

Abstract
Protein–metabolite networks are central to biological systems, but are incompletely understood. Here, we report a screen to catalog protein–lipid interactions in yeast. We used arrays of 56 metabolites to measure lipid-binding fingerprints of 172 proteins, including 91 with predicted lipid-binding domains. We identified 530 protein–lipid associations, the majority of which are novel. To show the data set's biological value, we studied further several novel interactions with sphingolipids, a class of conserved bioactive lipids with an elusive mode of action. Integration of live-cell imaging suggests new cellular targets for these molecules, including several with pleckstrin homology (PH) domains. Validated interactions with Slm1, a regulator of actin polarization, show that PH domains can have unexpected lipid-binding specificities and can act as coincidence sensors for both phosphatidylinositol phosphates and phosphorylated sphingolipids.
Keywords: interactome, lipid–array, network, pleckstrin homology domains, sphingolipids
Introduction
Biological function emerges from the concerted action of numerous interacting biomolecules. Deciphering the molecular mechanisms behind cellular processes requires the systematic charting of the multitude of interactions between all cellular components. While protein–protein and protein–DNA networks have been the subject of many systematic surveys, other critically important cellular components, such as lipids, have to date rarely been studied in large-scale interaction screens.
Lipids are one of the most abundant classes of cellular metabolites, with a wide range of structural and functional diversity. Their metabolism and transport account for about 5% of all coding genes in eukaryotes (van Meer, 2005). They are essential building blocks of biological membranes and some also function as anhydrous stores of energy. Besides these house-keeping functions, growing numbers of lipids are known to operate as signaling molecules, including phosphatidylinositol phosphates (PtdInsPs) and sphingolipids. Many of these functions, such as the recruitment of proteins to the membrane via binding to PtdInsPs, are conserved from yeast to human. Lipids are unevenly distributed among the various cell membranes. Their correct partitioning relies on a tight spatial organization of the enzymes involved in lipid metabolism, which suggests extensive lipid–lipid and protein–lipid interactions. The importance of these interactions is evident from the variety of protein domains that have evolved to bind particular lipids (Lemmon, 2008) and from the large list of disorders arising from altered protein–lipid interactions. Human pathologies, such as cancer and bipolar disorder, have been linked to mutations in genes involved in PtdInsPs synthesis (Lee et al, 2007) or in domains specialized in their recognition, such as the pleckstrin homology (PH) domain (Di Paolo and De Camilli, 2006; Carpten et al, 2007). These interactions are attractive targets for pharmaceutical drug development. For instance, small molecule inhibitors of phosphatidylinositide 3-kinases are currently in clinical trials as anti-cancer drugs (Raynaud et al, 2007).
The current understanding of protein–lipid recognition comes from the study of a limited number of lipids, principally PtdInsPs (Zhu et al, 2001), and lipid-binding domains (LBDs) in isolation (Dowler et al, 2000; Yu and Lemmon, 2001; Yu et al, 2004). For some signaling lipids, such as sphingolipids, intracellular targets and molecular mechanisms are only partially understood (Hannun and Obeid, 2008). The importance of lipids in biological processes and their under-representation in current biological networks suggest the need for systematic, unbiased biochemical screens (Dippold et al, 2009). Here, we describe the use of miniaturized arrays for the study of protein–lipid interactions and report the lipid-binding profiles for 172 soluble proteins. The screen successfully recovers known protein–lipid-binding events and uncovers many others not yet reported, several of which we validated using a variety of other techniques. Many of the new interactions promise to illuminate greatly the current understanding of lipids in signaling and other cell processes.
Results
Profiling of protein–lipid interactions in Saccharomyces cerevisiae
To screen protein–lipid interactions, we adapted a lipid overlay assay (Kanter et al, 2006) (Figure 1). We developed miniaturized nitrocellulose arrays that contained duplicated sets of 51 lipids and their metabolic intermediates that cover the main lipid classes in yeast as defined in the KEGG database of metabolic pathways (Kanehisa et al, 2008). For comparison, we also included five non-physiological analogs that are not synthesized in yeast (Supplementary Table S1A). We focused on the lipids that are exposed to cytosolic proteins and excluded complex sphingolipids, such as inositol phosphoceramide, mannosyl-inositolphosphoceramide and mannosyl-diinositolphosphoceramide, which localize in the extra-cellular leaflet of yeast membranes. We used the arrays to determine the binding profiles of 172 soluble proteins, expressed as carboxy-terminal tandem-affinity-purification (TAP)-tag fusion proteins in S. cerevisiae (Gavin et al, 2006). Bound proteins were immunodetected with an antibody that recognizes the TAP tag (see Materials and methods; Supplementary information). The selection included 91 proteins that were available from the collection of TAP fusions and which contained one or several possible LBDs as defined by SMART (Letunic et al, 2006), Pfam (Finn et al, 2008) or SuperFamily (Gough et al, 2001). The set of 91 covered 78% of all yeast proteins predicted or known to have an LBD. We also selected 32 soluble lipid-regulated proteins and enzymes involved in lipid metabolism, along with a set of 49 arbitrarily chosen soluble proteins (unclassified) (Figure 1; Supplementary Table S1B).
Figure 1.

Identification of protein–lipid interactions in S. cerevisiae using lipid–arrays and live-cell imaging. (A) Charting protein–lipid interactions with lipid–arrays. (B) Summary of the different lipids analyzed. Lipids are grouped according to their metabolic pathways. Numbers of different lipids analyzed for each pathway are indicated. (C) Summary of the different proteins analyzed. Proteins are grouped according to their LBDs. For each category, the numbers of proteins analyzed are indicated. (D) Summary of the validation procedure. The number of proteins analyzed in each assay is represented. Asterisk points to the series of 29 proteins that bound PtdInsPs (and not sphingolipids) and that we used as control.
We applied standardized protocols that gave an average reproducibility of 74%, measured on the repeated analysis of 26 different TAP fusions (Supplementary Table S2). We also expressed a subset of proteins in a heterologous system, Escherichia coli (Figure 1), which provides additional evidence for the interactions found in yeast. This also approximates the fraction of the direct interactions, that is not mediated by endogenous yeast proteins that will be absent in E. coli. Importantly, as many mechanisms might account for failure to recapitulate binding in E. coli (protein mis-folding or incorrect post-translational modifications), reproducibility in E. coli provides a lower limit for the fraction of direct interactions. Bacterially expressed proteins recovered 58% of the associations initially observed with TAP fusions produced in yeast (P≪0.000001; Supplementary Table S2). Assuming that the assay reproducibility is the same in yeast and E. coli, this suggests that a minimum of 78% (58%/74%) of the total observed interactions in yeast are direct.
We captured interactions with reported dissociation constants ranging from the high nanomolar to the mid-micromolar range (Supplementary Tables S1B and S3). We detected the weak interactions taking place between the yeast tricalbin Tcb3 and PtdInsPs at low calcium concentration (Schulz and Creutz, 2004), interactions that were specifically enhanced by the addition of calcium (data not shown).
Data filtering and specificity of the lipid–array assay
We considered several potential sources of false negatives and false positives in the lipid–array assay (see Supplementary information): biases owing to (i) desorption of the lipids from the nitrocellulose membranes, (ii) promiscuous lipids or proteins and (iii) non-specific interactions solely due to the TAP tag, to hydrophobic or electrostatic nature of some proteins or lipids. Based on the first two considerations, we eliminated eight most water-soluble metabolites (ethanolamine phosphate, CDP-choline, CDP-ethanolamine, CoA, acetyl-CoA, acetoacetyl-CoA, 3-OH-3methylglutaryl-CoA and L-serine) that showed little or no binding to proteins, and might have desorbed from the nitrocellulose membrane, and four promiscuous lipids (phosphatidic acid, phosphatidylserine, phosphatidylglycerol and desmosterol) that bound to >34% of the proteins screened (Supplementary Figure S1). The cut-offs were set to ensure best coverage of the literature-derived reference data set (see below). Regarding the third possibility, the level of expression of individual TAP fusions was not found to correlate with lipid-binding frequencies (data not shown). The set of proteins that bound lipids was not biased for hydrophobic (Supplementary Figure S2) or abundant (Ghaemmaghami et al, 2003) (data not shown; see Supplementary information) proteins and binding frequency was not dependent on lipid hydrophobicity or ionic state (Supplementary Figure S1). Overall, this illustrates that the assay did not produce general biases due to the presence of the TAP tag or the hydrophobic or electrostatic nature of some proteins or lipids. Rather, the binding profiles of related metabolites, such as different intermediates in a metabolic pathway or the five non-physiological analogs, revealed that discrete changes in lipid head groups affected protein specificity (Supplementary Figures S3 and S4). As might be expected, non-physiological lipid analogs tend generally to bind to fewer proteins than their natural counterparts. Lipids with similar structures do tend to share target proteins, but some discrete changes in lipid head groups can confer distinct protein-binding specificities. For example, for two metabolites of phosphatidylethanolamine, phosphatidyl-N-methylethanolamine and phosphatidyl-N-dimethylethanolamine, the presence of methyls in the head group affected protein binding (Supplementary Figure S3A). Similarly, we observed mutually exclusive binding for dihydrosphingosine-1P (DHS-1P) and a non-physiological analog unsaturated at position 4–5, sphingosine-1P (Supplementary Figure S3B). A double bond at this position affects the degree of freedom of the head group, likely accounting for the different binding properties. Head group phosphorylation also contributed to binding specificities of sphingolipids (Supplementary Figure S3B) and PtdInsPs. Phosphorylation of the latter at position three on the inositol ring conferred distinctive protein-binding profiles (Supplementary Figure S4) (Lemmon, 2008). Consistent with previous observations (Narayan and Lemmon, 2006), this suggests that lipids arrayed on nitrocellulose membranes have their hydrophilic head groups accessible for biomolecular interactions.
Assessment of overall data quality
After data filtering (i.e. removal of the four promiscuous and the eight most water-soluble lipids; see above), we obtained 530 interactions, among 124 proteins and 30 lipids (Figure 2; Supplementary Table S2B). Among all lipids studied, PtdInsPs were the most frequent binders (Figure 2; Supplementary Figure S5): 79% of the lipid-regulated proteins and 58% of the proteins with an LBD interacted with one or more PtdInsPs. Proteins with an LBD generally bound lipids more frequently: 66% were bound to more than one lipid. Proteins with an LBD bound to a median of three lipids, whereas unclassified proteins bound to one (P=0.032; Supplementary Figure S6).
Figure 2.

Protein–lipid map in yeast. The table displays all interactions color coded according to the number of experimental supports: reproducibility with proteins expressed in homologous and heterologous systems, binding to artificial (liposome) and biological (live-cell imaging) membranes and genetic interactions. The data set is provided as Supplementary information (Supplementary Tables S2C and S4; see also Supplementary Data 1). The bar charts show the fraction of novel interactions per lipid or protein classes for which supporting evidence is available. The total numbers of novel interactions are displayed next to each bar. The pie charts represent the fraction of conserved proteins. PtdInsP, phosphatidylinostitol phosphate; GPL, glycerophospholipid; SL, sphingolipid; FA, fatty acid; GL, glycerolipid; S&P, sterol and prenol.
We further assessed the quality of the data by comparing the results with known protein–lipid interactions (Figure 3A). We measured the false negative rate (fraction of true interactions missed) by comparison with a set of 40 protein–lipid interactions obtained from the literature and the STITCH database (Kuhn et al, 2008) (Supplementary Table S3A and B). The lipid–array data recovered 60% of this literature-derived reference data set (P≪0.000001). Missed interactions include those requiring additional binding events unlikely to occur in vitro. For example, in higher eukaryotes, the interaction between C1 domains and diacylglycerol entails both deep insertion of the domain in the membranes and the binding of basic residues to phosphatidylserine head groups (Kazanietz et al, 1995). For comparison, we also used a set of 31 interactions between enzymes or transporters in lipid metabolism and their substrates or products. Only 29% of those were captured, illustrating the method's limited ability to recover labile enzyme/substrate-binding events as well as interactions that imply binding in the hydrophobic pocket of lipid transfer proteins. We also compared with a published set of interactions measured in yeast using proteome arrays and PtdInsPs (Zhu et al, 2001). Of the 152 proteins common to both analyses, 77 interacted with PtdInsPs in either data set, our screen identified 76. The study of Zhu et al found five, of which four were also found in our analysis (5.2% overlap). It is important to emphasize that the Zhu et al data set (150 lipid-binding proteins) recovers none of the interactions from the literature-derived reference data set and that it is largely devoid of interactions involving LBDs. Instead, it is enriched in hydrophobic and often unknown proteins (Supplementary Figure S2), suggesting that this different assay has captured an interaction space different from that charted here.
Figure 3.

Assessment of the lipid–array data quality. (A, B) Comparison with sets of literature curated and genetic interactions. (A) Summary of the two reference data sets and strategies used to assess quality. Genetic coverage is defined as the percentage of physical protein–lipid interactions covered by the data set of genetic interactions. (B) Estimation of accuracy based on PtdInsPs metabolic pathway. The left column shows the number of proteins in the literature-derived reference set that are covered (dark blue) or not (magenta) by the data set of genetic interactions. The right column shows the fraction of proteins devoid of LBD and that did not bind PtdInsPs in the lipid–array that interact genetically with enzymes involved in the synthesis of these lipids (light blue, background coverage by genetics). The central column shows the estimation of accuracy (fraction of true positives) in the lipid–array data set assuming that genetic coverage results from the combination of true positives (that will have the same genetic coverage as the literature-derived reference data set, in dark blue) and false positive (with a background coverage, in light blue). The estimated fraction of true positive that is not covered by genetics is shown in magenta. Numbers on top of the columns are overall genetic coverage for each set of proteins (*P=0.015 and **P=0.035).
A fraction of genetic networks are known to coincide with physical interaction networks (Kelley and Ideker, 2005; Fraser and Plotkin, 2007), a property we exploited as an estimate of accuracy. From SGD (http://www.yeastgenome.org) and the literature (Nash et al, 2007; Costanzo et al, 2010), we assembled a list of 328 genetic interactions (for example, synthetic or suppressive genetic interactions; Figure 3A) between 96 enzymes involved in lipid metabolism and the 172 analyzed proteins (Supplementary Table S3C). We defined positive overlaps (between genetic and physical interactions) as those in which a lipid from a physical protein–lipid interaction resided inside a pathway containing one or more genes sharing a genetic interaction with the protein (Figure 3A; see also Materials and methods; Supplementary information). If the lipid–array and the literature-derived reference data sets are comparable in terms of quality and biological relevance, they should be similarly covered by genetic interactions (Figure 3B). Using the genetic coverage of the literature-derived reference data set, mainly implying PtdInsPs (Supplementary Table S3A and B), we extrapolated the fraction of true interactions (accuracy) across all lipid classes in our data to 61.4% (see Materials and methods; Supplementary information). We found that the agreement between the lipid–array and the genetic data sets is significant (P<0.01). In particular, the literature-derived reference data set and the proteins interacting with PtdInsPs on the lipid–array show a similar threefold (P=0.015) and twofold (P=0.035) enrichment in genetically consistent interactions, respectively. This is consistent with the view that the lipid–array interactions were often functionally informative. Overall, the set of lipid–array interactions shows similar quality in terms of false positives and false negatives as those previously reported for large protein–protein interaction sets (von Mering et al, 2002; Tarassov et al, 2008).
Multiple validations of the protein–lipid data set
To determine whether the protein–lipid pairs measured in vitro could represent true interactions in vivo, we related the in vitro binding profiles to physiologically derived in vivo data. We first integrated genetic interactions (see above); the lipid–array data set provides a molecular hypothesis for 136 genetic interactions previously identified (41% of the genetic data set; P<0.01). This is considerably more than could be inferred from the literature-derived reference data set that contributed a basis for only 14 interactions (4.2% of genetic data set). For 10 proteins (representing 34 protein–lipid pairs) selected because they represented intriguing novelties or specificities, we used more physiological assays (Table I): protein recruitment to liposomal (Supplementary Figure S7A–E) and biological (Supplementary Figure S7F and G) membranes. We could verify 24 interactions involving eight proteins Ecm25, Ira2, Slm1, Ypk1, Rvs161, Rvs167, Las17 and Pkh2 (Table I). For example, we confirmed the interaction initially observed between Pkh2, a serine/threonine protein kinase required for endocytosis, and PtdIns, PtdIns(4)P and PtdIns(4,5)P2, but not PtdIns(3)P (Supplementary Figure S7A). Pkh2 recruitment to liposomal and biological membranes requires specific PtdInsPs and involves a predicted globular domain at the C-terminus of Pkh2 that might act as a new type of LBD. For one lipid, PtdIns(4,5)P2, specificity was further assessed. Specifically, the soluble analog of PtdIns(4,5)P2, inositol(1,4,5)P3, inhibited binding of the C-terminal domain of Pkh2 to PtdIns(4,5)P2-containing liposomes. Interestingly, for the human homolog of Pkh2, the kinase PDK1, a C-terminal PH domain fulfills a similar binding function. We also confirmed the selective binding of Las17, a member of the WASP/WAVE family that regulates the Arp2/3 complex and actin function, to PtdIns(3,5)P2 and PtdIns(3,4,5)P3 (a non-physiological analog in yeast), and not PtdIns(4,5)P2 (Supplementary Figure S7B). In human, similar binding profiles have been reported for WAVE2, another member of the WASP/WAVE family (Oikawa et al, 2004), illustrating the conservation of lipid binding across considerable evolutionary distances. As a substantial fraction (45%) of the analyzed proteins were conserved in humans (Figure 2), the protein–lipid data set will have functional implications for higher eukaryotes and thus for human biology.
Table 1. Summary of protein–lipid interactions selected for validation.
| Protein name | Lipid | Validation assay | |
|---|---|---|---|
| aProtein–lipid interactions that could not be confirmed. | |||
| Liposomes, binding to liposomes; mss4ts, perturbation of cellular PtdInsPs using an mss4ts mutant; myriocin, perturbation of cellular sphingolipids using myriocin. | |||
| Ecm25 | PtdIns | Liposomes | |
| Ecm25 | PtdIns(3,4,5)P3 | Liposomes | |
| Ira2 | PtdIns(3,4,5)P3 | Liposomes | |
| Las17 | PtdIns(3,5)P2 | Liposomes/mss4ts | |
| Las17 | PtdIns(3,4,5)P3 | Liposomes | |
| Pkh2 | PtdIns | Liposomes | |
| Pkh2 | PtdIns(4)P | Liposomes/mss4ts | |
| Pkh2 | PtdIns(3,4)P2 | Liposomes | |
| Pkh2 | PtdIns(3,5)P2 | Liposomes/mss4ts | |
| Pkh2 | PtdIns(4,5)P2 | Liposomes/mss4ts | |
| Slm1 | PtdIns(4,5)P2 | Liposomes/mss4ts | |
| Rvs167 | PtdIns(4,5)P2 | Liposomes/mss4ts | |
| Slm1 | DHS-1P | Liposomes/myriocin | Pkh1/2 independent |
| Rvs167 | DHS-1P | Liposomes/myriocin | Pkh1/2 independent |
| Slm1 | PtdIns(4)P | mss4ts | |
| Rvs167 | PtdIns(4)P | mss4ts | |
| Rvs161 | PtdIns(3)P | mss4ts | |
| Ypk1 | PtdIns(4)P, PtdIns(4,5)P2 | mss4ts | |
| Ecm25a | PHS-1P, Sph, Sph-1P | Myriocin | |
| Las17 | PHS-1P, Sph-1P | Myriocin | Pkh1/2 independent |
| Mss4a | DHS-1P | Myriocin | Pkh1/2 dependent |
| Rvs167 | PHS-1P | Myriocin | Pkh1/2 independent |
| Ypk1 | DHCer, PHCer-C18 | Myriocin | Pkh1/2 independent |
| Spo14a | PHS, Sph, DHCer, PHCer-C8, PHCer-C18, Cer | Myriocin | |
We integrated the validations in a scoring system that ranks all interactions by considering the number of experimental supporting observations (Figure 2). This also included further validation of the selected set of 49 proteins that bound sphingolipids (see below). Overall, 54% of interactions were supported by additional evidence (Supplementary Tables S2C and S4; see also Supplementary Data 1).
Discovery of cryptic LBDs
Overall, 68% of all interactions were novel (i.e. absent from the literature-derived reference) or unexpected from either protein sequences or known LBDs specificities (Figure 2). For example, we found Ecm25, a RhoGAP of unknown function, associated to several different lipids, with a binding profile usually indicative of the presence of an LBD (Supplementary Figure S6; Supplementary Table S2B). Using sequence searches for remote homologs of known LBDs, we found that Ecm25 has a cryptic CRAL/TRIO domain that was previously undetected (Figure 4; see Materials and methods; Supplementary information). Another example is the RasGAP Ira2. The partial structure of the human ortholog, the tumor suppressor neurofibromatosis type 1, revealed the presence of an unexpected bipartite lipid-binding module that consists of both a CRAL/TRIO (Sec14) and a PH-like domains (D’Angelo et al, 2006). Our observation of Ira2 binding to PtdInsPs (Supplementary Table S2B) is consistent with a structure-based alignment that reveals that the CRAL/TRIO and PH-like domains are probably conserved in Ira2 (D’Angelo et al, 2006). These predictions were further tested in a more physiological assay measuring protein recruitment to liposomal membranes. Using recombinant, purified domains, we could confirm that the cryptic Ecm25-CRAL/TRIO and the Ira2-CRAL/TRIO/PH domains alone are responsible for lipid binding (Figure 4B; Table I; see also Supplementary Figure S7C). This illustrates that some LBDs have only weak sequence similarity to canonical examples found in databases like SMART (Letunic et al, 2006) or Pfam (Finn et al, 2008). Their annotation requires supporting biochemical measurements such as those in the data set presented here. This extended repertoire of protein–lipid interactions can be used as the basis for more detailed mechanistic or structural studies.
Figure 4.

Identification of a new cryptic lipid-binding domain in Ecm25. (A) Structure-based alignment of Ecm25 and five known CRAL/TRIO domain. Conserved hydrophobic sites are colored blue, polar sites are red, invariant residues are boxed, sites occupied by small residues are magenta and the ligand-binding sites of the structures are colored gray. Secondary structure elements are numbered according to the canonical C-terminal CRAL/TRIO domain structure of Sec14 (PDB:1aua), and grayed out when disrupted or absent in some of the structures. Sequences prefixed with ‘h’ are from human; all others are from S. cerevisiae. (B) The truncated Ecm25-CRAL/TRIO binds to artificial membranes in flotation assays. Liposomes contain 1 mM 1-palmitoyl-2-oleoyl-snglycero-3-phosphocholine (POPC) and phosphatidylserine (PS), PtdIns or PtdIns(3,4,5)P3, as indicated (3%, molar). Ecm25-CRAL/TRIO was analyzed by western blot.
New effectors of sphingolipids signaling validated in vivo by live-cell imaging
We extended the biological validation in vivo to the set of proteins that bound sphingolipids, a class of bioactive lipids that play important signaling functions in yeast and higher eukaryotes. The exact mode of action for these lipids remains elusive (Hannun and Obeid, 2008) and the data set points to series of new cellular targets. We identified 63 proteins that interacted with sphingoid long-chain bases (LCBs), ceramides or phosphorylated LCBs (Figure 5; Supplementary Table S5).
Figure 5.

Lipid–arrays and live-cell imaging identify new series of sphingolipid effectors. (A) The effect of the perturbation of sphingolipid metabolism on GFP-fusion localization was monitored in yeast cells treated with myriocin, myriocin and dihydrosphingosine (DHS) or untreated (Blank). (B) The proteins-binding sphingolipids in vitro are grouped according to their sphingolipid-binding specificities, and ordered by function. The symbol # points to proteins with a PH domain. Interactions are color coded as in Figure 2, but excluding live-cell imaging. The green boxes label the proteins selected for further analysis by live-cell imaging. Those sensitive to 2 h myriocin treatments are represented with filled green boxes. For each sphingolipid-binding group, the histogram next to the matrix summarizes the overall sensitivities to myriocin. For comparison, myriocin sensitivity of 32 controls is also shown in magenta. Non-physiological lipids in S. cerevisiae are in gray and italic. DHS-1P, dihydrosphingosine-1-phosphate; PHS-1P, phytosphingosine-1-phosphate; Sph-1P, sphingosine-1-phosphate; 3-ketoDHS, 3-ketodihydrosphingosine; DHS, dihydrosphingosine; PHS, phytosphingosine; Sph, sphingosine; DHCer, dihydroceramide; PHCer C8, phytoceramide C8; PHCer C18, phytoceramide C18.
These proteins included the five previously known sphingolipid effectors in yeast: the LCBs-responsive kinases Pkh1/Pkh2 and Ypk1/Ypk2 (Friant et al, 2001; Liu et al, 2005) (orthologs of the human PDK1 and SGK, respectively) that we found associated with LCBs or phosphorylated LCBs, as well as phospholipase D (Spo14), a known target of sphingolipids in mammals (Abousalham et al, 1997). The cellular functions of the proteins targeted by sphingolipids included endocytosis, cell polarity and lipid metabolism (Figure 5).
Using live-cell imaging, we determined the effect of perturbation of sphingolipid metabolism with the antibiotic myriocin on the cellular localization of 49 candidate sphingolipid targets fused to GFP. The specificity of the myriocin effect was assessed by the addition of a metabolite (DHS) that bypasses its inhibitory effect and restores sphingolipid synthesis (Figure 5A). As controls, we used a series of 29 proteins that bound PtdInsPs only, as well as three other proteins localized to the membrane and cell cortex (Figure 5B; Supplementary Figure S8A). We quantified the effects of myriocin using a standardized method for 32 proteins that showed similar punctate localization patterns (Supplementary Figure S8B; see Supplementary information). For the remaining 49 GFP fusions that had more diverse localization patterns, we assessed the effects qualitatively (Supplementary Table S6). Importantly, interactions induced upon cell stimulation or stress, as well as those that might affect protein properties other than localization (e.g. activity), are not traceable in this assay. Nevertheless, proteins that bound sphingolipids in vitro were nearly four times more frequently sensitive to myriocin treatment than the set of controls (P<0.009; Figure 5) and the effect of myriocin was not mimicked by the inhibition of the known effectors of sphingolipid in yeast, the Pkh1/Pkh2 pathway (Supplementary Figure S7G). Overall, this is consistent with proteins that bound sphingolipids in vitro also being direct sphingolipid targets in vivo.
Examples involving proteins conserved in higher eukaryotes were frequent (Figure 2). The two actin-associated proteins, Rvs161 and Rvs167, homolog of the mammalian amphiphysins, form a protein complex involved in endocytosis. They both possess a Bin/Amphiphysin/Rvs-homology domain that generates and senses membrane curvature by binding negatively charged lipid head groups (Lemmon, 2008). In vitro, we found Rvs167 associated with PtdInsPs and phosphorylated LCBs; interactions that were confirmed using artificial membranes (Supplementary Figure S7D). Consistent with these interactions having a role in vivo, inhibition of either PtdInsPs or sphingolipid synthesis specifically perturbed Rvs161 and Rvs167 association with punctate structures at the plasma membrane (Figure 5A; Supplementary Figure S7F and G). Protein levels were unaffected by the treatments (data not shown). These results provide a molecular explanation for several genetic interactions previously reported between amphiphysins and enzymes in sphingolipid metabolism (Desfarges et al, 1993) and support a direct targeting role of sphingolipids in endocytosis that might be conserved in higher eukaryotes.
Discovery of new ligands for PH domains
Despite the importance of sphingolipids in signaling processes, only a few domains, such as START or Saposins, have been reported to specifically bind these lipids in higher eukaryotes, and none of them have been found in yeast. Interestingly, almost 60% of proteins binding to phosphorylated LCBs in our assay also contained a PH domain and bound PtdInsPs (Figure 6A). Overall, 18 proteins with PH domains, out of the 29 that bound lipids on the array, bound phosphorylated PtdInsPs and LCBs (Figure 6B). These associations to phosphorylated LCB were often physiological; the proteins that bound both PtdInsPs and phosphorylated LCB were four times more sensitive to myriocin treatment than those that bound only PtdInsPs (42 versus 10%; P<0.02). This suggests that some PH domains might have unanticipated ligands and also have a function in sphingolipid recognition.
Figure 6.

The PH domain of Slm1 cooperatively binds PtdInsPs and phosphorylated sphingolipids. (A) A significant fraction of proteins-binding phosphorylated LCBs also contained a PH domain. For each sphingolipid-binding group, the pie chart shows the fraction of proteins with a PH domain (*P<0.00001). (B) Lipid-binding specificities of PH-domain containing proteins; more than half of those binding PtdInsPs also interacted with LCB-P. Proteins that did not show any binding were omitted. (C, D) Recruitment of Slm1-PH and PLCδ-PH to liposome membranes of different compositions: 3% (molar) PtdIns(4,5)P2 and/or 3% (molar) DHS-1P. (C) Quantitative measurement of liposome binding by isothermal titration calorimetry. Points represent heat evolved per mol of injected protein. ND: not determined. (D) Monitoring of Slm1-PH and PLCδ-PH co-eluting with liposomes by size exclusion chromatography and western blot. (E) Role of PtdInsPs and sphingolipids in the recruitment of Slm1 and PLCδ-PH to biological membranes in vivo. On the left, Slm1 fused to GFP (Slm1-GFP) co-localizes with eisosomes (Pil1-RFP). Interference with PtdIns(4,5)P2 metabolism using a temperature-sensitive mutant of the phosphatidylinositol 4-phosphate 5-kinase (mss4ts). Intensity plot profiles were extracted from the area represented by the respective white lines. On the right, localization of PLCδ-PH fused to GFP. Cell treatments are as in Figure 5A.
To test this hypothesis further in more physiological assays, that is the binding to artificial membranes in vitro and to cellular membranes in vivo, we selected two PH domains with different specificities: that from Slm1 (Slm1-PH), a component of the TORC2 signaling pathway (Fadri et al, 2005) that bound both DHS-1P and PtdInsPs (Supplementary Table S2B), and the more prototypic PH domain of PLCδ (PLCδ-PH) known to recognize PtdIns(4,5)P2 (Lemmon et al, 1995). As expected from its known specificity, the efficient recruitment of the PLCδ-PH to both artificial (Kd=0.2 μM; Figure 6C and D; see also Supplementary Figure S9) and biological (Figure 6E; Supplementary Figure S8C) membranes required only PtdIns(4,5)P2. In contrast, Slm1-PH showed an unusual behavior. Its targeting to liposomal membranes depends on the specific presence of both PtdIns(4,5)P2 and DHS-1P (Kd=1.8 μM; Figure 6C and D; see also Supplementary Figure S9). Other similar, negatively charged lipids, such as phosphatidylserine, have no effect illustrating specificity for DHS-1P (Supplementary Figure S7E). Finally, in vivo, both PtdIns(4,5)P2 and sphingolipid metabolisms are required for Slm1 association with specific membrane microdomains, the eisomomes (Figure 6E; Supplementary Figure S8D; Supplementary Movies S1 and S2). Other Slm1 locations such as its assembly in very dynamic membrane domains, distinct from the eisosomes, were apparently unaffected by metabolic perturbations (Supplementary Movies S1 and S2).
Overall, the observations above suggest cooperative lipid-binding by Slm1-PH. Consistent with these observations, the structure of Slm1-PH, which we solved by X-ray crystallography at 2 Å resolution, suggests the presence of an additional positively charged cavity in Slm1-PH (Figure 7A; Supplementary Table S7). The two putative Slm1-PH-binding pockets considerably vary in size. Manual docking with DHS-1P and PtdIns(4,5)P2 supports the view that the two lipids with head groups of different sizes (PtdIns(4,5)P2 is substantially bigger than DHS-1P) can simultaneously bind Slm1-PH. Interestingly, the aliphatic chains of the two lipids point toward two conserved hydrophobic residues (F481 and L482), surprisingly exposed in the loop that separates the two putative binding sites. We also identified two positively charged residues, often conserved among PH domains that point to the canonical binding site in PLCδ-PH (Figure 7A and B; Supplementary Figure S10) and were found to each contribute to one putative binding site in Slm1-PH (Figure 7A). Mutation of these residues to alanine affects the number of positive charges in each predicted Slm1-PH-binding site and specifically destabilizes the membrane association of Slm1 with the eisosome (Figure 7C). These point mutations also cause significant (P<0.01) cumulative defects in Slm1 function: yeast growth and actin polarization (Figure 7D). The stronger and apparently additive defect of the double mutant suggest that both positively charged sites are required for proper Slm1 functioning.
Figure 7.

The 2.0 Å X-ray structure of Slm1-PH reveals two putative binding sites required for Slm1 function in vivo. (A) Comparison of the structures of Slm1-PH with that of PLCδ-PH (Protein Data Bank (PDB) entry 1mai). The two structures are represented in the same orientations. PLCδ-PH binds lipid within a positively charged canonical site conserved in Slm1 (orange circle). Slm1 presents an additional positively charged cavity (gray circle). Two key residues (R477 and R478) were identified that contribute to both positively charged binding sites in Slm1. Side chain of analogous residues in PLCδ (K30 and K32) point exclusively to the canonical binding site. Electronegative regions are colored red and electropositive regions blue. (B) Sequence alignment of Slm1-PH and PLCδ-PH. Mutated amino acids in Slm1 (R477 and R478) and their analogous in PLCδ are boxed in black. Conserved residues are blue if positively charged and green if hydrophobic. (C) The two putative Slm1-PH-binding sites are required for Slm1 membrane association with the eisosome. Kymograph representations of Slm1-GFP and Pil1-RFP (1 frame per 1.5 s). Green lines above the kymograph represent the association dynamics of Slm1-GFP with the Pil1-RFP (red lines). Mutations affecting clusters of positive charges in one (R477A or R478A) or both (R478A/R477A) resulted in specific destabilization of Slm1 association with Pil1-containing microdomains. (D) The two putative Slm1-PH-binding sites are required for Slm1 function in vivo. Strains carrying Slm1 mutations in one (R477A or R478A) or both (R477A/R478A) putative lipid-binding sites show cumulative phenotypic defects. Upper panels show growth defect in the presence of myriocin; lower panels during heat stress. The bar chart shows actin polarization defects following heat stress. * decrease for the double mutants is significant compared with wild-type (P<0.01), R477A (P<0.01) and R478A (P<0.01) single mutants. The mutations have no effect on Slm1 expression levels (western blot not shown).
Collectively, these results indicate that the PH domain of Slm1 might work as a coincidence sensor to integrate both PtdInsP and sphingolipid signaling pathways. It might contribute to the cross-talk between the signaling of these two lipids that has been previously inferred based on genetic interactions in yeast (Tabuchi et al, 2006). Although the structural and mechanistic details for this binding remain to be fully characterized, recent structural data similarly support the existence of a second, non-canonical binding site in certain PH domains (Ceccarelli et al, 2007). Our results also reinforce the emerging notion that cooperative mechanisms have important functions in PH domains functioning (Maffucci and Falasca, 2001). These mechanisms initially described between PtdInsPs and proteins can now be extended to new lipid classes, illustrating the benefit of unbiased and systematic analyses.
Lipid-binding profiles as predictor for protein recruitment to biological membranes
Having shown the accuracy and in vivo relevance of the detected lipid-binding profiles, we sought to use this information as a fingerprint for predicting other protein properties such as protein localization. In the past, pioneering attempts to use PtdInsP-binding profiles for predicting membrane targeting or sub-cellular localization had limited success (Yu et al, 2004; Park et al, 2008). While PtdInsP-binding certainly has an important function, targeting to biological membranes in vivo also requires additional, often cooperative interactions; this suggests roles for other ligands (Maffucci and Falasca, 2001) that might have been captured with the screen (see above).
We grouped proteins by first scoring the similarity between pairs of lipid-binding profiles using a metric that considers both lipid and protein promiscuity, and used these scores for complete linkage clustering. Proteins sharing similar lipid-binding profiles also showed a tendency to share attributes, such as the presence of PH domains or localization in punctate structures in membranes (Figure 8A). These include the phosphatidylinositol-4-phosphate 5-kinase Mss4, known to localize in the cytoplasm and the membrane (Huh et al, 2003). The Mss4-binding profile clustered with proteins localized in dotted structures, an observation we could confirm by high resolution live-cell imaging (Figure 8B) (Sun et al, 2007). Another example is a cluster of proteins that show similar localization at the yeast bud, all of which were also insensitive to myriocin treatment. This illustrates the promise for comprehensive lipid-binding profiles to contribute to the molecular rationale for protein localization or dynamic behavior at biological membranes.
Figure 8.

Lipid-binding fingerprints as predictors of protein localization, domains and functions. (A) Proteins clustered according to their lipid-binding profiles. The wedge shapes highlight some groups that are enriched (P<0.05) in a particular annotation. The black oblong represents the group of proteins that did not bind any lipid in this study. This group is deficient in proteins annotated by SGD (Nash et al, 2007) as lipid binding. The violet cluster of 12 proteins from Caf120 to Kes1 is also enriched for myriocin-insensitive proteins (P=0.01). (B) Live-cell imaging of Mss4 fused to GFP reveals localization in punctate membrane structures.
Discussion
Accurate representations of biological processes require systematic charting of the physical and functional links between all cellular components. There is a clear need to expand the current biomolecular networks beyond protein–protein or protein–nucleic acid interactions, and involve additional biomolecules. It is important to widen bottlenecks in biochemical characterization by large-scale approaches. This work shows the feasibility and benefits of large-scale analyses combining biochemical arrays and live-cell imaging for charting protein–lipid interactions.
The number of novel interactions discovered clearly shows that even major classes of metabolites, such as PtdInsPs and sphingolipids, have been insufficiently studied, calling for further system-wide analyses. Many of the binding events reported could not be inferred from sequence comparison and/or the presence of canonical LBDs, arguing that the sites and modalities of protein–lipid recognition are still largely elusive. Even for known LBDs, our data suggest additional binding to new ligands. The observation that some PH domains might also orchestrate the input from other signaling and metabolic pathways involving sphingolipids adds to the view that PH domains can recognize other ligands besides PtdInsPs (Lemmon, 2004). Indeed, only 10% of the 234 human PH domain-containing proteins show strong and specific binding to PtdInsPs (Lemmon, 2008). Our data supports the notion that binding to several different lipids could well represent an attribute shared by other LBDs. Integration of different signals would ensure efficient, but also regulated targeting to biological membranes. Overall, the study has shown the importance of extending molecular interaction space from proteome- to metabolome-wide efforts and of systematic classifications of bioactive molecules based on their binding profiles. The data provided here represents an excellent resource to enhance the understanding of lipids function in eukaryotic systems.
Materials and methods
Lipid–arrays
The protocol to produce lipid–arrays was developed from Kanter et al (2006). Briefly, 1 mM solutions of lipids were prepared in adequate solvent mixtures. Using an argon flow, 0.1 μl of each lipid was sprayed on a nitrocellulose membrane (Hybond-C Extra, GE Healthcare) with an ATS4S spotter (CAMAG). We also spotted a nitrobenzoxadiazole-labeled phosphatidylglycerol (Sigma) at different positions on the array and monitored the quality of the spotting procedure by scanning at 432 nm excitation (GenePix 4000B, Molecular Devices). The three different solvent mixtures used (chloroform, chloroform:methanol 1:1 and chloroform:methanol:water–HCl 1:1:0.2) were also sprayed as blank controls. All the samples were spotted in duplicate. The arrays were stored at 4°C under argon atmosphere and protected from light.
Lipid overlay assay
The S. cerevisiae strains expressing the desired TAP-tagged protein were grown at 30°C to an OD600 of 3.5–3.8. Pelleted cells were disrupted by glass beads beating. Cell extracts were obtained by a 30 min centrifugation at 22 000 r.p.m. at 4°C and filtration (HPF Millex®—0.45 μm). The lipid overlay assay was adapted from Dowler et al (2000). The arrays were blocked for 1 h in 2 ml of blocking buffer (3% fatty-acid-free BSA, 150 mM NaCl, 10 mM Tris pH 7.4). The arrays were then incubated for 1 h in the presence of cell extracts, washed and the bound TAP-tagged proteins were immunodetected with PAP or with V5-specific antibodies (Invitrogen).
Molecular biology and recombinant protein expression
All primers used are listed in Supplementary Table S8. TAP-tagged proteins selected for recombinant expression in E. coli (Supplementary Tables S1B and S2A) and the PH domain of Slm1 (Slm1-PH) were cloned in pET100-D/TOPO or pET101-D/TOPO vector (Invitrogen) following the manufacturer's instructions. Mutations in Slm1 were introduced using the QuikChange® lightning Site-Directed Mutagenesis kit (Stratagene). For detailed information on the cloning, mutagenesis, expression and purification of the recombinant proteins, as well as strains used in this study, see Supplementary information.
Live-cell imaging
Perturbation of sphingolipid metabolism with myriocin
The localization of endogenously expressed proteins was examined using yeast strains expressing GFP fusions (Huh et al, 2003). Cells attached on 35 mm glass bottom culture dishes coated with Concanavalin A were treated with 5 μM myriocin or 5 μM myriocin and 5 μM DHS(Sigma). The effect of myriocin was measured after 2 h treatment, which represents the minimal exposure time that induced, in our experimental setting, the delocalization of two proteins that bound sphingolipids in vitro: Mss4 and Slm1. Under these conditions, cells remained perfectly viable (data not shown) and other membrane resident were unaffected (Figure 6E; Supplementary Figure S8A). For a more detailed description of the procedure, see Supplementary information.
Imaging was performed with an Olympus IX81 microscope equipped with 100 × /NA 1.45 objective lens and Hamamatsu Orca-ER camera.
For 49 GFP fusions that did not localize in punctate structures, the effect of myriocin was assessed qualitatively. Those proteins were considered sensitive to myriocin if the effect was restored by DHS. Yhr131c did not fulfill this requirement. We quantified the effects of myriocin using a standardized method for 32 proteins that showed similar punctate localization patterns (see Supplementary information).
Perturbation of PtdIns(4,5)P2 metabolism
The mss4ts cells coding for the respective C-terminal GFP-tagged protein were grown and attached to dishes at 25°C, following the same protocol described above. Dishes were kept at the selected temperature (25 or 37°C) for 2 h and imaged immediately after. Same protocol was followed for PLCδ-PH-GFP. In this case, mss4ts strain was transformed with the plasmid coding for PLCδ-PH-GFP.
Perturbation of Pkh1/Pkh2 signaling pathway
The pkh1ts/Δpkh2 cells coding for the respective C-terminal GFP-tagged proteins were grown and attached to dishes at 25°C, following the same protocol described above. Dishes were kept at the selected temperature (25 or 37°C) for 1 h and imaged immediately after. At 37°C, pkh1ts/Δpkh2 cells are defective in actin polarization (Inagaki et al, 1999). One hour represents the first time point, in our experimental condition, in which we observed the delocalization of the actin-binding protein Abp1. Under these conditions, cells remained viable (data not shown).
Cell based assays to assess Slm1-PH function
Actin polarization assay was performed as previously described (Fadri et al, 2005; see Supplementary information). Yeast wild-type strain and strains carrying point mutations in Slm1-PH domain were grown on SC plates containing 500 ng ml–1 myriocin or equivalent amounts of methanol at 30°C for 3 days. Strains carrying Slm2 deletion (Δslm2) were grown in YPD plates at 25 or 37°C for 1 day.
Liposome preparation
A mixture of the lipids was prepared in chloroform:methanol:water, 1:1:0.07, containing 0.03% HCl. We added 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (Avanti Polar Lipids) to a final concentration of 3.8 mM. Where indicated, PtdIns, PtdIns(3)P, PtdIns(4)P, PtdIns(5)P, PtdIns(3,4)P2, PtdIns(3,5)P2, PtdIns(4,5)P2, PtdIns(3,4,5)P3, DHS-1P (Avanti Polar Lipids) and phosphatidylserine (Sigma) were also included. Lipid mixtures were dried under an argon stream followed by 30 min high vacuum. Dried mixtures were rehydrated in binding buffer (10 mM HEPES, 150 mM NaCl, pH 7.4) by mixing at 60°C for 2 h. Lipids were subjected to 5 min sonication and three snap-freeze/thaw cycles in liquid N2 and shaking at 60°C. Finally, small unilamellar vesicles were generated using a mini-extruder (Avanti Polar Lipids) and a membrane pore size of 0.1 μm.
Liposome-binding assays: flotation assay and size exclusion chromatography and western blot
Flotation assay was performed as previously described (Miller et al, 2002) (see Supplementary information). Size exclusion chromatography was performed on Pharmacia FPLC system by using Superdex 200 HR 10/30 column, equilibrated with binding buffer at the flow rate 0.25 ml × min−1. After 30 min incubation at 22°C with 8 μM Slm1-PH or PLCδ-PH, 250 μl of the different liposome solutions were injected. We collected 0.5 ml fractions that were then analyzed by SDS–PAGE and western blot. A V5-specific antibody produced in mouse (Invitrogen) was used to detect Slm1-PH. Total band intensity was integrated with Photoshop software and normalized versus the total amount of protein loaded. Presented results are the sum of all detected fractions of Slm1-PH or PLCδ-PH co-eluted with liposomes.
Isothermal titration calorimetry
Isothermal titration calorimetry (ITC) was performed using a VP-ITC Microcal calorimeter (Microcal). Injectant (Slm1-PH or PLCδ-PH) was dialyzed extensively against binding buffer before all titrations. The experiments were performed at 25°C. A typical titration consisted of injecting 6–12 μl aliquots of 47 μM protein into the different solutions of liposomes, at intervals of 5 min to ensure that the titration peak returned to the baseline. The ITC data were corrected for the injectant dilution heat. To estimate Kd, we used the concentration of binding sites on liposome surface as a fitting parameter, assuming that the interactions occur in a stoichiometry of 1:1. The analysis was performed with the Origin 5.0 software.
Slm1-PH crystallization and structure determination
Crystals were grown at 20°C by vapor diffusion using the sitting-drop method. For crystallization, 0.5 μl of protein solution (9 mg ml–1) were mixed with 0.5 μl of precipitant solution (2 M (NH4)2SO4, 2% PEG 400, 0.1 M Hepes pH 7.5). A single crystal was cryo-protected in mother liquor supplemented with 30% glycerol and flash frozen in liquid nitrogen at 100 K. Diffraction data were collected at beamline ID14-2 of the European Synchrotron Radiation Facility (ESRF, Grenoble France) using an ADSC Q4r CCD detector, and subsequently processed with XDS (Kabsch, 2010). The structure was solved by molecular replacement with the program PHASER (McCoy et al, 2007) using a search model obtained from the PDB entry 1btk (Hyvonen and Saraste, 1997) after conversion to polyalanine and removal of poorly conserved regions among PH domains. The search model included the following residues in the PDB entry 1btk: 5–14, 25–42, 53–57, 63–65, 101–104, 111–134. The initial solution was completed by iterative cycles of manual building in COOT (Emsley and Cowtan, 2004) and refinement using PHENIX (Adams et al, 2002), yielding a final model with R and Rfree values of 22.1 and 27.1, respectively (Supplementary Table S7). The stereochemistry of the final model was checked with PROCHECK (Laskowski et al, 1993). The atomic coordinates and structure factors have been deposited in the Protein Data Bank under accession code 3nsu.
In Figure 7, the electrostatic potential calculated with APBS (Baker et al, 2001) is represented on the solvent-accessible surface. Blue and red indicate positive (+4 kT/e) and negative (−4 kT/e) potential, respectively. Images were generated using Pymol (DeLano, 2002).
Estimation of accuracy based on interactions with PtdInsPs pathway
We thought to use the genetic coverage of the literature-derived reference data set to extrapolate the fraction of true interactions (accuracy) in our data (see below). We reasoned that if the lipid–array and the literature-derived reference data set are comparable in terms of quality and biological relevance, they should be similarly covered by genetic interactions. As the literature-derived reference data set mainly consists of PtdInsPs, we used accuracy measured for this lipid class as an approximation for the entire data set. For this analysis, intermediate cutoff was used for Costanzo et al (2010) data set along with data from SGD and literature (Supplementary Tables S2C and S4; see also Supplementary Data 1).
For different sets of proteins, we measured the fraction that interacts genetically with enzymes involved in the synthesis of PtdInsPs (Figure 3B): (i) proteins that bound PtdInsPs in the literature-derived reference data set (10/16=62.5%=reference genetic coverage); (ii) proteins that bound PtdInsPs in the lipid–array (40/86=46.5%=experimental genetic coverage); (iii) a set of proteins defined as those proteins devoid of LBD and that did not bind PtdInsPs in the lipid–array (4/19=21.1%=background genetic coverage). We observed that the literature-derived reference data set has significantly more genetic interactions than the background genetic coverage (P=0.015). The same was true for proteins that bound PtdInsPs in the lipid–array versus the background genetic coverage (P=0.035). Interestingly, the lipid–array data did not show any significant difference when compared with the literature-derived reference data set (P=0.18). Fisher's exact test was used to measure significance.
We can now interpolate the fraction of true interactions (accuracy) expected in the PtdInsPs lipid–array data set. The coverage of genetic interactions in our data set results from two different components: interactions of ‘true positive’ (x) and ‘false positive’ (1−x) proteins. Assuming that the ‘false positive’ will have a genetic coverage equal to the background genetic coverage and that ‘true positive’ will have a genetic coverage equal to the reference genetic coverage, we predict that 61.4% of the proteins are ‘true positives’ (see below). If all of the 86 proteins that bound PtdInsPs in the lipid–array are equally likely to be among the ‘true positives’, the ‘true positive’ rate among our protein–lipid interactions will also be 61.4% (Figure 3B).
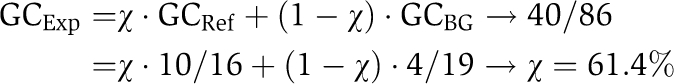 |
where
χ=‘true positive’ in the lipid–array data set (accuracy).
(1−χ)=‘false positive’ in the lipid–array data set.
GCExp=experimental genetic coverage.
GCRef=reference genetic coverage.
GCBG=background genetic coverage.
Prediction of a CRAL/TRIO in Ecm25
The putative CRAL/TRIO domain of Ecm25 was detected by running HHsearch (Soding, 2005) for all yeast proteins against the SCOP 1.69 database. For detailed information on the sequence-based alignment of the non-redundant set of structures annotated by Pfam as having a CRAL/TRIO domain, see Supplementary information.
Clustering of proteins and lipids according to their binding profiles
For every protein, we calculated the fraction f1 of all the lipids with which it interacted and the fraction f0 of all the lipids with which it did not interact. Likewise, for every lipid, we calculated the fraction f1 of all the proteins with which it interacted and the fraction f0 of all the proteins with which it did not interact. Then at every position (i, j) in the interaction matrix, we have a score s1i,j for an interaction between protein i and lipid j=log(f1i)+log(f1j), and a score s0i,j for no interaction=log(f0i)+log(f0j). Thus, an interaction between a promiscuous protein and a promiscuous lipid has a lower score than an interaction between a highly selective protein and lipid. We then scored the similarity between the lipid-binding profiles of all pairs of proteins i1 and i2 by summing the scores for every lipid j in the profile, where the score for lipid j=
 |
We then clustered the proteins by complete linkage using the program OC (Barton, 2002), on the basis of these scores. We followed the same procedure to cluster the lipids on the basis of their protein-binding profiles. The calculation of the binomial probability for a significant deficiency or enrichment with a particular attribute and correction for testing for a particular feature in multiple places is described in Supplementary information.
For detailed description on other bioinformatic procedures (e.g. multiple sequence alignment), see Supplementary information.
Supplementary Material
Acknowledgments
We are grateful to Giulio Superti-Furga for comments on the manuscript, and to Thorsten Brach and Marko Kaksonen's group; Damien Devos, Eduardo Garcia and Rob B Russell's group; Gavin's group; Eileen Furlong's group; the EMBL GeneCore and Protein Expression and Purification Core facilities; Michael Knop, Sofia Rybina and Eric Karsenti; Scott Emr; Howard Riezman and Mark Lemmon for expert help and the sharing of reagents. We acknowledge ESRF and EMBL staff for beamline assistance. This work is partially funded by Federal Ministry of Education and Research (BMBF) in the framework of the National Genome Research Network (NGFN) to ACG (BMBF NGNF IG-Cellular Systems Genomics, 01GS0865). OG is a fellow of the Ministerio de ciencia e innovación, Spain. KM is a fellow of the Danish Natural Science Research Council (09-064986/FNU). The protein interactions from this publication have been submitted to the IntAct database (pmid: 19850723) and assigned the identifier EBI-2933237.
Author contributions: OG, MKa and ACG designed the experiments. OG, JGJ, CM, CAG, PBA, KM, JT, VR performed the experiments. OG, MJB, SB, LJJ, MKu, RBR analyzed the data. OG, MJB, PB, RBR, ACG wrote the paper. OG, CFT, CWM determined Slm1-PH structure. Original idea was formulated by OG and ACG.
Footnotes
The authors declare that they have no conflict of interest.
References
- Abousalham A, Liossis C, O’Brien L, Brindley DN (1997) Cell-permeable ceramides prevent the activation of phospholipase D by ADP-ribosylation factor and RhoA. J Biol Chem 272: 1069–1075 [DOI] [PubMed] [Google Scholar]
- Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC (2002) PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr 58: 1948–1954 [DOI] [PubMed] [Google Scholar]
- Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci USA 98: 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton GJ (2002) OC—a cluster analysis program, University of Dundee, Scotland, UK www.compbio.dundee.ac.uk/downloads/oc
- Carpten JD, Faber AL, Horn C, Donoho GP, Briggs SL, Robbins CM, Hostetter G, Boguslawski S, Moses TY, Savage S, Uhlik M, Lin A, Du J, Qian YW, Zeckner DJ, Tucker-Kellogg G, Touchman J, Patel K, Mousses S, Bittner M, et al. (2007) A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature 448: 439–444 [DOI] [PubMed] [Google Scholar]
- Ceccarelli DF, Blasutig IM, Goudreault M, Li Z, Ruston J, Pawson T, Sicheri F (2007) Non-canonical interaction of phosphoinositides with pleckstrin homology domains of Tiam1 and ArhGAP9. J Biol Chem 282: 13864–13874 [DOI] [PubMed] [Google Scholar]
- Costanzo M, Baryshnikova A, Bellay J, Kim Y, Spear ED, Sevier CS, Ding H, Koh JL, Toufighi K, Mostafavi S, Prinz J, St Onge RP, VanderSluis B, Makhnevych T, Vizeacoumar FJ, Alizadeh S, Bahr S, Brost RL, Chen Y, Cokol M et al. (2010) The genetic landscape of a cell. Science 327: 425–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Angelo I, Welti S, Bonneau F, Scheffzek K (2006) A novel bipartite phospholipid-binding module in the neurofibromatosis type 1 protein. EMBO Rep 7: 174–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano WL (2002) The PyMOL Molecular Graphics System http://www.pymol.org
- Desfarges L, Durrens P, Juguelin H, Cassagne C, Bonneu M, Aigle M (1993) Yeast mutants affected in viability upon starvation have a modified phospholipid composition. Yeast 9: 267–277 [DOI] [PubMed] [Google Scholar]
- Di Paolo G, De Camilli P (2006) Phosphoinositides in cell regulation and membrane dynamics. Nature 443: 651–657 [DOI] [PubMed] [Google Scholar]
- Dippold HC, Ng MM, Farber-Katz SE, Lee SK, Kerr ML, Peterman MC, Sim R, Wiharto PA, Galbraith KA, Madhavarapu S, Fuchs GJ, Meerloo T, Farquhar MG, Zhou H, Field SJ (2009) GOLPH3 bridges phosphatidylinositol-4- phosphate and actomyosin to stretch and shape the Golgi to promote budding. Cell 139: 337–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowler S, Currie RA, Campbell DG, Deak M, Kular G, Downes CP, Alessi DR (2000) Identification of pleckstrin-homology-domain-containing proteins with novel phosphoinositide-binding specificities. Biochem J 351: 19–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60: 2126–2132 [DOI] [PubMed] [Google Scholar]
- Fadri M, Daquinag A, Wang S, Xue T, Kunz J (2005) The pleckstrin homology domain proteins Slm1 and Slm2 are required for actin cytoskeleton organization in yeast and bind phosphatidylinositol-4,5-bisphosphate and TORC2. Mol Biol Cell 16: 1883–1900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RD, Tate J, Mistry J, Coggill PC, Sammut SJ, Hotz HR, Ceric G, Forslund K, Eddy SR, Sonnhammer EL, Bateman A (2008) The Pfam protein families database. Nucleic Acids Res 36: D281–D288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser HB, Plotkin JB (2007) Using protein complexes to predict phenotypic effects of gene mutation. Genome Biol 8: R252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friant S, Lombardi R, Schmelzle T, Hall MN, Riezman H (2001) Sphingoid base signaling via Pkh kinases is required for endocytosis in yeast. EMBO J 20: 6783–6792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavin AC, Aloy P, Grandi P, Krause R, Boesche M, Marzioch M, Rau C, Jensen LJ, Bastuck S, Dumpelfeld B, Edelmann A, Heurtier MA, Hoffman V, Hoefert C, Klein K, Hudak M, Michon AM, Schelder M, Schirle M, Remor M et al. (2006) Proteome survey reveals modularity of the yeast cell machinery. Nature 440: 631–636 [DOI] [PubMed] [Google Scholar]
- Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O’Shea EK, Weissman JS (2003) Global analysis of protein expression in yeast. Nature 425: 737–741 [DOI] [PubMed] [Google Scholar]
- Gough J, Karplus K, Hughey R, Chothia C (2001) Assignment of homology to genome sequences using a library of hidden Markov models that represent all proteins of known structure. J Mol Biol 313: 903–919 [DOI] [PubMed] [Google Scholar]
- Hannun YA, Obeid LM (2008) Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol 9: 139–150 [DOI] [PubMed] [Google Scholar]
- Huh WK, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, O’Shea EK (2003) Global analysis of protein localization in budding yeast. Nature 425: 686–691 [DOI] [PubMed] [Google Scholar]
- Hyvonen M, Saraste M (1997) Structure of the PH domain and Btk motif from Bruton's tyrosine kinase: molecular explanations for X-linked agammaglobulinaemia. EMBO J 16: 3396–3404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki M, Schmelzle T, Yamaguchi K, Irie K, Hall MN, Matsumoto K (1999) PDK1 homologs activate the Pkc1-mitogen-activated protein kinase pathway in yeast. Mol Cell Biol 19: 8344–8352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W (2010) XDS. Acta Crystallogr D Biol Crystallogr 66: 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, Katayama T, Kawashima S, Okuda S, Tokimatsu T, Yamanishi Y (2008) KEGG for linking genomes to life and the environment. Nucleic Acids Res 36: D480–D484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanter JL, Narayana S, Ho PP, Catz I, Warren KG, Sobel RA, Steinman L, Robinson WH (2006) Lipid microarrays identify key mediators of autoimmune brain inflammation. Nat Med 12: 138–143 [DOI] [PubMed] [Google Scholar]
- Kazanietz MG, Wang S, Milne GW, Lewin NE, Liu HL, Blumberg PM (1995) Residues in the second cysteine-rich region of protein kinase C delta relevant to phorbol ester binding as revealed by site-directed mutagenesis. J Biol Chem 270: 21852–21859 [DOI] [PubMed] [Google Scholar]
- Kelley R, Ideker T (2005) Systematic interpretation of genetic interactions using protein networks. Nat Biotechnol 23: 561–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn M, von Mering C, Campillos M, Jensen LJ, Bork P (2008) STITCH: interaction networks of chemicals and proteins. Nucleic Acids Res 36: D684–D688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA, Moss DS, Thornton JM (1993) Main-chain bond lengths and bond angles in protein structures. J Mol Biol 231: 1049–1067 [DOI] [PubMed] [Google Scholar]
- Lee JY, Engelman JA, Cantley LC (2007) Biochemistry. PI3K charges ahead. Science 317: 206–207 [DOI] [PubMed] [Google Scholar]
- Lemmon MA (2004) Pleckstrin homology domains: not just for phosphoinositides. Biochem Soc Trans 32: 707–711 [DOI] [PubMed] [Google Scholar]
- Lemmon MA (2008) Membrane recognition by phospholipid-binding domains. Nat Rev Mol Cell Biol 9: 99–111 [DOI] [PubMed] [Google Scholar]
- Lemmon MA, Ferguson KM, O’Brien R, Sigler PB, Schlessinger J (1995) Specific and high-affinity binding of inositol phosphates to an isolated pleckstrin homology domain. Proc Natl Acad Sci USA 92: 10472–10476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I, Copley RR, Pils B, Pinkert S, Schultz J, Bork P (2006) SMART 5: domains in the context of genomes and networks. Nucleic Acids Res 34: D257–D260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Zhang X, Lester RL, Dickson RC (2005) The sphingoid long chain base phytosphingosine activates AGC-type protein kinases in Saccharomyces cerevisiae including Ypk1, Ypk2, and Sch9. J Biol Chem 280: 22679–22687 [DOI] [PubMed] [Google Scholar]
- Maffucci T, Falasca M (2001) Specificity in pleckstrin homology (PH) domain membrane targeting: a role for a phosphoinositide-protein co-operative mechanism. FEBS Lett 506: 173–179 [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ (2007) Phaser crystallographic software. J Appl Crystallogr 40: 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller E, Antonny B, Hamamoto S, Schekman R (2002) Cargo selection into COPII vesicles is driven by the Sec24p subunit. EMBO J 21: 6105–6113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayan K, Lemmon MA (2006) Determining selectivity of phosphoinositide-binding domains. Methods 39: 122–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nash R, Weng S, Hitz B, Balakrishnan R, Christie KR, Costanzo MC, Dwight SS, Engel SR, Fisk DG, Hirschman JE, Hong EL, Livstone MS, Oughtred R, Park J, Skrzypek M, Theesfeld CL, Binkley G, Dong Q, Lane C, Miyasato S et al. (2007) Expanded protein information at SGD: new pages and proteome browser. Nucleic Acids Res 35: D468–D471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oikawa T, Yamaguchi H, Itoh T, Kato M, Ijuin T, Yamazaki D, Suetsugu S, Takenawa T (2004) PtdIns(3,4,5)P3 binding is necessary for WAVE2-induced formation of lamellipodia. Nat Cell Biol 6: 420–426 [DOI] [PubMed] [Google Scholar]
- Park WS, Heo WD, Whalen JH, O’Rourke NA, Bryan HM, Meyer T, Teruel MN (2008) Comprehensive identification of PIP3-regulated PH domains from C. elegans to H. sapiens by model prediction and live imaging. Mol Cell 30: 381–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raynaud FI, Eccles S, Clarke PA, Hayes A, Nutley B, Alix S, Henley A, Di-Stefano F, Ahmad Z, Guillard S, Bjerke LM, Kelland L, Valenti M, Patterson L, Gowan S, de Haven Brandon A, Hayakawa M, Kaizawa H, Koizumi T, Ohishi T et al. (2007) Pharmacologic characterization of a potent inhibitor of class I phosphatidylinositide 3-kinases. Cancer Res 67: 5840–5850 [DOI] [PubMed] [Google Scholar]
- Schulz TA, Creutz CE (2004) The tricalbin C2 domains: lipid-binding properties of a novel, synaptotagmin-like yeast protein family. Biochemistry 43: 3987–3995 [DOI] [PubMed] [Google Scholar]
- Soding J (2005) Protein homology detection by HMM-HMM comparison. Bioinformatics 21: 951–960 [DOI] [PubMed] [Google Scholar]
- Sun Y, Carroll S, Kaksonen M, Toshima JY, Drubin DG (2007) PtdIns(4,5)P2 turnover is required for multiple stages during clathrin- and actin-dependent endocytic internalization. J Cell Biol 177: 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabuchi M, Audhya A, Parsons AB, Boone C, Emr SD (2006) The phosphatidylinositol 4,5-biphosphate and TORC2 binding proteins Slm1 and Slm2 function in sphingolipid regulation. Mol Cell Biol 26: 5861–5875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarassov K, Messier V, Landry CR, Radinovic S, Serna Molina MM, Shames I, Malitskaya Y, Vogel J, Bussey H, Michnick SW (2008) An in vivo map of the yeast protein interactome. Science 320: 1465–1470 [DOI] [PubMed] [Google Scholar]
- van Meer G (2005) Cellular lipidomics. EMBO J 24: 3159–3165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Mering C, Krause R, Snel B, Cornell M, Oliver SG, Fields S, Bork P (2002) Comparative assessment of large-scale data sets of protein-protein interactions. Nature 417: 399–403 [DOI] [PubMed] [Google Scholar]
- Yu JW, Lemmon MA (2001) All phox homology (PX) domains from Saccharomyces cerevisiae specifically recognize phosphatidylinositol 3-phosphate. J Biol Chem 276: 44179–44184 [DOI] [PubMed] [Google Scholar]
- Yu JW, Mendrola JM, Audhya A, Singh S, Keleti D, DeWald DB, Murray D, Emr SD, Lemmon MA (2004) Genome-wide analysis of membrane targeting by S. cerevisiae pleckstrin homology domains. Mol Cell 13: 677–688 [DOI] [PubMed] [Google Scholar]
- Zhu H, Bilgin M, Bangham R, Hall D, Casamayor A, Bertone P, Lan N, Jansen R, Bidlingmaier S, Houfek T, Mitchell T, Miller P, Dean RA, Gerstein M, Snyder M (2001) Global analysis of protein activities using proteome chips. Science 293: 2101–2105 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.