 6 A) and glycogen (PAS; Fig. 6 B) of the same cell. Fig. 6 C indicates that the molecules that contribute the most to the identification of CMs are localized in the center of this cell, at the same location where the PAS staining also shows a significantly higher concentration of glycogen. Fig. 6 D shows a typical Raman spectrum measured at a position with a high abundance of both glycogen and myofibrils (top spectrum, blue square). The Raman spectrum shows a very close similarity to the PC1 and pure glycogen spectra in Fig. 5. On the other hand, the immunostaining for α-actinin (Fig. 6 A) suggests that myofibrils are distributed over the entire cell, with an accumulation at the central region. The bottom Raman spectrum in Fig. 6 D (green star), measured at a position that is rich in myofibrils but has a lower glycogen concentration, is similar to that of myosin and troponin, with a strong band at 938 cm−1 but no band at 860 cm−1. Therefore, the closer similarity between the Raman map and glycogen staining combined with the band assignment identification suggests that glycogen is the main contributor to the Raman spectral marker of hESC-derived CMs, whereas the proteins found in myofibrils have a lower contribution. It is well known that glycogen is an integral part of cells in general. Therefore, we cannot claim that glycogen is a general unique identifier of the CM phenotype. However, the results obtained in this study demonstrate that if hESCs are differentiated as described, glycogen can provide a high accuracy for identification of CMs. Further studies will be necessary to achieve a complete assignment of the Raman bands, which should include relevant biomolecules isolated directly from hESC-derived CMs. However, such an analysis was beyond the scope of this work.
Figure 6.
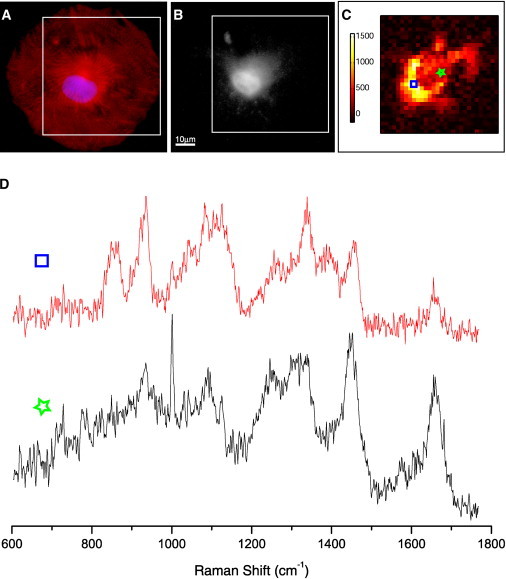
(A) Immunostaining for cardiac phenotype (α-actinin, red) and cell nucleus (DAPI, purple). (B) PAS staining for glycogen. (C) Raman spectral image corresponding to the 938 cm−1 band obtained from a typical CM. (D) Selected Raman spectra at positions indicated by the blue square and green star (acquisition time: 1 s per spectrum).
Fig. 6 also highlights another important point: the high intracellular chemical variability of the CMs, which led to high variations in the Raman spectra measured at various locations inside the cells. For example, although the two spectra presented in Fig. 6 D correspond to two positions only ∼12 μm apart inside the cell, their spectral features are completely different. Given that typical cells have diameters of ∼50–60 μm, and the laser spot is ∼2.3 μm, it was important to ensure that the cells were raster-scanned through the laser spot (step size smaller than the laser spot) and all spectra were averaged to obtain a Raman spectrum that was representative of each individual cell. Although measuring only single spectra at single locations inside cells by Raman microspectrometers with lasers focused to micrometer sizes may require shorter acquisition times, this approach is not appropriate for these cells due to their high spectral variability.
Conclusions
We have demonstrated that RMS combined with multivariate analysis (PCA) can be used with high accuracy for label-free phenotypic identification of CMs derived from hESCs. During measurements, the cells were maintained under sterile physiological conditions and experimental conditions were optimized to ensure that the cells incurred no damage. We developed a beating-frequency analysis to study individual cells, and the results showed that laser irradiation at 785 nm did not cause changes in the beating frequency.
We developed a PCA model using Raman spectra of 50 CMs and 40 non-CMs within the same heterogeneous populations as derived from hESCs. The phenotype of each individual cell was confirmed as CM or non-CM by immunofluorescence staining for α-actinin. The PCA model using the scores of the first PC (65% variance captured) showed that even for highly specific regimes (target 100% specificity), RMS can discriminate CMs from other phenotypes with >97% specificity and >96% sensitivity. Furthermore, these values did not vary for three CV algorithms (70/30%, 80/20%, and LOOCV), demonstrating the robustness of the technique.
The Raman bands identified in the PCA model indicated that the spectral differences between CMs and non-CMs were mainly attributed to glycogen and myofibrils. To advance this assignment, we compared Raman spectral images corresponding to the selected Raman bands identified in the PCA loadings with the immunostaining of myofibrils and glycogen. The similarity between the Raman spectral images and glycogen-rich regions suggests that glycogen makes a higher contribution to the spectral markers than the myofibrils. However, further studies are under way to compare the spectral markers with the Raman spectra of myofibril proteins purified directly from hESC-derived CMs.
Although this study focuses on hESC-derived CMs, the results demonstrate the potential of RMS for enabling noninvasive phenotypic identification of hESC progeny. Such label-free, noninvasive analytical techniques can be further developed to aid in the separation and purification of cell populations as required for medical applications such as cell therapy and regenerative medicine. However, the long acquisition times as described in this work are not yet practical for such applications. Therefore, further developments in sampling methods, such as laser beam expansion using low numerical objectives or line scanning, are required to reduce measurement times and increase the cell-sorting speed. In addition, the ability to noninvasively monitor time-dependent molecular changes in live hESCs during differentiation may also have a huge beneficial impact on the refinement and standardization of protocols to efficiently differentiate pluripotent SCs toward desired phenotypes.
Acknowledgments
This study was supported by the Biotechnology and Biological Sciences Research Council (BB/G010285/1).
Contributor Information
Chris Denning, Email: chris.denning@nottingham.ac.uk.
Ioan Notingher, Email: ioan.notingher@nottingham.ac.uk.
Supporting Material
References
- 1.Klimanskaya I., Rosenthal N., Lanza R. Derive and conquer: sourcing and differentiating stem cells for therapeutic applications. Nat. Rev. Drug Discov. 2008;7:131–142. doi: 10.1038/nrd2403. [DOI] [PubMed] [Google Scholar]
- 2.Marot D., Knezevic M., Novakovic G.V. Bone tissue engineering with human stem cells. Stem Cell Res. Ther. 2010;1:10. doi: 10.1186/scrt10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fang B., Li Y., Zhao R.C. Human adipose tissue-derived adult stem cells can lead to multiorgan engraftment. Transplant. Proc. 2010;42:1849–1856. doi: 10.1016/j.transproceed.2010.01.058. [DOI] [PubMed] [Google Scholar]
- 4.Thomson J.A., Itskovitz-Eldor J., Jones J.M. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- 5.Burridge P.W., Anderson D., Denning C. Improved human embryonic stem cell embryoid body homogeneity and cardiomyocyte differentiation from a novel V-96 plate aggregation system highlights interline variability. Stem Cells. 2007;25:929–938. doi: 10.1634/stemcells.2006-0598. [DOI] [PubMed] [Google Scholar]
- 6.Reubinoff B.E., Pera M.F., Fong C.Y., Trounson A., Bongso A. Embryonic stem cell lines from human blastocysts: somatic differentiation in vitro. Nat. Biotechnol. 2000;18:399–404. doi: 10.1038/74447. [DOI] [PubMed] [Google Scholar]
- 7.Fukuda H., Takahashi J., Hashimoto N. Fluorescence-activated cell sorting-based purification of embryonic stem cell-derived neural precursors averts tumor formation after transplantation. Stem Cells. 2006;24:763–771. doi: 10.1634/stemcells.2005-0137. [DOI] [PubMed] [Google Scholar]
- 8.David R., Groebner M., Franz W.M. Magnetic cell sorting purification of differentiated embryonic stem cells stably expressing truncated human CD4 as surface marker. Stem Cells. 2005;23:477–482. doi: 10.1634/stemcells.2004-0177. [DOI] [PubMed] [Google Scholar]
- 9.Anderson D., Self T., Denning C. Transgenic enrichment of cardiomyocytes from human embryonic stem cells. Mol. Ther. 2007;15:2027–2036. doi: 10.1038/sj.mt.6300303. [DOI] [PubMed] [Google Scholar]
- 10.Huber I., Itzhaki I., Gepstein L. Identification and selection of cardiomyocytes during human embryonic stem cell differentiation. FASEB J. 2007;21:2551–2563. doi: 10.1096/fj.05-5711com. [DOI] [PubMed] [Google Scholar]
- 11.Notingher I., Hench L.L. Raman microspectroscopy: a noninvasive tool for studies of individual living cells in vitro. Exp. Rev. Med. Dev. 2006;3:215–234. doi: 10.1586/17434440.3.2.215. [DOI] [PubMed] [Google Scholar]
- 12.Puppels G.J., de Mul F.F.M., Jovin T.M. Studying single living cells and chromosomes by confocal Raman microspectroscopy. Nature. 1990;347:301–303. doi: 10.1038/347301a0. [DOI] [PubMed] [Google Scholar]
- 13.Krafft C., Salzer R., Schieker M. Differentiation of individual human mesenchymal stem cells probed by FTIR microscopic imaging. Analyst (Lond.) 2007;132:647–653. doi: 10.1039/b700368d. [DOI] [PubMed] [Google Scholar]
- 14.Kuimova M.K., Chan K.L., Kazarian S.G. Chemical imaging of live cancer cells in the natural aqueous environment. Appl. Spectrosc. 2009;63:164–171. doi: 10.1366/000370209787391969. [DOI] [PubMed] [Google Scholar]
- 15.Matthäus C., Chernenko T., Diem M. Label-free detection of mitochondrial distribution in cells by nonresonant Raman microspectroscopy. Biophys. J. 2007;93:668–673. doi: 10.1529/biophysj.106.102061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Uzunbajakava N., Lenferink A., Otto C. Nonresonant Raman imaging of protein distribution in single human cells. Biopolymers. 2003;72:1–9. doi: 10.1002/bip.10246. [DOI] [PubMed] [Google Scholar]
- 17.Uzunbajakava N., Lenferink A., Otto C. Nonresonant confocal Raman imaging of DNA and protein distribution in apoptotic cells. Biophys. J. 2003;84:3968–3981. doi: 10.1016/S0006-3495(03)75124-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krafft C., Knetschke T., Salzer R. Identification of organelles and vesicles in single cells by Raman microspectroscopic mapping. Vib. Spectrosc. 2005;38:85–93. [Google Scholar]
- 19.Wood B.R., Chernenko T., Lacham-Kaplan O. Shedding new light on the molecular architecture of oocytes using a combination of synchrotron Fourier transform-infrared and Raman spectroscopic mapping. Anal. Chem. 2008;80:9065–9072. doi: 10.1021/ac8015483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wood B.R., McNaughton D. Micro-Raman characterization of high- and low-spin heme moieties within single living erythrocytes. Biopolymers. 2002;67:259–262. doi: 10.1002/bip.10120. [DOI] [PubMed] [Google Scholar]
- 21.Takai Y., Masuko T., Takeuchi H. Lipid structure of cytotoxic granules in living human killer T lymphocytes studied by Raman microspectroscopy. Biochim. Biophys. Acta. 1997;1335:199–208. doi: 10.1016/s0304-4165(96)00138-9. [DOI] [PubMed] [Google Scholar]
- 22.Swain R.J., Kemp S.J., Stevens M.M. Spectral monitoring of surfactant clearance during alveolar epithelial type II cell differentiation. Biophys. J. 2008;95:5978–5987. doi: 10.1529/biophysj.108.136168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Owen C.A., Selvakumaran J., Stevens M.M. In vitro toxicology evaluation of pharmaceuticals using Raman micro-spectroscopy. J. Cell. Biochem. 2006;99:178–186. doi: 10.1002/jcb.20884. [DOI] [PubMed] [Google Scholar]
- 24.Pyrgiotakis G., Bhowmick T.K., Moudgil B.M. Cell (A549)-particle (Jasada Bhasma) interactions using Raman spectroscopy. Biopolymers. 2008;89:555–564. doi: 10.1002/bip.20947. [DOI] [PubMed] [Google Scholar]
- 25.Notingher I., Green C., Hench L.L. Discrimination between ricin and sulphur mustard toxicity in vitro using Raman spectroscopy. J. R. Soc. Interface. 2004;1:79–90. doi: 10.1098/rsif.2004.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hamada K., Fujita K., Kawata S. Raman microscopy for dynamic molecular imaging of living cells. J. Biomed. Opt. 2008;13:044027. doi: 10.1117/1.2952192. [DOI] [PubMed] [Google Scholar]
- 27.Zoladek, A., F.C. Pascut, …, I. Notingher. 2010. Non-invasive time-course imaging of apoptotic cells by confocal Raman micro-spectroscopy. J. Raman Spectrosc.http://onlinelibrary.wiley.com/doi/10.1002/jrs.2707/abstract.
- 28.Notingher I., Bisson I., Hench L.L. In situ spectral monitoring of mRNA translation in embryonic stem cells during differentiation in vitro. Anal. Chem. 2004;76:3185–3193. doi: 10.1021/ac0498720. [DOI] [PubMed] [Google Scholar]
- 29.Notingher I., Bisson I., Hench L.L. In-situ study of embryonic stem cells differentiation by Raman micro-spectroscopy. Vib. Spectrosc. 2004;35:199–203. [Google Scholar]
- 30.Schulze H.G., Konorov S.O., Turner R.F. Assessing differentiation status of human embryonic stem cells noninvasively using Raman microspectroscopy. Anal. Chem. 2010;82:5020–5027. doi: 10.1021/ac902697q. [DOI] [PubMed] [Google Scholar]
- 31.Kim B.S., Lee C.C., Tarantal A.F. Growth, differentiation, and biochemical signatures of rhesus monkey mesenchymal stem cells. Stem Cells Dev. 2008;17:185–198. doi: 10.1089/scd.2007.0076. [DOI] [PubMed] [Google Scholar]
- 32.Chan J.W., Lieu D.K., Li R.A. Label-free separation of human embryonic stem cells and their cardiac derivatives using Raman spectroscopy. Anal. Chem. 2009;81:1324–1331. doi: 10.1021/ac801665m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Puppels G.J., Olminkhof J.H., Greve J. Laser irradiation and Raman spectroscopy of single living cells and chromosomes: sample degradation occurs with 514.5 nm but not with 660 nm laser light. Exp. Cell Res. 1991;195:361–367. doi: 10.1016/0014-4827(91)90385-8. [DOI] [PubMed] [Google Scholar]
- 34.Liang H., Vu K.T., Berns M.W. Wavelength dependence of cell cloning efficiency after optical trapping. Biophys. J. 1996;70:1529–1533. doi: 10.1016/S0006-3495(96)79716-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Neuman K.C., Chadd E.H., Block S.M. Characterization of photodamage to Escherichia coli in optical traps. Biophys. J. 1999;77:2856–2863. doi: 10.1016/S0006-3495(99)77117-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dick E., Rajamohan D., Denning C. Evaluating the utility of cardiomyocytes from human pluripotent stem cells for drug screening. Biochem. Soc. Trans. 2010;38:1037–1045. doi: 10.1042/BST0381037. [DOI] [PubMed] [Google Scholar]
- 37.Tu A.T. John Wiley and Sons; New York: 1982. Raman Spectroscopy in Biology: Principles and Applications. [Google Scholar]
- 38.Wolleben C.D., Jaspers S.R., Miller T.B., Jr. Use of adult rat cardiomyocytes to study cardiac glycogen metabolism. Am. J. Physiol. 1987;252:E673–E678. doi: 10.1152/ajpendo.1987.252.5.E673. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.