

Abstract
A prime goal in systems biology is the comprehensive use of existing high-throughput genomic datasets to gain a better understanding of chromatin organization and genome function. In this report, we use chromatin immunoprecipitation (ChIP) data that map protein-binding sites on the genome, and Hi-C data that map interactions between DNA fragments in the genome in an integrative approach. We first reanalyzed the contact map of the human genome as determined with Hi-C and found that long-range interactions are highly nonrandom; the same DNA fragments are often found interacting together. We then show using ChIP data that these interactions can be explained by the action of the CCCTC-binding factor (CTCF). These CTCF-mediated interactions are found both within chromosomes and in between different chromosomes. This makes CTCF a major organizer of both the structure of the chromosomal fiber within each individual chromosome and of the chromosome territories within the cell nucleus.
Keywords: chromosome conformation, chromatin, computational biology, genome architecture, networks
Introduction
Recent progress in high-throughput sequencing has opened new avenues in studying genome structure and its implications on gene regulation. Lieberman-Aiden et al (2009) recently presented the first contact map of the human genome. They obtained this map using Hi-C, a method enabling the examination of the spatial proximity of DNA fragments in the nucleus. These results confirmed the existence of chromosome territories and showed that open and closed chromatin compartments are spatially segregated. They determined the distribution of the genomic distances between interacting DNA fragments within chromosomes and proposed that this distribution is compatible with a fractal globular organization of the chromosomal fiber.
Another interesting outcome of Hi-C experiments is that the interactions are highly nonrandom; the same DNA fragments are often found to interact with each other. In this study, we address this question in detail and question whether these specific interactions can be explained by the action of the CCCTC-binding factor (CTCF). CTCF is a highly conserved protein from fly to human and was recently presented as the ‘master weaver' of the genome (Phillips and Corces, 2009). Chromosome conformation capture (3C) techniques have highlighted its role in organizing long DNA loops within chromosomes at specific loci (Phillips and Corces, 2009; Zlatanova and Caiafa, 2009; Ohlsson et al, 2010). Evidence of CTCF-mediated intra- and inter-chromosomal interactions has also been obtained using 4C (an advanced 3C technique) on the mouse Igf2/H19 locus (Kurukuti et al, 2006; Ling et al, 2006; Zhao et al, 2006). In addition to this architectural role, this versatile protein is found to be involved in gene regulation (Phillips and Corces, 2009; Zlatanova and Caiafa, 2009; Ohlsson et al, 2010). Over 13 000 CTCF-binding sites (CTCF sites) on the human genome have been identified using chromatin immunoprecipitation (ChIP) on Chip, enabling the characterization of the specific binding sequence (Kim et al, 2007). This library of binding sites has been enriched using ChIP followed by deep sequencing (ChIP-Seq; Barski et al, 2007) and computational predictions (Xie et al, 2007), yielding an extensive inventory of over 40 000 locations (Bao et al, 2008). We set out to determine whether fragments found to interact in the Hi-C experiments are associated with a CTCF site. We show that the presence of CTCF sites is highly correlated with the ability of fragments to make strong interactions, both within the same chromosome and between different chromosomes.
Results and discussion
We based our analysis on a Hi-C experiment (Lieberman-Aiden et al, 2009) conducted using human lymphoblastoid cell line (GM06990). The restriction enzyme used (HindIII) cuts the human genome in ∼800 000 fragments; ∼37 000 of which bear at least one CTCF site. The experimental procedure yields an inventory of binary interactions between all the fragments. Eight million interaction reads were produced. Almost all the fragments in the genome are found in at least one of these reads and some fragments are found in many interaction reads.
The first question we have addressed is whether observing the same interaction many times in the experiment confers nonrandomness. To answer this question, we first noticed that the results from the experiment can be represented by a network in which each node is a DNA fragment, and each link represents an interaction between two fragments (Figure 1A). Taking any two random nodes from this network, they can be either unlinked, or linked by one link, or even linked by many links. The number of links emanating from a node is called the node degree. Nodes with a high degree correspond to fragments, which are found to interact a lot in the experiment, and we can expect that such high-degree nodes will have many links in common. To statistically quantify the significance of the number of interactions between two fragments (i.e. the number of links between two nodes), we created samples of randomized networks (n=100), which preserve the linkage characteristics of the original network, that is, the number of nodes, links, and node degrees. We subsequently inspected the number of interactions between any two nodes arisen due to pure chance and contrast that to the actual observed value. We observe significantly higher numbers of interactions between nodes in the observed data than those in the randomized networks. Figure 1B shows the distributions of the number of links between each nodes pairs for both the actual network and the randomized networks. The two distributions are found to be significantly different (Kolmogorov–Smirnov test, P<2.2 × 10–16). The same difference is found when considering only interchromosomal interactions (Supplementary Figure 1). This means that the nonrandomness of an interaction between two fragments is not only due to the genomic proximity between those two fragments. At this point, we decided to test the hypothesis that these nonrandom interactions are due to specific factors, the most widely known being CTCF.
Figure 1.

Comparison between the interaction network obtained in the Lieberman-Aiden et al (2009) experiment and a randomized interaction network. (A) Schematic drawing of an interaction network: nodes represent interacting fragments and each link between two nodes corresponds to one interaction read. (B) Distribution (Log10 scale) of the number of reads obtained between each pair of nodes in the actual data (red triangles) and in a randomized network (green dots, error bars correspond to a 95% confidence interval computed on 100 random networks). The statistical difference between those two distributions was assessed using the Kolmogorov–Smirnov test on the same plot normalized by the total number of interaction pairs. The obtained P-value was lower than 2.2 × 10–16.
We therefore set out to determine whether the fragments that are found in many interaction reads are more likely to have a CTCF-binding site. We took the following approach:
First, we removed all binary interactions from the data that were present only once as some of these may well be attributed to noise in the experiment.
Second, we set a threshold n and considered only the fragments that are present in at least n interaction reads.
Lastly, for each value of n ranging from 1 to 100, we computed the corresponding number of fragments (Figure 2A, black line) and the percentage of those fragments that contain at least one CTCF site (Figure 2B, black line).
Figure 2.

CTCF presence is correlated with the most frequently observed interactions in the human genome. (A) Number of fragments that are present in at least n interaction reads in the Hi-C experiments on lymphoblastoid cell line (log scale on the y-axis). In black, all interactions are considered. In green, only inter-chromosomal interactions are considered. (B) The percentage of interacting fragments that contain at least one CTCF site is presented as a function of n. In black, all interactions are considered. In green, only interchromosomal interactions are considered.
From Figure 2A, we estimate that about 200 000 fragments are found in at least two interaction reads; however, only ∼100 are found in at least 100 reads. Interestingly, the decline in the number of fragments for increasing n is not monotonic but clearly has two different components: a fast one for n<10 and a slow one for n>10. In other words, two different kinds of fragments can be distinguished: strongly interacting fragments that correspond to the slow component and weakly interacting fragments that correspond to the fast component. Strong interactions can either result from a stable interaction in a subpopulation of cells or a weaker, but more frequent interaction in a majority of cells. We then computed the proportion of fragments containing CTCF-binding sites for increasing n and found that strongly interacting fragments are enriched in CTCF sites with respect to weakly interacting fragments (Figure 2B, black line). As n becomes higher than 20, the percentage of fragments containing CTCF reaches ∼40%. These results strongly support the proposed role of CTCF as a major factor in mediating long-range interactions among distant DNA elements (Phillips and Corces, 2009; Zlatanova and Caiafa, 2009; Ohlsson et al, 2010) and show that hundreds of such interactions are formed within the nucleus of human lymphoblastoid cells.
We then repeated the same analysis considering only interchromosomal interactions. The results are presented in Figure 2A and B with green lines. Out of the ∼200 000 fragments found to interact with another fragment, ∼100 000 are involved in interchromosomal interactions (Figure 2A, green line). The same high proportion of interchromosomal interactions holds for the strong interactions found in the Hi-C experiment. To verify whether these strong interchromosomal interactions are mediated through CTCF, we computed the percentage of fragments containing CTCF sites involved in these interactions (Figure 2B, green line). We observed that as n increases, the percentage of fragments containing CTCF sites continues to increase eventually reaching ∼60%. These results suggest that strong interchromosomal interactions found in the human genome can be mediated by CTCF. These results point toward CTCF being a key interactor in mediating chromosome–chromosome interactions and in organizing chromosome territories in the cell nucleus.
The genomic coordinates of CTCF-binding sites that we used to compute these correlations come from three different human data sets (Supplementary Table I). These data sets were obtained from different cell types and using different modus operandi. As shown in Figure 3A, the two experimental data sets (Barski et al, 2007; Kim et al, 2007) have an overlap of about 50%, whereas the computationally predicted positions (Xie et al, 2007) for CTCF sites have weaker correlation with experimentally determined positions. To check whether these three data sets contribute differently to the correlation we observed (Figure 2B), we computed the proportion of fragments containing CTCF-binding sites for increasing n for each data set separately (Figure 3B). To our surprise, only one (Barski et al, 2007) of these three data sets account for all the observed correlation. This difference might be explained either by the technique used (ChIP-Seq versus ChIP-on-Chip or computational predictions) or by the difference in cell type used in different experiments (Supplementary Table I). In fact, it is likely that both happen. First, differences in CTCF sites have been reported between fibroblast and erythroid cell lines by using the exact same protocol (Hou et al, 2010). Lymphoblastoid cells on which interactions were determined (Lieberman-Aiden et al, 2009) are more closely related to the CD4+ T lymphocytes used in the ChIP-Seq analysis (Barski et al, 2007) than to the fibroblast cells used in the ChiIP-on-Chip experiment (Kim et al, 2007). Second, deep sequencing that allows probing of the entire genome is used both in Hi-C and ChIP-Seq, whereas ChIP-on-Chip is only suitable to probe positions predetermined by the oligomers that are found on the microarray. We noticed that many interacting fragments were found on regions that were not covered by the microarray used in the experiment by Kim et al (2007).
Figure 3.

The correlation between strong chromosomal interactions and each of the three data sets taken from CTCFBSDB. In red: data set of Kim et al (2007), in green: data set of Barski et al (2007) and in blue data set of Xie et al (2007) (A) Venn diagram presenting number of fragments containing one or more CTCF-binding site for each data set and corresponding overlap. (B) The percentage of interacting fragments that contain at least one CTCF site is presented as a function of n. In black, all three data sets are combined. In colored, each data set is used separately.
To contextualize the correlation we found between strongly interacting fragments and the presence of CTCF, we repeated the same analysis with other DNA-binding factors. First, we used six ChIP-Seq data sets from two factors known to activate transcription (SRF and GABP) in three different cell lines: HeLa cells, lymphoblastoid cells and liver carcinoma cell (Valouev et al, 2008). The results presented in Figure 4A do not show similar correlation compared with the one seen with CTCF. This suggests that the correlation we found with CTCF is not simply due to the matching of experimental conditions between ChIP-seq and Hi-C protocols. Second, we mapped on the genome all 132 known DNA-binding factors that have a specific consensus binding sequence longer than 15 bp (see Materials and methods section) and used the top 50 000 genomic coordinates to repeat the same analysis for each factor.
Figure 4.

Correlation between strongly interacting fragments and the presence of specific DNA-binding factors. (A) CTCF versus other ChIP-Seq data sets. SRF and GABP genomic locations were mapped on three different cell types: in red, Hep G2 cells; in blue, HeLa cells and in green, lymphoblastoid cells. (B) Same analysis conducted with computationally predicted binding sites for transcription factors from the TRANSFAC database. The black line and error bars correspond to random sequences of 20 bp (see Materials and methods section).
Three conclusions can be drawn from the results (Figure 4B):
None of these factors' presence on a fragment correlates with the ability of this fragment to interact strongly as much as CTCF presence, as determined in the experiment of Zhao et al (2006), does.
For most of the factors, this correlation is comparable to the same correlation computed for a random 20 bp sequence (Figure 4B, in black). This is also the case for the correlation computed with the consensus sequence for CTCF determined from the experiment by Kim et al (2007). This is consistent with the fact that the experimentally determined positions for CTCF from Kim et al (2007) data didn't correlate with strong interactions.
For some of the factors, this correlation is greater than the same correlation computed for a random sequence. The three factors for which the correlation is the highest are: HNF4, PPARγ and Freac4. Interestingly, these three transcription factors are all known to be expressed in lymphocytes (Su et al, 2004; Jo et al, 2006; Humphreys et al, 2009) and to activate gene transcription. This agrees with the concept that these transcription factors would trigger the formation of transcription factories that recruit active genes, thus mediating strong interactions between the different genes expressed in lymphocytes.
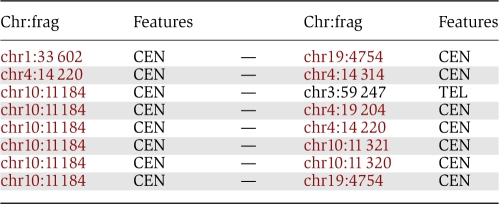
Lastly, we looked in more detail at the eight strongest interactions detected in lymphoblastoid cells (Table I). Seven of these interactions are found between fragments that both contain CTCF sites (for more details on the number of pairs of interacting fragments having both CTCF sites see Supplementary Figure 2). One interaction involves only one fragment containing CTCF sites. Five of these interactions are interchromosomal interactions. We observed that some fragments (such as chr10:11 184, chr4:14 220) are found to interact with multiple fragments on different chromosomes. These fragments contain several CTCF sites (nine for chr10:11 184 and four for chr4:14 220), suggesting that CTCF can mediate the formation of chromosomal hubs of interactions across chromosomes. Analyzing the 8 interactions listed in Table I, we identified one hub gathering fragments from chromosomes 1, 3, 4, 10, and 19. This hub involves centromeres and telomeres, suggesting that repeat sequences have a central function in genome folding and ordering as proposed by Kumar et al (2010). Many examples of repetitive DNA sequence clustering have indeed been reported (de Laat and Grosveld, 2007).
Table 1. List of the eight most frequent interactions found in the Hi-C experiment on lymphoblastoid cell line.
 |
In conclusion, our results show that the Hi-C data can be used together with ChIP data to characterize the role of CTCF as the master weaver of the human genome and to identify chromosomal hubs of interactions and factors participating in the formation of those hubs.
Materials and methods
Randomization of the interaction network
To create a randomized network, we used a random rewiring procedure on the original network described as follows:
For each node A, we rewired each emanating edge.
For each of these edges (A, B), we picked a random node B′ (A ≠ B′) in the network and rewired the edge to connect A to B′.
If B′ was different from B, B′ had one extra edge and B had one less edge. We then randomly removed an edge connecting B′ to A′ (A′ ≠ B) and created a new edge connecting A′ to B.
Repeat steps 1–3 until all edges have been rewired.
After each rewiring run, we inspected the pairs of nodes that were connected in the original network and gathered the number of interactions found between them in each randomized instance of the network. We can then compare the number found in the original network to the average of the randomized networks (see Supplementary Figure 3 for a schematic of the procedure).
Computing the correlation between strongly interacting fragments and CTCF-binding sites
Hi-C data sets were downloaded from Gene Expression Omnibus; GEO accession: GSE18199. CTCFs genomic locations were taken from CTCFBSDB (http://insulatordb.uthsc.edu/help.php).
Fragments obtained from Hi-C were labeled according to the presence of CTCF-binding sites. The CTCF-binding sites that span multiple contiguous fragments were assigned to each of those fragments. The percentage of fragments involved in at least n interaction reads was then computed for each n. We tested for two possible biases in our analysis: the fragments lengths and the repetitive sequences (see Supplementary Figures 4 and 5).
Computing the correlation between strongly interacting fragments and other transcription factor binding sites
The GABP and SRF data sets were downloaded from Gene Expression Omnibus, GEO accession: GSE8489. Position-specific scoring matrices (PSSM) were downloaded from TRANSFAC release 10.2 (Wingender et al, 1996).
We selected all human transcription factors which have a consensus sequence longer than 15 bp. This resulted in a total of 132 transcription factors. Each TF matrix was used to find binding sites on the human genome (assembly hg18) using PATSER (Hertz and Stormo, 1999), and the top scored 50 000 matches were retained and mapped on the fragments. This resulted in 29 634 to 46 778 fragments containing at least one TF-binding site.
The random hypothesis was assessed using 10 random sequences (with the same GC content as the human genome). The black line on Figure 4B presents the average and s.d. value of the percentages obtained using those 10 random sequences.
Supplementary Material
Supplementary table S1, Supplementary figures S1–5
Acknowledgments
We thank Rob White for careful reading of the paper. This study was supported by funds from EMBO and ANR (ASTF 277-2009 and ANR-09-PIRI-0024). IL and PL acknowledge financial support from the EC FP7 SOCIALNETS project, 217141 and Queens' College, Cambridge.
Footnotes
The authors declare that they have no conflict of interest.
References
- Bao L, Zhou M, Cui Y (2008) CTCFBSDB: a CTCF-binding site database for characterization of vertebrate genomic insulators. Nucleic Acids Res 36: D83–D87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K (2007) High-resolution profiling of histone methylations in the human genome. Cell 129: 823–837 [DOI] [PubMed] [Google Scholar]
- de Laat W, Grosveld F (2007) Inter-chromosomal gene regulation in the mammalian cell nucleus. Curr Opin Genet Dev 17: 456–464 [DOI] [PubMed] [Google Scholar]
- Hertz GZ, Stormo GD (1999) Identifying DNA and protein patterns with statistically significant alignments of multiple sequences. Bioinformatics 15: 563–577 [DOI] [PubMed] [Google Scholar]
- Hou C, Dale R, Dean A (2010) Cell type specificity of chromatin organisation mediated by CTCF and cohesin. Proc Natl Acad Sci USA 107: 3651–3656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphreys BD, Lin SL, Kobayashi A, Hudson TE, Nowlin BT, Bonventre JV, Valerius MT, McMahon AP, Duffield JS (2009) Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol 176: 85–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo SH, Yang C, Miao Q, Marzec M, Wasik MA, Lu P, Wang YL (2006) Peroxisome proliferator-activated receptor gamma promotes lymphocyte survival through its actions on cellular metabolic activities. J Immunol 177: 3737–3745 [DOI] [PubMed] [Google Scholar]
- Kim TH, Abdullaev ZK, Smith AD, Ching KA, Loukinov DI, Green RD, Zhang MQ, Lobanenkov VV, Ren B (2007) Analysis of the vertebrate insulator protein CTCF-binding sites in the human genome. Cell 128: 1231–1245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar RP, Senthilkumar R, Singh V, Mishra RK (2010) Repeat performance: how do genome packaging and regulation depend on simple sequence repeats? Bioessays 32: 165–174 [DOI] [PubMed] [Google Scholar]
- Kurukuti S, Tiwari VK, Tavoosidana G, Pugacheva E, Murrell A, Zhao Z, Lobanenkov V, Reik W, Ohlsson R (2006) CTCF binding at the H19 imprinting control region mediates maternally inherited higher-order chromatin conformation to restrict enhancer access to Igf2. Proc Natl Acad Sci USA 103: 10684–10689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, Sandstrom R, Bernstein B, Bender MA, Groudine M, Gnirke A, Stamatoyannopoulos J, Mirny LA, Lander ES, Dekker J (2009) Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326: 289–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling JQ, Li T, Hu JF, Vu TH, Chen HL, Qiu XW, Cherry AM, Hoffman AR (2006) CTCF mediates interchromosomal colocalization between Igf2/H19 and Wsb1/Nf1. Science 312: 269–272 [DOI] [PubMed] [Google Scholar]
- Ohlsson R, Lobanenkov V, Klenova E, Yang C (2010) Does CTCF mediate between nuclear organisation and gene expression? Bioessays 32: 37–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips JE, Corces VG (2009) CTCF: master weaver of the genome. Cell 137: 1194–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su AI, Wiltshire T, Batalov S, Lapp H, Ching KA, Block D, Zhang J, Soden R, Hayakawa M, Kreiman G, Cooke MP, Walker JR, Hogenesch JB (2004) A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci USA 101: 6062–6067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valouev A, Johnson DS, Sundquist A, Medina C, Anton E, Batzoglou S, Myers RM, Sidow A (2008) Genome-wide analysis of transcription factor binding sites based on ChIP-Seq data. Nat Methods 5: 829–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingender E, Dietze P, Karas H, Knuppel R (1996) TRANSFAC: a database on transcription factors and their DNA binding sites. Nucleic Acids Res 24: 238–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, Mikkelsen TS, Gnirke A, Lindblad-Toh K, Kellis M, Lander ES (2007) Systematic discovery of regulatory motifs in conserved regions of the human genome, including thousands of CTCF insulator sites. Proc Natl Acad Sci USA 104: 7145–7150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Tavoosidana G, Sjolinder M, Gondor A, Mariano P, Wang S, Kanduri C, Lezcano M, Sandhu KS, Singh U, Pant V, Tiwari V, Kurukuti S, Ohlsson R (2006) Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra- and interchromosomal interactions. Nat Genet 38: 1341–1347 [DOI] [PubMed] [Google Scholar]
- Zlatanova J, Caiafa P (2009) CCCTC-binding factor: to loop or to bridge. Cell Mol Life Sci 66: 1647–1660 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary table S1, Supplementary figures S1–5