

Abstract
Focal cortical injuries are accompanied by a reorganization of the adjacent neuronal networks. An increased synaptic plasticity has been suggested to mediate, at least in part, this functional reorganization. Previous studies showed an increased long-term potentiation (LTP) at synapses formed by ascending fibres projecting onto layers 2/3 pyramidal cells following lesions in rat visual cortex. This could be important to establish new functional connections within a vertical cortical column. Importantly, horizontal intracortical connections constitute an optimal substrate to mediate the functional reorganization across different cortical columns. However, so far little is known about their potential implication in the functional rewiring post-lesion. Here, we investigated possible alterations of synaptic plasticity of horizontal connections in layers 2/3 in an ‘ex vivo–in vitro’ model of focal laser lesion in rat visual cortex. LTP at these synapses was found to be enhanced post-lesion, whereas long-term depression (LTD) was impaired, revealing a metaplastic shift toward strengthening of these synapses. Furthermore, we disclosed a prolonged decay-time constant of NMDAR-dependent currents, which can contribute to the enhanced LTP. Taken together these data revealed that a laser lesion-induced focal damage of the visual cortex is accompanied by a facilitated potentiation of horizontal synaptic connections in the vicinity of the focal injury. This specific strengthening of synaptic plasticity at horizontal connections in layers 2/3 might be one important cellular mechanism to compensate focal injury-mediated dysfunction in the cerebral cortex.
Introduction
A focal injury of the cerebral cortex causes cell death and leads to neurological deficits, but the severity of this functional loss diminishes over time (Taub et al. 2002; Zepeda et al. 2003; Jablonka et al. 2010). Since neuronal regeneration is absent in most parts of the central nervous system, the observed functional recovery is probably based on a functional and structural reorganization of the neuronal networks surrounding the injured area. Evidence supporting this hypothesis came from in vivo experiments that revealed an increase in the size of receptive fields of intact neurons at the border of the lesion (Eysel & Schweigart, 1999) (for review: Kaas, 1991). In searching for the underlying cellular mechanisms, a previous electrophysiological in vitro study from our laboratory reported an injury-mediated increased long-term potentiation (LTP) of ascending fibres projecting onto layers 2/3 pyramidal neurons in the surround of a laser-induced lesion in the visual cortex of rats (Mittmann & Eysel, 2001). Similar changes in synaptic plasticity have been described in the surround of an experimentally induced focal cortical infarction in the somatosensory cortex of rats (Hagemann et al. 1998). Thus, modification in the efficacy of pre-existing vertical synaptic connections seems to be an important mechanism for the functional reorganization of surviving cortical areas in the surround of a focal brain injury. Lesion-induced plastic changes might also occur at intracortical horizontal fibres, which have not been studied so far. Modelling of neuronal networks also predicted a fundamental role of adapting lateral interactions for cortical reorganization post-lesion (Sirosh & Miikkulainen, 1994). However, experimental data on animal models are still missing.
Here we describe for the first time metaplastic changes at synapses of horizontal connections in layers 2/3 of rat visual cortex at a distance of 2–3 mm from the border of a laser-induced lesion. The facilitated synaptic plasticity at horizontal connections post-lesion is accompanied, similarly to the vertical fibres, by changes in the functional properties of postsynaptic NMDA receptors (NMDARs) (Huemmeke et al. 2004).
Methods
Ethical approval
The animal experiments performed in the present study comply with the policies and regulations of The Journal of Physiology and UK regulations (Drummond, 2009). Furthermore, the experimental protocols were carried out in accordance with the German regulations for experiments with vertebrate animals, and local ethics committee approval was obtained from the regional government.
Cortical lesion induction
Long–Evans rats (n= 56) at the age of 20–21 days were anaesthetized by an intraperitoneal injection of chloral hydrate (4%; 0.1 ml per 10 g) and a subcutaneous injection of lidocain (0.08%, 2 mg kg−1) above the visual cortex. Subsequently, the animals were held in a stereotaxic apparatus, and the skull was exposed and cautiously drilled above the right visual cortex parallel to the midline in a rectangular area of 1 mm width beginning right anterior at the lambda suture and extending 3 mm towards the Bregma without penetrating the dura mater. Cortical lesions were made under visual control with an 810 nm infrared diode laser (OcuLight SLx, Iris Medical Instruments, Mountain View, CA, USA) attached to a binocular operating microscope. Multiple, partially overlapping round lesions were performed about 2–2.5 mm lateral from the midline in order to form an elongated lesion of 1 mm mediolateral width and 3 mm anteroposterior length starting anterior to the lambda suture in the visual cortex (areas V1M, V2ML V2MM) (Paxinos & Watson, 1986). Age-matched sham-operated animals were treated similarly. However, after opening of the skull no laser-lesions were induced.
Immunohistochemistry
For immunohistochemical analysis, slices were briefly incubated in phosphate-buffered saline (PBS, pH 7.3, 4°C) and subsequently fixed in 4% paraformaldehyde, diluted in 0.1 m PBS for 24 h at 4°C. For cryoprotection the slices were subsequently immersed in 30% sucrose (in 0.1 m PBS) for at least 72 h at 4°C. Coronal sections of 30 μm thickness were cut on a freezing microtome and collected in PBS. Some slices were used for standard Nissl and biocytin staining reactions. To detect a lesion-induced gliosis reaction, some slices were incubated overnight with a marker for astrocytes, the primary antibody ‘anti-glial fibrillary acidic protein (GFAP) polyclonal’ (1:2000; DakoCytomation, Hamburg, Germany). Afterwards the slices were incubated in biotinylated goat–anti-rabbit IgG (1:200, Vectastain; Vector Laboratories, Inc., Burlingame, CA, USA) for 90 min followed by an incubation with ABC reagent (1:500, Vectastain) for 90 min. Finally, the immunoreactivity was visualized by transferring the slices to PBS containing 0.05% diaminobenzidine (DAB) and 0.01% H2O2.
Slices preparation
After a survival time of 2–6 days post-surgery, animals were deeply anaesthetized with ether and decapitated. Coronal slices containing the visual cortex (350 μm) were prepared from the lesioned hemisphere by use of a vibrating blade microtome (VT-1000-S; Leica Microsystems AG, Wetzlar, Germany). The tissue was kept at room temperature and incubated for 1 h in artificial cerebrospinal fluid (ACSF) containing (in mm): 125 NaCl, 25 NaHCO3, 2.5 KCl, 1.5 MgCl2, 2 CaCl2, 1.25 NaH2PO4, and 25 d-glucose and bubbled with 95% O2 and 5% CO2 (pH 7.4). Single slices were transferred into a submerged recording chamber superfused with standard ACSF at 32 ± 2°C bubbled with 95% O2 and 5% CO2. The recording chamber was mounted on an upright microscope (Olympus-BX50WI, Olympus, Japan) equipped with 2.5× and 40× water immersion type objectives.
Whole-cell patch clamp recordings
Whole-cell patch-clamp recordings were performed from layers 2/3 pyramidal neurons under visual guidance using DIC optics. Patch pipettes were pulled from borosilicate glass capillaries (GB 150F-8P, Science Products, Hofheim, Germany) and their resistance ranged from 4 to 6 MΩ.
Current clamp mode
The experiments on synaptic plasticity and the intrinsic membrane properties of the recorded neurons were performed in current-clamp mode. The intracellular solution contained (in mm): 140 potassium gluconate, 8 KCl, 2 MgCl2, 4 Na2-ATP, 0.3 Na2-GTP, 10 sodium phosphocreatine and 10 Hepes. The pH was set to 7.3 with KOH. Only neurons with resting membrane potential of at least −65 mV were used for further investigations. The input resistance was continuously monitored during the recordings by injection of a hyperpolarizing current pulse (intensity: 50 pA; duration: 250 ms) after delivering each test stimulus. Only cells with an input resistance changing <20% during the experiment were used for further analysis. Extracellular presynaptic stimulations were performed by placing a glass electrode (resistance ∼3 MΩ) 500 μm lateral to the patch-clamped neuron in the same layer to stimulate horizontal fibres running in layers 2/3. The presynaptic stimulation induced a monosynaptic response with a peak latency of 5 ± 0.5 ms following the stimulation artefact. To prevent polysynaptic activity, we used a low stimulation intensity (80–120 μA, no difference between the two groups), which evoked a relatively small postsynaptic response. Thus, we excluded any parallel activation of adjacent ascending fibres in the synaptic signals. Baseline excitatory postsynaptic potentials (EPSPs) were recorded every 20 s for at least 5 min they had a mean amplitude of 2.5 ± 0.5 mV and 3.5 ± 0.5 mV before LTP and LTD induction, respectively, and were similar in both experimental groups. LTP and LTD were induced by applying a theta-burst stimulation (TBS) protocol and a low-frequency stimulation (LFS) protocol, respectively. The TBS protocol consisted of five synaptic trains (at 20 s intervals) of five bursts (at 5 Hz) each providing four stimuli at 100 Hz. Bursts were paired with intracellular depolarization delayed by 10 ms (900 ± 200 pA, 45 ms duration) through the recording electrode. Similar protocols have been used to induce NMDAR-dependent LTP in the visual cortex (Kirkwood & Bear, 1994a; Yoshimura et al. 2003; Huemmeke et al. 2004). The LFS protocol consisted of 10 min of synaptic stimulation at a frequency of 1 Hz paired with intracellular depolarization delayed by 10 ms (500 ± 100 pA, 45 ms duration). Both type of stimulations induced spikes in all the recorded neurons. The lesion-induced changes in long-term synaptic plasticity were analysed by averaging the EPSP amplitudes evoked by the last 30 stimuli in each recorded neuron. These data were then compared between the two experimental groups.
Voltage clamp mode
All other experiments were conducted in voltage-clamp mode. The intracellular solution contained (in mm): 125 caesium gluconate, 5 CsCl, 10 EGTA, 2 MgCl2, 2 Na2-ATP, 0.4 Na2-GTP, 10 Hepes and 5 QX-314. The pH was set to 7.3 with CsOH. Only cells with a holding current <200 pA were used for further recordings. Access resistance was controlled before and after each recording. The cells were discarded if the parameter changed more than 20%.
AMPA-receptor (AMPAR) mediated currents were isolated by bath application of the GABAA receptor antagonist picrotoxin (50 μm, Biozol, Eching, Germany) and the NMDAR blocker d-(–)-2-amino-5-phosphonopentanoic acid (d-AP5, 25 μm) (Tocris/Biotrend, Cologne, Germany) and acquired at a holding potential of −80 mV. NMDAR-mediated currents were isolated by bath application of picrotoxin and the AMPAR-antagonist 6,7-dinitroquinoxaline-2,3-dione (DNQX) (20 μm) (Tocris/Biotrend), and they were recorded at a holding potential of +40 mV.
Paired-pulse ratio
Pairs of synaptic stimulations with inter-stimulus-intervals (ISIs) ranging from 20 to 200 ms were used to analyse the paired pulse ratio (PPR). The stimulus intensity was always set to evoke a first EPSC of ∼60 pA. This prevented significant differences in the initial amplitude of EPSCs between the different experimental groups.
Data acquisition and analysis
Electrical signals were recorded with an Axoclamp-2B amplifier (Molecular Devices, Sunnyvale, CA, USA). Data were digitized at 5 kHz and filtered at 2 kHz using a Digidata-1400 system with pCLAMP 10 software (Molecular Devices). The same software was used for off-line analysis. The decay-time constant of the NMDAR-mediated currents was calculated by fitting the decaying current to the following monoexponential function:
Statistics
Statistical significance was tested with an independent Student's t test. Data are presented as means ±s.e.m. P values <0.05 were considered to be significant.
Results
The cortical slices were selected from region Bregma – 6 mm to Bregma – 7.6 mm. Here the laser-lesion was primarily located in visual cortical areas V1M, V2MM and V2ML. Nissl-staining revealed a necrotic lesion centre with a diameter of 0.6 ± 0.2 mm in mediolateral extent. The necrotic area reached down to cortical layer IV/V (Fig. 1A). Immunohistochemical staining for GFAP showed an astrogliosis which was mostly restricted to distances of 300–400 μm from the border of the lesion (Fig. 1C and D). The visual cortex of sham-operated rats did not reveal any histological tissue damage. All electrophysiological recordings were performed from visually identified pyramidal neurons in layers 2/3 at distances of 2–3 mm away from the border of the lesion. This specific region has been previously characterized by changes in synaptic plasticity of vertically ascending synaptic inputs (Dohle et al. 2010) and an increase in resting and stimulus evoked [Ca2+]i (Barmashenko et al. 2003). All neurons labelled with biocytin could be identified as characteristic pyramidal shaped-body neurons (Fig. 1B).
Figure 1. Histochemistry of the focal laser-lesion in rat visual cortex.
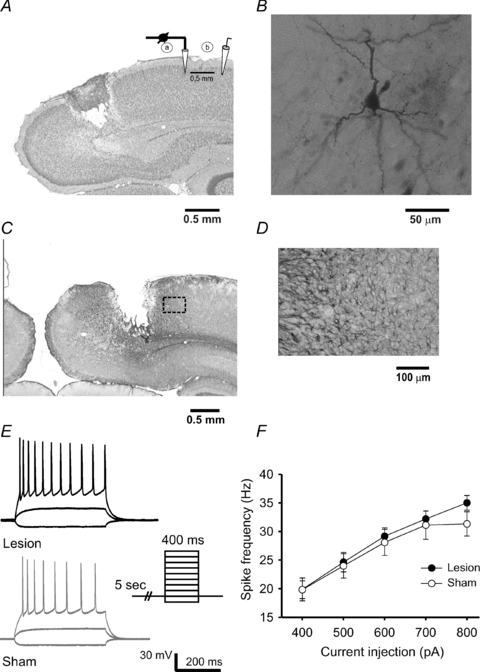
A, Nissl stained coronal section of the visual cortex from a 25-day-old rat at 4 days post-lesion. Note the position of the recording patch-clamp electrode (a) in layers 2/3 and the stimulation electrode (b) lateral in the same layers. B, pyramidal neuron located in cortical layers 2/3 of the visual cortex and intracellularly labelled with biocytin. C, another slice was labelled with the glial marker GFAP to disclose the astrogliosis reaction that was restricted to 500 μm from the border of the lesion. D, the same slice as shown in C revealed at higher magnification the limited astrogliosis. E, The intrinsic membrane and firing properties of the pyramidal neurons were not changed by the lesion. F, summary plot of the spike frequency in response to an increasing current injection into the recorded neurons.
Unaltered intrinsic properties of pyramidal neurons in layers 2/3 post-lesion
Since both synaptic efficacy and intrinsic excitability of neurons can undergo activity-dependent plasticity, we investigated potential changes in the intrinsic properties of the neurons surrounding the lesion. The resting membrane potential was not significantly altered. The firing properties of the neurons were studied by applying square pulses of hyper- and depolarizing currents through the patch-clamp electrode. Neurons of both groups started firing action potentials during injection of 300–400 pA of depolarizing current, and all the neurons (lesion: 10 cells from 4 animals; sham: 10 cells from 4 animals) showed regular frequency-adapting spikes which are characteristic for cortical pyramidal neurons (Fig. 1E) (Connors & Gutnick, 1990; Daoudal & Debanne, 2003). No significant differences were found in the spike frequency between the two animal groups (Fig. 1F). The input resistance was also not significantly altered by the lesion (lesion: 109.90 ± 11.86 MΩ, 10 cells from 4 rats; sham: 94.22 ± 7.38 MΩ, 9 cells from 4 rats).
Lesion-induced metaplasticity of L2/3–L2/3 synapses
So far it is unknown, whether horizontal inputs onto neurons located in layers 2/3 at the border of a focal lesion express any changes in synaptic plasticity. Thus, we evaluated potential changes of LTP and LTD at horizontal connections in layers 2/3 located lateral to the border of the lesion. The TBS protocol, when applied in sham-operated animals, induced a relatively small LTP of the signal amplitude in eight neurons. One neuron expressed a moderate short-term potentiation, which decayed within the first 20 min, and two other neurons did not show any potentiation, but rather a slight depression (Fig. 2E). The average of relative EPSP amplitude reached 125.3 ± 15.3% (11 cells from 9 animals). In contrast, the same protocol led to a significantly higher potentiation (P < 0.05) in neurons from lesion treated animals (174.5 ± 20.4%, 16 cells from 10 rats) (Fig. 2A and B). In this group only one neuron out of 16 failed to show any plastic changes, while three other cells showed short term potentiation (Fig. 2E). Next we tested whether the strength of LTD would also be affected by the lesion. The LFS protocol reliably induced LTD in all neurons from sham-operated rats. In contrast, the protocol differently affected the EPSP amplitudes in neurons from lesion-treated animals (Fig. 2F). About 50% of the neurons revealed a LFS induced LTD, while the other 50% gave rise to LTP.
Figure 2. The focal laser-lesion facilitated the induction of LTP and impaired LTD at synapses of neurons in L2/3–L2/3 located lateral at the lesion border.
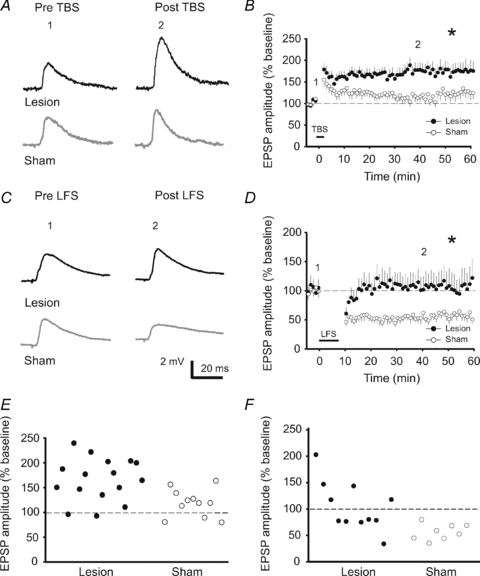
A, representative voltage traces of EPSPs recorded 2 min before and 40 min after TBS in lesion-treated (black traces) and sham-operated (grey traces) animals. B, time course of the changes in the relative EPSP amplitudes induced by the TBS. Note the enhanced potentiation of EPSPs post-lesion. C, representative traces of EPSPs recorded 2 min before and 40 min after induction of the LFS. D, time course of the changes in the relative EPSP amplitudes. Note the missing induction of LTD post-lesion. E and F, graph showing the mean relative EPSP amplitudes for each individual neuron recorded at 20–30 min following TBS (E) or LFS (F).
While the strength of synaptic depression in the LTD-expressing neurons was not different between the lesion and sham group, the summary diagram of all cells treated with a LFS protocol revealed a significant difference (lesion-treated rats: 100.4 ± 15.4%, 11 cells from 4 animals; sham: 60.8 ± 6.7%, 8 cells from 5 rats; P < 0.05) (Fig. 2C and D). Thus, overall the synapses at horizontal connections at the border of the lesion show a metaplastic shift towards a strengthening of these synapses.
Prolonged decay-time constants of NMDAR-mediated currents
Next, we investigated the potential underlying mechanisms of the lesion-induced metaplasticity of horizontal inputs at the L2/3–L2/3 synapses. Postsynaptically, we observed a significantly prolonged decay-time constant of NMDAR-mediated currents post-lesion (lesion: 185.7 ± 10.8 ms, 11 cells from 3 rats; sham: 148.9 ± 12.8 ms, 10 cells from 3 animals; P < 0.05) (Fig. 3A and B).
Figure 3. Prolonged decay-time constants of NMDAR-mediated currents (EPSCs) post-lesion.
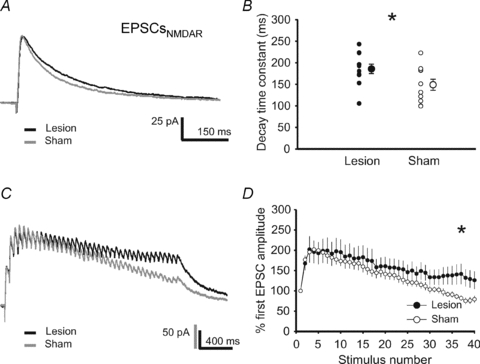
A, representative traces of NMDAR-mediated EPSCs recorded at a holding potential of +40 mV from lesion-treated (black trace) and sham-operated (grey trace) animals. B, the decay-time constants of these EPSCs were significantly prolonged in the vicinity of the lesion. C, representative current traces of NMDA-mediated EPSCs in response to high frequency presynaptic stimulation (40 pulses at 33 Hz). D, the mean relative amplitude of NMDAR-mediated EPSCs upon high-frequency synaptic stimulation was higher in the vicinity of the lesion as compared to sham-operated controls.
In accordance, repetitive high-frequency synaptic stimulation (40 pulses, 33 Hz) evoked NMDAR-dependent EPSCs that showed a relatively slower decay in the signal amplitude over time as compared to controls (Fig. 3C and D). This effect is very unlikely to be mediated by alterations of the presynaptic function, e.g. change in glutamate release or increased glutamate spillover, because (1) the paired-pulse ratio of AMPAR-mediated EPSCs was not altered for all tested inter-stimulus intervals (20–200 ms) (sham: 11 cells from 3 animals; lesion: 9 cells from 3 animals) (Fig. 4A and B), and (2) the temporal dynamics of AMPAR-mediated currents induced by the same high-frequency synaptic stimulation (40 pulses at 33 Hz) showed no changes post-lesion (sham: 15 cells from 4 animals; lesion: 12 cells from 3 animals) (Fig. 4C and D). Since NMDARs are permeable to calcium, the prolonged decay-time constant of NMDARs might enhance Ca2+-influx into the postsynaptic neuron, thereby facilitating the induction of synaptic plasticity.
Figure 4. No changes in PPR and synaptic fatigue of AMPAR-mediated currents (EPSCs) post-lesion.
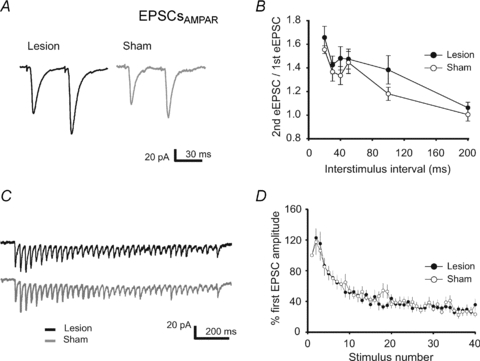
A, representative traces of AMPAR-mediated EPSCs recorded at −80 mV evoked by pairs of synaptic stimulations with an inter-stimulus interval (ISI) of 30 ms from lesion-treated (black trace) and sham-operated (grey trace) animals. B, summary diagram of the mean paired-pulse ratio (PPR) of EPSCs evoked by two synaptic stimulations with an inter-stimulus interval of 20–200 ms. PPR was not different between the 2 groups. C, representative current traces of AMPAR-mediated EPSCs in response to high frequency presynaptic stimulation (40 pulses at 33 Hz). D, the decay of the mean relative amplitudes of AMPAR-mediated EPSCs was similar in both groups.
Discussion
The present study disclosed an enhanced LTP and reduced LTD of L2/3–L2/3 synapses at horizontal connections in the vicinity of a focal laser lesion in rat visual cortex. This indicates a lesion-induced shift toward strengthening of these synapses disclosing the metaplastic capacity of these intracortical horizontal fibres at the border of a focal injury.
In general, LTP and LTD have been well studied by others at synapses of ascending fibres in layers 2/3 of the visual cortex (Artola & Singer, 1987; Kirkwood & Bear, 1994b) and in our own laboratory (Mittmann & Eysel, 2001; Barmashenko et al. 2003). Some studies reported that synaptic plasticity can be induced also at horizontal connections in superficial cortical layers in the rat motor (Hess et al. 1996) and sensory cortex (Marik & Hickmott, 2009; Hickmott, 2010). Here we present evidence that these intracortical, horizontal fibres are also able to undergo synaptic metaplastic changes in the context of a focal brain injury in rat visual cortex.
Generally, the strength of the postsynaptic response determines whether a synapse undergoes depression or facilitation. Furthermore, the threshold defining whether LTP or LTD is evoked has been reported to vary as a function of the history of a synapse (Bienenstock et al. 1982). For the visual cortex it has been shown that a reduced visual input, e.g. under conditions of light deprivation, shifted this threshold in favour of LTP induction (Kirkwood et al. 1996). Our data suggest that the threshold for LTP induction at intracortical fibres might be also shifted post-lesion. This could explain why the relatively weak LFS protocol reliably induced a depression of EPSPs in sham-operated animals, while it led to a bidirectional change of EPSPs (some of the neurons responded with LTP, others with LTD) in lesion-treated animals, and why the high-frequency stimulation (TBS) gave rise to an increased level of LTP post-lesion. Interestingly, the lesion-induced shift in synaptic plasticity was accompanied by an increase in the variability of the EPSP amplitudes following the LFS protocol. This larger variability was observed within each individual, which suggests that a particular subset of neurons might be susceptible to the lesion-induced changes.
The metaplastic alterations post-lesion were accompanied by a change in the kinetics of synaptically evoked NMDAR-mediated currents. The slower decay-time constant of NMDARs could be mediated by changes in the subunit composition of NMDARs, especially through a relative increase of NMDARs containing the NR2B subunit (Monyer et al. 1994; Flint et al. 1997). This resembles the developmental status of a more juvenile brain, where neuronal networks show relatively high plastic properties. In line with this is a previous study from our laboratory that showed a lesion-induced enhancement of LTP at synapses of ascending fibres projecting from layer 4 to layers 2/3. Here the increased LTP could be reduced to the level of control animals by application of ifenprodil, a specific blocker of NMDARs containing the NR2B subunit (Huemmeke et al. 2004). Since NMDAR-channels are permeable to calcium, a prolonged decay-time constant post-lesion could promote a higher calcium influx into the postsynaptic cell. This is supported by a previous study for our laboratory, which reported a lesion-mediated increase of LTP from ascending fibres associated with an enhanced stimulus-evoked calcium influx (Barmashenko et al. 2003). Both the dynamics and the magnitude of peak calcium transients are critically important to define whether a synapse will undergo potentiation or depression (Lisman, 1989). Thus, it is likely that an increased calcium influx could be responsible for the lesion-induced metaplastic shift in favour of LTP.
However, other mechanisms might also affect the lesion-induced metaplastic shift. For example, studies on several cortical lesion models recorded an impaired intracortical GABAergic inhibition in the vicinity of the injury (Mittmann et al. 1994; Buchkremer-Ratzmann et al. 1996; Schiene et al. 1996). Such cortical disinhibition can facilitate processes of synaptic plasticity, as has been shown in models of a pharmacological reduction of GABAergic transmission, leading to an increased LTP in slices (Artola & Singer, 1990; Kirkwood & Bear, 1994a) and to a reactivation of ocular dominance plasticity in the adult visual cortex in vivo (Harauzov et al. 2010). Yet, we failed to observe any functional changes of IPSCs recorded during a paired or repetitive presynaptic stimulations at the horizontal connections of L2/3–L2/3 synapses (B. Imbrosci & T. Mittmann, unpublished observations). Still, an impaired GABAergic transmission might be expressed at other synaptic inputs, thereby indirectly influencing the induction of LTP/LTD at horizontal connections. Future studies are needed to investigate this hypothesis.
Taken together, the facilitated synaptic plasticity, which we previously observed at ascending (Barmashenko et al. 2003; Huemmeke et al. 2004) and in the present work at lateral projections could both be essential to guarantee a proper cortical rewiring post-lesion. Anatomical studies conducted in cat and tree shrew visual cortex emphasize the exuberance of horizontal connections leading to the conclusion that not all, but only a defined subset of inputs activate a target cell in response to a specific visual stimulation (Gilbert & Wiesel, 1979; Rockland & Lund, 1982). Horizontal intracortical fibres seem therefore to constitute a very flexible framework offering numerous connections with variable weights, the modification of which could have a strong impact on the processing of visual inputs. Assuming a similar distribution of intracortical connections in rat visual cortex, upon visual stimulation, some subthreshold horizontal inputs could be unmasked by the described facilitated synaptic potentiation.
In conclusion the present study extends our knowledge on the reorganization processes in reaction to a traumatic brain injury. In general, focal lesions in the visual cortex lead to an initial functional loss due to the damaged cortical tissue. However, these functional deficits could be compensated, at least in part, by an increased synaptic plasticity of the cortical networks in the vicinity of the injury. The present study provides evidence for a metaplastic shift at specific intracortical horizontal connections towards a strengthening of these connections at the border of a focal lesion. Interestingly, an increased synaptic plasticity has been also observed with a similar spatial and temporal profile at ascending cortical fibres post lesion (Mittmann & Eysel, 2001). This suggests a robust potential of the visual cortex to reorganize. Furthermore, it discloses a spatial and temporal window for functional reorganization, which might be useful for future training and rehabilitation therapies following a focal injury in the cerebral cortex.
Acknowledgments
We thank Petra Küsener for excellent technical assistance. This work is supported by a grant of the EU (CORTEX) and by the DFG (Mi 432-1).
Glossary
Abbreviations
- AMPAR
AMPA receptor
- EPSC
excitatory postsynaptic current
- EPSP
excitatory postsynaptic potential
- GABAR
γ-aminobutyric acid receptor
- ISI
inter-stimulus interval
- LFS
low frequency stimulation
- LTD
long-term depression
- LTP
long-term potentiation
- NMDAR
NMDA receptor
- PPR
paired pulse ratio
- TBS
theta burst stimulation
Author contributions
B.I.: conception and design of experiments, analysis and interpretation of data, drafting the article. U.T.E.: conception of experiments, critically revising the article for important intellectual content. T.M.: conception and design of experiments, interpretation of data, critically revising the article for important intellectual content. All authors approved the final version for publication. The experiments were performed in the Faculty of Medicine, Institute of Physiology, Ruhr-University Bochum.
References
- Artola A, Singer W. Long-term potentiation and NMDA receptors in rat visual cortex. Nature. 1987;330:649–652. doi: 10.1038/330649a0. [DOI] [PubMed] [Google Scholar]
- Artola A, Singer W. The involvement of N-methyl-D-aspartate receptors in induction and maintenance of long-term potentiation in rat visual cortex. Eur J Neurosci. 1990;2:254–269. doi: 10.1111/j.1460-9568.1990.tb00417.x. [DOI] [PubMed] [Google Scholar]
- Barmashenko G, Eysel UT, Mittmann T. Changes in intracellular calcium transients and LTP in the surround of visual cortex lesions in rats. Brain Res. 2003;990:120–128. doi: 10.1016/s0006-8993(03)03447-4. [DOI] [PubMed] [Google Scholar]
- Bienenstock EL, Cooper LN, Munro PW. Theory for the development of neuron selectivity: orientation specificity and binocular interaction in visual cortex. J Neurosci. 1982;2:32–48. doi: 10.1523/JNEUROSCI.02-01-00032.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchkremer-Ratzmann I, August M, Hagemann G, Witte OW. Electrophysiological transcortical diaschisis after cortical photothrombosis in rat brain. Stroke. 1996;27:1105–1109. doi: 10.1161/01.str.27.6.1105. [DOI] [PubMed] [Google Scholar]
- Connors BW, Gutnick MJ. Intrinsic firing patterns of diverse neocortical neurons. Trends Neurosci. 1990;13:99–104. doi: 10.1016/0166-2236(90)90185-d. [DOI] [PubMed] [Google Scholar]
- Daoudal G, Debanne D. Long-term plasticity of intrinsic excitability: learning rules and mechanisms. Learn Mem. 2003;10:456–465. doi: 10.1101/lm.64103. [DOI] [PubMed] [Google Scholar]
- Dohle CI, Eysel UT, Mittmann T. Spatial distribution of long-term potentiation in the surround of visual cortex lesions in vitro. Exp Brain Res. 2010 doi: 10.1007/s00221-009-1964-5. in press. [DOI] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eysel UT, Schweigart G. Increased receptive field size in the surround of chronic lesions in the adult cat visual cortex. Cereb Cortex. 1999;9:101–109. doi: 10.1093/cercor/9.2.101. [DOI] [PubMed] [Google Scholar]
- Flint AC, Maisch US, Weishaupt JH, Kriegstein AR, Monyer H. NR2A subunit expression shortens NMDA receptor synaptic currents in developing neocortex. J Neurosci. 1997;17:2469–2476. doi: 10.1523/JNEUROSCI.17-07-02469.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert CD, Wiesel TN. Morphology and intracortical projections of functionally characterised neurones in the cat visual cortex. Nature. 1979;280:120–125. doi: 10.1038/280120a0. [DOI] [PubMed] [Google Scholar]
- Hagemann G, Redecker C, Neumann-Haefelin T, Freund HJ, Witte OW. Increased long-term potentiation in the surround of experimentally induced focal cortical infarction. Ann Neurol. 1998;44:255–258. doi: 10.1002/ana.410440217. [DOI] [PubMed] [Google Scholar]
- Harauzov A, Spolidoro M, DiCristo G, De Pasquale R, Cancedda L, Pizzorusso T, Viegi A, Berardi N, Maffei L. Reducing intracortical inhibition in the adult visual cortex promotes ocular dominance plasticity. J Neurosci. 2010;30:361–371. doi: 10.1523/JNEUROSCI.2233-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess G, Aizenman CD, Donoghue JP. Conditions for the induction of long-term potentiation in layer II/III horizontal connections of the rat motor cortex. J Neurophysiol. 1996;75:1765–1778. doi: 10.1152/jn.1996.75.5.1765. [DOI] [PubMed] [Google Scholar]
- Hickmott PW. Synapses of horizontal connections in adult rat somatosensory cortex have different properties depending on the source of their axons. Cereb Cortex. 2010;20:591–601. doi: 10.1093/cercor/bhp125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huemmeke M, Eysel UT, Mittmann T. Lesion-induced enhancement of LTP in rat visual cortex is mediated by NMDA receptors containing the NR2B subunit. J Physiol. 2004;559:875–882. doi: 10.1113/jphysiol.2004.069534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jablonka JA, Burnat K, Witte OW, Kossut M. Remapping of the somatosensory cortex after a photothrombotic stroke: dynamics of the compensatory reorganization. Neuroscience. 2010;165:90–100. doi: 10.1016/j.neuroscience.2009.09.074. [DOI] [PubMed] [Google Scholar]
- Kaas JH. Plasticity of sensory and motor maps in adult mammals. Annu Rev Neurosci. 1991;14:137–167. doi: 10.1146/annurev.ne.14.030191.001033. [DOI] [PubMed] [Google Scholar]
- Kirkwood A, Bear MF. Hebbian synapses in visual cortex. J Neurosci. 1994a;14:1634–1645. doi: 10.1523/JNEUROSCI.14-03-01634.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkwood A, Bear MF. Homosynaptic long-term depression in the visual cortex. J Neurosci. 1994b;14:3404–3412. doi: 10.1523/JNEUROSCI.14-05-03404.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkwood A, Rioult MC, Bear MF. Experience-dependent modification of synaptic plasticity in visual cortex. Nature. 1996;381:526–528. doi: 10.1038/381526a0. [DOI] [PubMed] [Google Scholar]
- Lisman J. A mechanism for the Hebb and the anti-Hebb processes underlying learning and memory. Proc Natl Acad Sci U S A. 1989;86:9574–9578. doi: 10.1073/pnas.86.23.9574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marik SA, Hickmott PW. Plasticity of horizontal connections at a functional border in adult rat somatosensory cortex. Neural Plast. 2009;2009:294192. doi: 10.1155/2009/294192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittmann T, Eysel UT. Increased synaptic plasticity in the surround of visual cortex lesions in rats. Neuroreport. 2001;12:3341–3347. doi: 10.1097/00001756-200110290-00039. [DOI] [PubMed] [Google Scholar]
- Mittmann T, Luhmann HJ, Schmidt-Kastner R, Eysel UT, Weigel H, Heinemann U. Lesion-induced suppression of inhibitory function in rat neocortex in vitro. Neuroscience. 1994;60:891–906. doi: 10.1016/0306-4522(94)90270-4. [DOI] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12:529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. 2nd edn. London: Academic Press; 1986. [Google Scholar]
- Rockland KS, Lund JS. Widespread periodic intrinsic connections in the tree shrew visual cortex. Science. 1982;215:1532–1534. doi: 10.1126/science.7063863. [DOI] [PubMed] [Google Scholar]
- Schiene K, Bruehl C, Zilles K, Qu M, Hagemann G, Kraemer M, Witte OW. Neuronal hyperexcitability and reduction of GABAA-receptor expression in the surround of cerebral photothrombosis. J Cereb Blood Flow Metab. 1996;16:906–914. doi: 10.1097/00004647-199609000-00014. [DOI] [PubMed] [Google Scholar]
- Sirosh J, Miikkulainen R. The Neurobiology of Computation: Proceedings of the Annual Computational Neuroscience Meeting. Monterey, CA, USA: Kluwer Academic Publishers; 1994. Modeling cortical plasticity based on adapting lateral interaction; pp. 1–5. [Google Scholar]
- Taub E, Uswatte G, Elbert T. New treatments in neurorehabilitation founded on basic research. Nat Rev Neurosci. 2002;3:228–236. doi: 10.1038/nrn754. [DOI] [PubMed] [Google Scholar]
- Yoshimura Y, Ohmura T, Komatsu Y. Two forms of synaptic plasticity with distinct dependence on age, experience, and NMDA receptor subtype in rat visual cortex. J Neurosci. 2003;23:6557–6566. doi: 10.1523/JNEUROSCI.23-16-06557.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zepeda A, Vaca L, Arias C, Sengpiel F. Reorganization of visual cortical maps after focal ischemic lesions. J Cereb Blood Flow Metab. 2003;23:811–820. doi: 10.1097/01.WCB.0000075010.31477.1E. [DOI] [PubMed] [Google Scholar]