

Abstract
Exercise training induces mitochondrial biogenesis, but the time course of molecular sequelae that accompany repetitive training stimuli remains to be determined in human skeletal muscle. Therefore, throughout a seven-session, high-intensity interval training period that increased 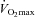 (12%), we examined the time course of responses of (a) mitochondrial biogenesis and fusion and fission proteins, and (b) selected transcriptional and mitochondrial mRNAs and proteins in human muscle. Muscle biopsies were obtained 4 and 24 h after the 1st, 3rd, 5th and 7th training session. PGC-1α mRNA was increased >10-fold 4 h after the 1st session and returned to control within 24 h. This ‘saw-tooth’ pattern continued until the 7th bout, with smaller increases after each bout. In contrast, PGC-1α protein was increased 24 h after the 1st bout (23%) and plateaued at +30–40% between the 3rd and 7th bout. Increases in PGC-1β mRNA and protein were more delayed and smaller, and did not persist. Distinct patterns of increases were observed in peroxisome proliferator-activated receptor (PPAR) α and γ protein (1 session), PPAR β/δ mRNA and protein (5 sessions) and nuclear respiratory factor-2 protein (3 sessions) while no changes occurred in mitochondrial transcription factor A protein. Citrate synthase (CS) and β-HAD mRNA were rapidly increased (1 session), followed 2 sessions later (session 3) by increases in CS and β-HAD activities, and mitochondrial DNA. Changes in COX-IV mRNA (session 3) and protein (session 5) were more delayed. Training also increased mitochondrial fission proteins (fission protein-1, >2-fold; dynamin-related protein-1, 47%) and the fusion protein mitofusin-1 (35%) but not mitofusin-2. This study has provided the following novel information: (a) the training-induced increases in transcriptional and mitochondrial proteins appear to result from the cumulative effects of transient bursts in their mRNAs, (b) training-induced mitochondrial biogenesis appears to involve re-modelling in addition to increased mitochondrial content, and (c) the ‘transcriptional capacity’ of human muscle is extremely sensitive, being activated by one training bout.
(12%), we examined the time course of responses of (a) mitochondrial biogenesis and fusion and fission proteins, and (b) selected transcriptional and mitochondrial mRNAs and proteins in human muscle. Muscle biopsies were obtained 4 and 24 h after the 1st, 3rd, 5th and 7th training session. PGC-1α mRNA was increased >10-fold 4 h after the 1st session and returned to control within 24 h. This ‘saw-tooth’ pattern continued until the 7th bout, with smaller increases after each bout. In contrast, PGC-1α protein was increased 24 h after the 1st bout (23%) and plateaued at +30–40% between the 3rd and 7th bout. Increases in PGC-1β mRNA and protein were more delayed and smaller, and did not persist. Distinct patterns of increases were observed in peroxisome proliferator-activated receptor (PPAR) α and γ protein (1 session), PPAR β/δ mRNA and protein (5 sessions) and nuclear respiratory factor-2 protein (3 sessions) while no changes occurred in mitochondrial transcription factor A protein. Citrate synthase (CS) and β-HAD mRNA were rapidly increased (1 session), followed 2 sessions later (session 3) by increases in CS and β-HAD activities, and mitochondrial DNA. Changes in COX-IV mRNA (session 3) and protein (session 5) were more delayed. Training also increased mitochondrial fission proteins (fission protein-1, >2-fold; dynamin-related protein-1, 47%) and the fusion protein mitofusin-1 (35%) but not mitofusin-2. This study has provided the following novel information: (a) the training-induced increases in transcriptional and mitochondrial proteins appear to result from the cumulative effects of transient bursts in their mRNAs, (b) training-induced mitochondrial biogenesis appears to involve re-modelling in addition to increased mitochondrial content, and (c) the ‘transcriptional capacity’ of human muscle is extremely sensitive, being activated by one training bout.
Introduction
The numerous improvements in health and exercise performance associated with endurance exercise training are closely related to a progressive increase in the capacity of skeletal muscle to oxidize fatty acids and carbohydrates (Holloszy & Booth, 1976; Goodyear & Kahn, 1998; Hood, 2001; Sirikul et al. 2006). These enhancements are due in part to a greater volume of the mitochondrial reticulum. Mitochondrial biogenesis occurs rapidly with increased mitochondrial protein content observed after as little as six to seven training sessions in humans (Chesley et al. 1996; Spina et al. 1996; Gibala et al. 2006; Talanian et al. 2007). The expansion of the mitochondrial reticulum in skeletal muscle is due to a complex intracellular system of sensing homeostatic challenges imposed by contractions which initiate transcriptional programmes essential to increasing metabolic proteins (Hood, 2001; Booth & Neufer, 2006).
Mitochondrial biogenesis in skeletal muscle is believed to result from the cumulative effects of transient increases in mRNA transcripts encoding mitochondrial proteins after successive exercise sessions (Williams & Neufer, 1996; Pilegaard et al. 2000; Fluck & Hoppeler, 2003; Booth & Neufer, 2006). This process requires the coordinated expression of both nuclear and mitochondrial (mtDNA) genomes through factors dedicated to specific families of genes encoding distinct categories of mitochondrial proteins (Hood, 2001; Scarpulla, 2002). In the past decade considerable work has shown that coordinating these various processes is regulated, in part, by the peroxisome proliferator-activated receptor γ co-activator (PGC)-1α (Puigserver et al. 1998; Hood, 2001; Scarpulla, 2006) and possibly PGC-1β (Arany et al. 2007). This transcriptional co-activator influences the transcriptional activity of nuclear respiratory factors (NRFs) (Wu et al. 1999; Scarpulla, 2002; Kelly & Scarpulla, 2004; Gleyzer et al. 2005) and peroxisome proliferator-activated receptors (PPARs) (Gilde & Van Bilsen, 2003; Huss & Kelly, 2004) as well as the mitochondrial transcription factor A (Tfam) through NRF activation (Wu et al. 1999). NRFs promote the expression of nuclear genes which encode for proteins in the electron transport chain (Scarpulla, 2002) whereas PPARs regulate genes for lipid transport and metabolism and lipid catabolism (Gilde & Van Bilsen, 2003). Tfam regulates the expression of the mtDNA which encodes fundamental respiratory proteins and other factors involved in mtDNA transcription and replication (Gordon et al. 2001; Hood, 2001). By interacting with these transcription factors, PGC-1α directs a coordinated up-regulation of muscle mitochondrial content and the capacity for substrate metabolism and oxidative phosphorylation.
The likely involvement of PGC-1 in mediating training-induced mitochondrial biogenesis is supported by numerous observations. Both acute exercise (Pilegaard et al. 2003; Norrbom et al. 2004; Watt et al. 2004; Cartoni et al. 2005; Russell et al. 2005; Hellsten et al. 2007; Mortensen et al. 2007) and several weeks of endurance training (Russell et al. 2003; Short et al. 2003; Kuhl et al. 2006) increase PGC-1α mRNA, with an increase in PGC-1α protein after >6 weeks of training in association with greater mitochondrial enzyme activities (Russell et al. 2003; Kuhl et al. 2006; Burgomaster et al. 2008). A single bout of exercise also increases PGC-1 α mRNA and protein (Mathai et al. 2008) in human muscle and stimulates PGC-1α translocation into the nucleus in rat skeletal muscle (Wright et al. 2007). Concurrently, this results in greater binding of NRF-1 and -2 to DNA (Wright et al. 2007) and binding to the nuclear transcription factor MEF-2 in human skeletal muscle (McGee & Hargreaves, 2004). Collectively, these findings suggest that activation of PGC-1α in response to acute exercise mediates the early stages of mitochondrial biogenesis and that increases in PGC-1α protein expression may serve to amplify the transcriptional response to subsequent exercise challenges or sustain the greater mitochondrial volume already achieved through training (Pilegaard et al. 2003; Wright et al. 2007). In other words, a greater PGC-1 protein content may boost the ‘transcriptional sensitivity’ of the cell to further exercise challenges. These potential benefits of increased transcriptional protein expression may also apply to other transcription factors, especially considering that both acute and chronic exercise increases the expression of many of the NRFs (Short et al. 2003; Cartoni et al. 2005), PPARs (Horowitz et al. 2000; Russell et al. 2003; Watt et al. 2004; Russell et al. 2005) and Tfam (Bengtsson et al. 2001; Pilegaard et al. 2003; Short et al. 2003; Garcia-Roves et al. 2006; Norrbom et al. 2010) in human skeletal muscle.
The improved oxidative capacity of skeletal muscle following training may also be due to alterations in mitochondrial morphology in addition to a greater mitochondrial content. Mitochondria can exist as a fused tubular network or ‘reticulum’, and as fragmented globules (Hoppeler et al. 1973, 1985; Hoppeler, 1986; Kirkwood et al. 1986, 1987; Ogata & Yamasaki, 1997). Importantly, fragmented mitochondria appear to possess lower maximal capacities to oxidize carbohydrates and fatty acids (Bach et al. 2003; Pich et al. 2005). The balance between reticular or globular states appears to be partly regulated by proteins which promote mitochondrial fusion (mitofusins, MFN-1 and -2) and fission (dynamin-related protease-1, DRP-1; fission protein-1, Fis-1) (Joseph et al. 2006; Cerveny et al. 2007; Westermann, 2008). A single exercise bout can increase the mRNA of MFN-1 and -2 (Cartoni et al. 2005), while training increased the amount of reticular mitochondria in rat muscle (Kirkwood et al. 1987). Collectively, these observations suggest that mitochondrial fusion may be important for increasing mitochondrial oxidative capacity in human skeletal muscle during training.
At present there is a paucity of information with respect to the sequences of molecular events that occur in human muscle when mitochondrial biogenesis is induced with exercise training. Therefore, the purposes of this study were to (1) examine the time course of responses in the protein contents of PGC-1α and β, NRF-2, PPARα, β/δ and γ and Tfam in human skeletal muscle following repeated exercise sessions, (2) compare these responses to the time-dependent increases in mitochondrial enzymes, (3) examine the mRNA responses for the PGC-1 and PPAR isoforms, Tfam and select genes encoding mitochondrial proteins throughout training, and (4) determine the temporal responses of mitochondrial fusion (MFN-1 and -2) and fission proteins (Fis-1 and DRP-1) during training. We hypothesized that increased mitochondrial proteins would be preceded by initial increases in the content of PGC-1α protein and occur concurrent with early increases in PGC-1β, NRFs, PPARs and Tfam proteins. We also hypothesized that initial increases in mRNA encoding mitochondrial proteins would occur before the first increase in PGC-1α protein, as has been observed previously in rats (Wright et al. 2007). We also expected that adaptive increases in transcriptional and metabolic proteins follow the accumulation of transient increases in mRNA after successive exercise sessions. Finally, we hypothesized that training would increase the ratio of fusion to fission proteins.
Methods
Subjects
Nine male subjects volunteered to participate in this study. Their age, height, weight and BMI (mean ±s.e.m.) were 23.0 ± 0.7 year, 179.3 ± 2.0 cm, 82.1 ± 3.9 kg and 25.4 ± 0.8. Body mass was not different after training (82.3 ± 3.7 kg). Prior to the study, subjects were not involved in structured training programmes, but participated in some form of light aerobic activity (cycling, jogging) for ∼1 h day−1, 2–3 days week−1. Subjects were advised to avoid any other physical activity beginning 72 h before the initial 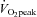 test (see below) and throughout the 2 week study to isolate the effects of the training stimulus on the Pre and each post-training (Post) biopsy. Subjects were medication free and were advised to refrain from taking supplements other than multivitamins. The experimental protocol and associated risks were explained orally and in writing to all subjects before written consent was obtained. The study was approved by the Research Ethics Boards of the University of Guelph and McMaster University and conformed to the Declaration of Helsinki.
test (see below) and throughout the 2 week study to isolate the effects of the training stimulus on the Pre and each post-training (Post) biopsy. Subjects were medication free and were advised to refrain from taking supplements other than multivitamins. The experimental protocol and associated risks were explained orally and in writing to all subjects before written consent was obtained. The study was approved by the Research Ethics Boards of the University of Guelph and McMaster University and conformed to the Declaration of Helsinki.
Study design
Each subject completed seven sessions of cycle ergometer high intensity interval training over a ∼2 week period Fig. 1). A resting biopsy was sampled before the first training session as well as 4 h and 24 h after the 1st, 3rd, 5th and 7th training sessions (9 biopsies total, Fig. 1). Subjects completed a 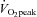 test 72 h before the Pre biopsy and 48 h after the Post biopsy by completing a continuous incremental cycling test to exhaustion on an electronic cycle ergometer (Lode Instrument, Groningen, The Netherlands). Pulmonary
test 72 h before the Pre biopsy and 48 h after the Post biopsy by completing a continuous incremental cycling test to exhaustion on an electronic cycle ergometer (Lode Instrument, Groningen, The Netherlands). Pulmonary 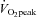 was measured using a metabolic measurement system (Vmax Series 229, Sensormedics Corporation, Yorba Linda, CA, USA). Subjects refrained from alcohol and caffeine consumption throughout the entire study including 48 h prior to the first
was measured using a metabolic measurement system (Vmax Series 229, Sensormedics Corporation, Yorba Linda, CA, USA). Subjects refrained from alcohol and caffeine consumption throughout the entire study including 48 h prior to the first 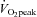 test. Subjects were asked to consume their regular diet throughout the entire study. Analysis of dietary records verified that all subjects consumed an average of 2786 ± 491 kcal (54 ± 2% carbohydrates; 24 ± 3% fat; 22 ± 2% protein) 24 h before the Pre biopsy. The average dietary intake of all 24 h periods prior to each Post session biopsy was 3325 ± 381 kcal (53 ± 1% carbohydrates, 27 ± 1% fat; 20 ± 1% protein).
test. Subjects were asked to consume their regular diet throughout the entire study. Analysis of dietary records verified that all subjects consumed an average of 2786 ± 491 kcal (54 ± 2% carbohydrates; 24 ± 3% fat; 22 ± 2% protein) 24 h before the Pre biopsy. The average dietary intake of all 24 h periods prior to each Post session biopsy was 3325 ± 381 kcal (53 ± 1% carbohydrates, 27 ± 1% fat; 20 ± 1% protein).
Figure 1.
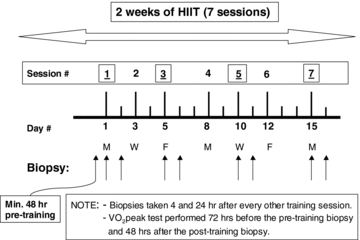
Study design for 2 weeks of high intensity interval training (HIIT)
High-intensity interval training protocol (HIIT)
Subjects conducted their first training session 48 h after the first resting biopsy. Subjects trained on a cycle ergometer (Monark 894 E, Vansbro, Sweden) at a power output that elicited ∼90%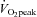 , 3 days week−1 for 2 weeks. For each training session, subjects completed 10 exercise intervals lasting 4 min and separated by 2 min of rest. During the 1st and 2nd training sessions, the power output was adjusted to the highest intensity that could be tolerated for a complete set of 10 intervals. Each subject's heart rate (HR) reached a steady state during the final 2 min of intervals 6–10, averaging 183 ± 2.0 beats min−1 (∼93% of HRmax). This HR was maintained during the remainder of the seven training sessions by increasing the power output during the exercise intervals and sessions. During the rest periods, subjects remained on the ergometer and were allowed to consume water or a sports drink ad libitum. Four subjects consumed water and five consumed sports drink throughout each training session. The volume consumed was consistent each day (∼750 ml). No differences were observed in any measurements between subjects who consumed water and those who consumed sports drink. All training sessions were supervised by one of the investigators. The mean power output during the training sessions increased by 18% between the 1st and 7th training session (203 ± 11 to 239 ± 11 W, P < 0.05).
, 3 days week−1 for 2 weeks. For each training session, subjects completed 10 exercise intervals lasting 4 min and separated by 2 min of rest. During the 1st and 2nd training sessions, the power output was adjusted to the highest intensity that could be tolerated for a complete set of 10 intervals. Each subject's heart rate (HR) reached a steady state during the final 2 min of intervals 6–10, averaging 183 ± 2.0 beats min−1 (∼93% of HRmax). This HR was maintained during the remainder of the seven training sessions by increasing the power output during the exercise intervals and sessions. During the rest periods, subjects remained on the ergometer and were allowed to consume water or a sports drink ad libitum. Four subjects consumed water and five consumed sports drink throughout each training session. The volume consumed was consistent each day (∼750 ml). No differences were observed in any measurements between subjects who consumed water and those who consumed sports drink. All training sessions were supervised by one of the investigators. The mean power output during the training sessions increased by 18% between the 1st and 7th training session (203 ± 11 to 239 ± 11 W, P < 0.05). 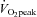 increased by 12% after training (3.74 ± 0.15 to 4.18 ± 0.17 l min−1, P < 0.05).
increased by 12% after training (3.74 ± 0.15 to 4.18 ± 0.17 l min−1, P < 0.05).
Percutaneous muscle biopsy procedure
Subjects arrived at the laboratory and rested on a bed. A single incision was made over the vastus lateralis muscle of one leg under local anaesthesia (2% lidocaine, no epinephrine) for muscle biopsy sampling. With the subject on the bed, a resting muscle sample was taken. Muscle samples were immediately frozen in liquid N2 and stored until analysis. Successive muscle biopsies were alternated between legs.
Muscle enzyme activities
A small piece of frozen wet muscle (6–10 mg) was removed from each biopsy under liquid N2 for the spectrophotometric determination of mitochondrial citrate synthase (CS) and β-hydroxyacyl-CoA dehydrogenase (β-HAD) maximal activities (37°C), as described previously (Srere, 1969; Bergmeyer, 1974). An aliquot of each muscle enzyme homogenate was then extracted with 0.5 m perchloric acid (HClO4) containing 1 mm EDTA and neutralized with 2.2 m KHCO3. The supernatants from the extracts were used for the enzymatic spectrophotometric determination of total creatine (Cr) (Bergmeyer, 1974). Enzyme measurements were normalized to the highest total Cr content measured from all biopsies from each subject to correct for the diluting effects of blood contamination. There was no change in the average uncorrected total Cr content in the Pre and Post samples which allowed all Pre and Post biopsies from one subject to be used for creatine corrections of enzymatic activities without concern for training influences on total Cr, as previously reported (Perry et al. 2008).
Western blot analyses
An aliquot of wet muscle (30 mg) was homogenized with a Teflon pestle and glass mortar as described previously (Holloway et al. 2008; Perry et al. 2008). Protein concentrations were determined using a BCA assay (Sigma-Aldrich, St Louis, MO, USA). Forty micrograms of denatured protein was loaded for Western blotting and resolved by SDS-PAGE on 6–12% polyacrylamide gels, depending on the molecular weight of the protein, and transferred to a PVDF membrane. Membranes were blocked with 7.5% BSA and immunoblotted overnight (4°C) with antibodies specific for each protein. Commercially available monoclonal antibodies were used to detect COX-IV (Molecular Probes/Invitrogen), PPARγ (isoforms 1 and 2; R&D Systems, Minneapolis, MN, USA), PPARα, PPARβ/δ, MFN-1, MFN-2, Fis-1 (Abnova, Hornby, ON, Canada), and DRP-1 (BD Biosciences, Mississauga, ON, Canada). Commercially available polyclonal antibodies were used to detect PGC-1α (Calbiochem, La Jolla, CA, USA), PGC-1β (Abnova, Hornby, ON, Canada), Tfam (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and NRF-2 (Spring Biosciences, Fremont, CA, USA). NRF-1 content was not determined due to inadequate blot qualities obtained with commercial antibodies. Ponceau staining of total protein per lane was used to verify consistent loading. A human muscle crude-membrane standard was loaded on each gel to correct for differences in blotting efficiency between gels. Membranes were incubated for 1 h at room temperature with the corresponding secondary antibody and the immunoreactive proteins were detected with enhanced chemiluminescence and quantified by densitometry (ChemiGenius 2 Bioimaging system, SynGene, Cambridge, UK). The specificity of the PGC-1α antibody has previously been established (Benton et al. 2008a; Benton et al. 2008b; Benton et al. 2010; Buddo & Benton, unpublished observations; Holloway et al. 2008; Holloway et al. 2010. Gurd et al. 2009). Representative blots are shown in Fig. 2.
Figure 2. Representative Western blots.
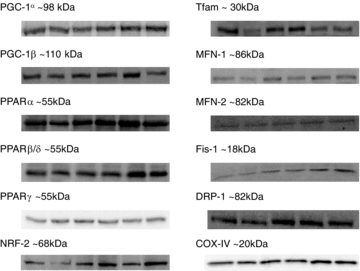
Bands (left to right) are Pre, 1st-4 h, 1st-24 h, 3rd-24 h, 5th-24 h and 7th-24 h time points. MFN-1 and DRP-1 do not include 1st-4 h.
mRNA
The mRNA content for the isoforms of PGC-1 (α and β) and PPAR (α, β/δ and γ), Tfam, CS, β-HAD and COX-IV were determined using procedures described previously (Handschin et al. 2007a; Benton et al. 2008a). RNA was isolated from ∼20 mg wet muscle using TRIzol reagent (Invitrogen) and an RNeasy mini kit (Qiagen). Total RNA was treated with RNase-free DNase (Qiagen) during the column purification as outlined by the manufacturer. Reverse transcription was conducted using a First Strand cDNA synthesis kit for reverse transcription-PCR (avian myeloblastosis virus) (Roche Applied Science) using random primers. Real time PCR was performed using a 7500 real time PCR system (Applied Biosystems) using Platinum SYBR Green qPCR Supermix-UDG (Invitrogen). Relative mRNA levels were calculated using the ΔΔCT method using 7500 System SDS software version 1.2.1.22 (Applied Biosystems). 18S ribosomal RNA was used as an endogenous control. A comparison of the 18S CT values between all biopsy time-points within each subject indicated 18S mRNA did not change throughout training. Dissociations curves verified that a single PCR product was amplified. Primer sequences are in Table 1.
Table 1.
Primer sequences for RT-PCR
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| CS | 5′-CCTGCCTAATGACCCCATGTT-3′ | 5′-CATAATACTGGAGCAGCACCCC-3′ |
| β-HAD | 5′-GAGGAGGACTTGAGGTTGCCA-3′ | 5′-CTCCTGCTCCTGGTAAGGCC-3′ |
| COX-IV | 5′-AGAAGCACTATGTGTACGGCCC-3′ | 5′-GGTTCACCTTCATGTCCAGCAT-3′ |
| PGC-1α | 5′-CACTTACAAGCCAAACCAACAACT-3′ | 5′-CAATAGTCTTGTTCTCAAATGGGGA-3′ |
| PGC-1β | 5′-TCTCGCTGACACGCAGGGT-3′ | 5′-GCACCACTGCAGCTCCCC-3′ |
| PPARβ/δ | 5′-ACGGCGCCCTTTGTGATC-3′ | 5′-GGTAGAAGACGTGCACGCTGA-3′ |
| PPARα | 5′-GACAAGTGCGACCGCAGCT-3′ | 5′-CGAATCGCGTTGTGTGACATC-3′ |
| PPARγ | 5′-ACCAGCTGAATCCAGAGTCCG-3′ | 5′-AGATCGCCCTCGCCTTTG-3′ |
| Tfam | 5′-CACCGCAGGAAAAGCTGAAG-3′ | 5′-TTCGTCCTCTTTAGCATGCTGA-3′ |
| 18S | 5′-GACTCAACACGGGAAACCTCAC-3′ | 5′-ATCGCTCCACCAACTAAGAACG-3′ |
| Genomic (SLC2A4) | 5′-CTGCTCCTGGGCCTCACAG-3′ | 5′-CCCCTCGAGATTCTGGATGAT-3′ |
| Mitochondrial (ND5) | 5′-CGGCTGAGAGGGCGTAGG-3′ | 5′-GATGAAACCGATATCCGCCGA-3′ |
CS, citrate synthase; β-HAD, β-hydroxyacyl coenzyme A dehydrogenase; COX-IV, cytochrome oxidase subunit IV; PGC-1α, peroxisome proliferator activated receptor γ co-activator-1α; PGC-1β, peroxisome proliferator activated receptor γ co-activator-1β; PPARβ/δ, peroxisome proliferator activated receptor β/δ; PPARα, peroxisome proliferator activated receptor α; PPARγ, peroxisome proliferator activated receptor α; Tfam, mitochondrial transcription factor A.
Mitochondrial DNA (mtDNA)
mtDNA content was determined using real time PCR as previously described (Benton et al. 2008a). Total DNA was isolated using DNeasy blood and tissue kit (Qiagen). mtDNA primers were designed using the human mitochondrial genome sequence (GenBankTM accession number NC_001807) within the NADH dehydrogenase subunit 5 gene (ND5). Primers measuring genomic content were designed within exon 6 of the solute carrier family 2 (facilitated glucose transporter), member 4 (SLC2A4, GenBankTM accession number NM_001042). mtDNA content was calculated by the ΔΔCT method using genomic DNA content as an internal standard with 7500 System SDS software version 1.2.1.22 (Applied Biosystems). Primer sequences are provided in Table 1. The efficiency for all primer sets for mRNA and mtDNA was 100 ± 2.5%.
Statistics
Results are expressed as mean ± standard error of the mean (s.e.m.). The level of significance was established at P < 0.05 for all statistics. A one-way ANOVA with repeated measures was used to test for difference in training power outputs and heart rates as well as all muscle measurements across the seven training sessions. When a significant F-ratio was obtained, post hoc analyses were completed using a LSD post hoc analysis. Student's t test for paired data was used to assess the effects of training on  .
.
Results
Mitochondrial biogenesis
mtDNA
mtDNA increased with training. However, this was not observed until 24 h after the 3rd session (+65%, P < 0.05) and continued to increase further until 24 h after the 5th session (+118%, P < 0.05; Table 2).
Table 2.
mtDNA, Tfam and NRF2 responses during 2 weeks of high-intensity interval training in human skeletal muscle
| Pre | 1st-4 h | 1st-24 h | 3rd-4 h | 3rd-24 h | 5th-4 h | 5th-24 h | 7th-4 h | 7th-24 h | |
|---|---|---|---|---|---|---|---|---|---|
| mtDNA | 1 | 1.2 ± 0.15 | 1.3 ± 0.1 | 1.1 ± 0.1 | 1.7 ± 0.1* | 1.8 ± 0.2* | 2.2 ± 0.4*§ | 1.1 ± 0.1‡a,c | 1.7 ± 0.4* |
| Tfam mRNA | 1 | 1.5 ± 0.2*† | 1.2 ± 0.1*b | 1.6 ± 0.2*† | 1.4 ± 0.2* | 1.6 ± 0.2*† | 1.4 ± 0.2* | 1.6 ± 0.3*†c | 1.5 ± 0.3*† |
| Tfam protein | 1 | 0.7 ± 0.1*†‡c | 1.0 ± 0.1 | ND | 1.1 ± 0.1 | ND | 1.1 ± 0.1 | ND | 0.7 ± 0.1*†‡c |
| NRF-2 protein | 1 | 1.0 ± 0.3 | 1.2 ± 0.1 | ND | 1.5 ± 0.3*#c | ND | 1.0 ± 0.2 | ND | 1.7 ± 0.3*#†c |
Values are relative to pre-training contents which were set to 1 and are presented as means ±s.e.m. mtDNA, mitochondrial DNA; Tfam, mitochondrial transcription factor A; NRF-2, nuclear respiratory factor 2, n= 8–9.
Significantly different from Pre (P < 0.05)
significantly different from 1st-4 h
significantly different from 1st-24 h
significantly different from 3rd-24 h
significantly different from 7th-24 h
significantly different from all 4 h time points
significantly different from 5th-24 h
significantly different from all 24 h time points (P < 0.05).
ND, no data.
Mitochondrial enzymes
CS mRNA (48%) and β-HAD mRNA (17%) were increased 4 and 24 h after the 1st session (P < 0.05; Fig. 3A and B), and continued to increase until after the third exercise session (P < 0.05). Thereafter, CS mRNA remained up-regulated (Fig. 3A) while β-HAD mRNA declined until the 7th training session (P < 0.05; Fig. 3B). The rapid increases in β-HAD (4 h) and CS mRNA (24 h) after the initial exercise session occurred well before the first observed increases in the maximal activities of β-HAD (8%, 24 h after the 3rd session) and CS (17%, 4 h after the 3rd session) (P < 0.05; Fig. 3D and E).
Figure 3. Maximal activities and protein content of mitochondrial enzymes in skeletal muscle throughout 2 weeks of high-intensity interval training.
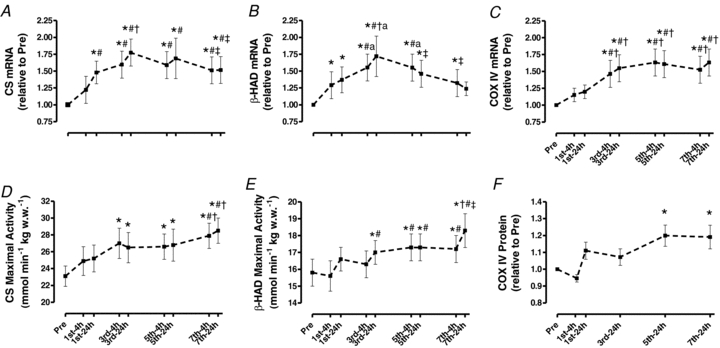
A, citrate synthase (CS). B, β-hydroxyacyl CoA dehydrogenase (β-HAD). C, COX-IV. Values are means ±s.e.m. for 9 subjects. *Significantly different from Pre, #significantly different from 1st-4 h, †significantly different from 1st-24 h, ‡significantly different from 3rd-24 h (P < 0.05).
In contrast to the training induced responses in β-HAD and CS mRNAs, up-regulation of COXIV mRNA occurred considerably later (29%, P < 0.05; 4 h after 3rd session). Thereafter, it remained elevated for the remainder of training (Fig. 3C). The first increase in COX-IV protein occurred 24 h after the 5th training session (20%, P < 0.05; Fig. 3F).
Transcription proteins
PGC-1α: PGC-1α mRNA increased by >10-fold within 4 h after the initial exercise session and returned to Pre levels 24 h into recovery (P < 0.05; Fig. 4A). Each subsequent session repeatedly stimulated increases in PGC-1α mRNA 4 h after the session followed by a return to the Pre level by 24 h (P < 0.05). However, the magnitude of the 4 h increase after each training session was progressively decreased, resulting in a ‘stair-case’ type response over seven training sessions.
Figure 4. Skeletal muscle PGC-1α and PGC-1β mRNA (A and B) and protein content (C and D) throughout 2 weeks of high-intensity interval training.
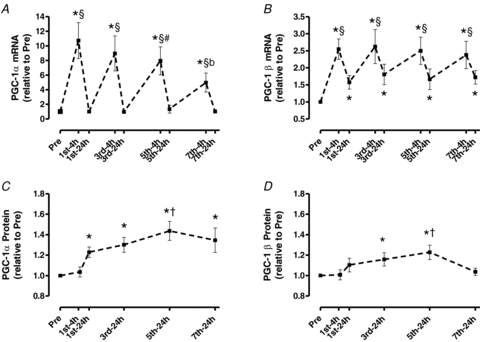
Values are means ±s.e.m. for 8–9 subjects. *Significantly different from Pre, §significantly different from all 24 h time points, #significantly different from 1st-4 h, bsignificantly different from all 4 h time points, †significantly different from 1st-24 h (P < 0.05).
PGC-1α protein expression occurred later (23%, 24 h after 1st session, P < 0.05; Fig. 4C) than PGC-1α mRNA. Moreover, unlike the reduced PGC-1α mRNA responses over seven training sessions (P < 0.05; Fig. 4A), PGC-1α protein expression continued to increase, attaining a plateau (+42%, P < 0.05) by the 5–7th training session (Fig. 4C).
PGC-1β
PGC-1β mRNA increased 2.6-fold 4 h after the 1st session and decreased by 24 h, but not to Pre levels (P < 0.05; Fig. 4B), as it remained up-regulated by 58% at 24 h. This pattern continued throughout training, with the content being consistently up-regulated (138–162%) 4 h after each training session, and decreasing at each 24 h time point, but staying 67–81% above the Pre level at all 24 h time points (P < 0.05).
Up-regulation of PGC-1β protein expression occurred later than PGC-1α protein expression (16%, 24 h after the 3rd session) and eventually declined to Pre level after the 7th session (P < 0.05, Fig. 4D).
PPAR isoforms
PPARβ/δ mRNA increased 50% 4 h after the 1st session and returned to the Pre level by 24 h (P < 0.05; Fig. 5A). This pattern continued throughout training. Surprisingly, no increases in PPARα or γ mRNA was observed at any time point (Fig. 5B and C).
Figure 5. Skeletal muscle PPARα, PPARβ/δ and PPARγ mRNA (A, B and C) and protein content (D, E and F) throughout 2 weeks of high-intensity interval training.
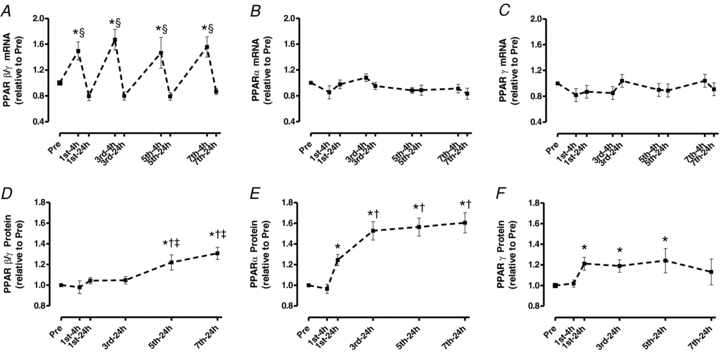
Values are means ±s.e.m. for 8–9 subjects. *Significantly different from Pre, §significantly different from all 24 h time points, †significantly different from 1st-24 h, ‡significantly different from 3rd-24 h (P < 0.05).
Up-regulation of PPARβ/δ protein expression occurred after the 5th session (21%, 24 h) and remained elevated (31%) after the 7th session (P < 0.05; Fig. 5D). PPARα (25%; 24 h) and PPARγ (18%; 24 h) protein were greater after the 1st session (P < 0.05). PPARα protein increased further after the 7th session (60%, P < 0.05; Fig. 5E) whereas PPARγ protein returned to Pre levels by the end of training (P < 0.05; Fig. 5F).
Tfam and NRF-2
Tfam mRNA increased rapidly (54%, 4 h after session 1) and remained elevated throughout training (P < 0.05; Table 2). Tfam protein content was essentially unchanged throughout training and NRF-2 protein increased by ∼50% after the 3rd training session (P < 0.05; Table 2).
Mitochondrial fusion and fission proteins
The protein contents of MFN-1 and -2 were not affected by training except for an increase in MFN-1 after the 7th training session (35%, P < 0.05; Fig. 6A and B). However, Fis-1 was up-regulated after the 3rd session (42%) and more than doubled after the 7th session (111%, P < 0.05; Fig. 6C). DRP-1 was increased after the 3rd session (47%) and remained up-regulated thereafter (P < 0.05; Fig. 6D).
Figure 6. Protein content of skeletal muscle MFN-1 (A), MFN-2 (B), Fis-1 (C) and DRP-1 (D) throughout 2 weeks of high-intensity interval training.
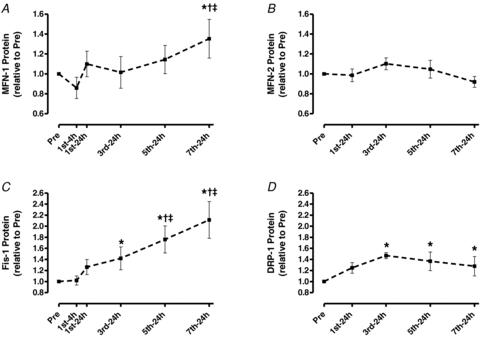
Values are means ±s.e.m. for 6–9 subjects. *Significantly different from Pre, †significantly different from 1st-24 h, ‡significantly different from 3rd-24 h (P < 0.05).
Discussion
It has been suggested that increased mitochondrial protein expression follows repeated training-induced transient increases in mRNA expression of selected genes, and that repeated bursts in mRNA expression are fundamental to the intracellular adaptive response to exercise training (Neufer & Dohm, 1993; O’Doherty et al. 1996; Williams & Neufer, 1996; Pilegaard et al. 2000; Hood, 2001; Booth & Neufer, 2006). Therefore, we examined the early time course of several potential transcriptional, translational and morphological processes that are involved in mitochondrial biogenesis during exercise training in human skeletal muscle. Importantly, the temporal resolution of this study revealed unique expression patterns of many mitochondrial and transcriptional mRNAs and proteins. In general, an increase in mRNA was observed before the initial increase in protein for a given mitochondrial enzyme and transcription protein. However, both the timing and magnitude of mRNA and protein responses varied considerably for transcription and mitochondrial proteins. Rapid up-regulation of PGC-1α, PGC-1β and PPAR β/δ mRNAs (<4 h) occurred transiently after one session and were repeatedly stimulated after each session. PGC-1α and PPARα protein expression were up-regulated rapidly (<24 h), while PGC-1β and PPAR β/δ and γ protein expression were delayed (3rd session). Similarly, despite rapid up-regulation of CS and B-HAD mRNAs (<24 h), the increase in protein expression, as suggested by their maximal activities, were also not apparent until after the 3rd session. These genes eventually became less sensitive to exercise challenges. In addition, the mitochondrial fission protein Fis-1 increased earlier during training (3rd session) and to a greater extent than the fusion protein MFN-1 (7th session).
These observations suggested that the rapid increase in the content of PGC-1α may have been responsible for the induction of PPARs and selected mitochondrial protein expression. Collectively, this investigation reveals numerous and precise time-dependent molecular events that are likely to have contributed to the early phases of mitochondrial biogenesis during training in human skeletal muscle.
Temporal responses in mitochondrial biogenesis during training
Several weeks of endurance exercise training increases the maximal activities and content of mitochondrial proteins involved in lipid and carbohydrate oxidation (Holloszy & Coyle, 1984). These responses have also been observed after only six to seven training sessions of moderate or high intensity exercise (Gulve & Spina, 1995; Chesley et al. 1996; Burgomaster et al. 2005; Talanian et al. 2007). The present study demonstrated that increases in the maximal activities of CS and β-HAD, as well as mtDNA content, can occur after only three high-intensity interval training sessions over a five day period. The preceding and sustained increases in CS and β-HAD mRNA support the hypothesis that the cumulative effect of transient bursts in gene expression encoding mitochondrial proteins are required for the adaptive protein responses to exercise training (Williams & Neufer, 1996; Pilegaard et al. 2000; Fluck & Hoppeler, 2003; Booth & Neufer, 2006). Furthermore, the initial increases in mRNA encoding of certain oxidative enzymes (CS and COX-IV) were more delayed after the 1st session relative to the mRNAs encoding some of the transcription proteins (PGC-1α, PGC-1β and PPAR β/δ). These late vs. early expression rates for oxidative enzymes and transcription proteins has also been reported recently in human skeletal muscle (Leick et al. 2009) and was previously suggested to be a reflection of the transient importance of certain transcriptional proteins in mediating an early adaptive response to exercise (Williams & Neufer, 1996; Booth & Neufer, 2006). Our data support this suggestion but further evidence is required to determine if this concept is true.
Temporal relationship between PGC-1 isoforms and mitochondrial biogenesis
PGC-1α
Up-regulation of PGC-1α protein appears to be extremely sensitive to intense exercise in human muscle given the initial increase was observed after only one session. This early increase preceded the initial increases in CS and β-HAD maximal activities (3rd session). However, this increase in PGC-1α occurred after the first increase in β-HAD mRNA and concurrent with the first CS mRNA elevation. This suggests that Pre levels of PGC-1α are sufficient to mediate the initial transcriptional phase of selected oxidative genes involved in mitochondrial biogenesis, but that early increases in PGC-1α protein content are likely to be important to support further adaptive phases during training.
The mRNA response of PGC-1α was attenuated as training progressed despite a continual increase in training power output. Pilegaard et al. (2003) observed the opposite pattern, with increased PGC-1α mRNA 2 h after single-leg kicking exercise being greater following training. While this discrepancy may be due to differences in exercise modality, our data suggest transcriptional responses to exercise challenges are attenuated as training adaptation progresses. Hence, blunted transcriptional responses may explain the plateau phenomenon typical after muscle has adapted to training. Attenuated mRNA responses after the last exercise session were also seen for CS and β-HAD.
The mechanism by which PGC-1α mediates the initial phase of mitochondrial biogenesis has been previously explored. Holloszy's group (Wright et al. 2007) observed a greater protein content of several mitochondrial enzymes immediately after prolonged exercise in rats (6 h of swimming) which preceded increased PGC-1α protein. Furthermore, increased PGC-1α protein was preceded by rapid translocation of PGC-1α into the nucleus followed by NRF-1 and -2 binding to DNA and increased CS mRNA. In contrast, Hargreaves's group (McGee & Hargreaves, 2004) did not observe an increase in nuclear PGC-1α protein immediately after cycling (1 h at 70%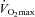 ) in human skeletal muscle, but did find indices of increased nuclear PGC-1α activity (co-activation of the transcription factor MEF-2) with no change in total PGC-1α protein content. However, Little et al. (2010) recently demonstrated acute exercise (90 min cycling at ∼65%
) in human skeletal muscle, but did find indices of increased nuclear PGC-1α activity (co-activation of the transcription factor MEF-2) with no change in total PGC-1α protein content. However, Little et al. (2010) recently demonstrated acute exercise (90 min cycling at ∼65%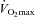 ) increased nuclear PGC-1α protein content also with no change in total PGC-1α protein. These observations suggest PGC-1α is activated in response to exercise in both rat and human skeletal muscle but the role of bolstering nuclear PGC-1α contents via nuclear translocation from the cytoplasmic pool remains equivocal in humans. Hence, activation of existing PGC-1α may explain the initial increases in CS and β-HAD mRNA prior to increases in PGC-1α protein in the present study. Furthermore, our report of no increase in PGC-1α protein 4 h after the 1st training session is similar to previous reports at the end of exercise in rats (Wright et al. 2007) and humans (McGee & Hargreaves, 2004), although an increase has been observed following 2 h of continuous exhaustive exercise in males (Mathai et al. 2008).
) increased nuclear PGC-1α protein content also with no change in total PGC-1α protein. These observations suggest PGC-1α is activated in response to exercise in both rat and human skeletal muscle but the role of bolstering nuclear PGC-1α contents via nuclear translocation from the cytoplasmic pool remains equivocal in humans. Hence, activation of existing PGC-1α may explain the initial increases in CS and β-HAD mRNA prior to increases in PGC-1α protein in the present study. Furthermore, our report of no increase in PGC-1α protein 4 h after the 1st training session is similar to previous reports at the end of exercise in rats (Wright et al. 2007) and humans (McGee & Hargreaves, 2004), although an increase has been observed following 2 h of continuous exhaustive exercise in males (Mathai et al. 2008).
Pilegaard et al. (Leick et al. 2008) reported that PGC-1α knock-out mice synthesized mitochondrial enzymes to a similar extent as control mice after 5 weeks of training. Given that exercise appears to activate PGC-1αin vivo (McGee & Hargreaves, 2004; Wright et al. 2007), these results suggest PGC-1α likely mediates training-induced mitochondrial biogenesis but alternative mechanisms are also involved. A possible corollary is that an absence of PGC-1α may simply impede the rate of mitochondrial biogenesis without affecting the maximal increase over time. Indeed, the early increase in PGC-1α protein in this study (<24 h) suggests greater PGC-1α content may enhance the rate of biogenesis in response to subsequent exercise challenges.
PGC-1β
Previous work reported that a single bout of exercise does not increase PGC-1β mRNA in human (Mortensen et al. 2007), mouse (Meirhaeghe et al. 2003) and rat (Koves et al. 2005) skeletal muscle. Our data contrast with these findings, as PGC-1β mRNA increased ∼2.6-fold after one exercise session (<4 h). This may be due to the high-intensity interval exercise employed in the present study which suggests this gene is more responsive to greater exercise challenges. The increase in PGC-1β protein followed by a return to Pre level by the 7th session suggests a prominent role for PGC-1β early during training. This may explain why others reported no change in PGC-1β mRNA in humans following 10 weeks of single leg-kick training (Mortensen et al. 2007).
In a broader context, the varied responses of PGC-1α and β over only seven sessions underscore the importance of assessing mRNA and protein over multiple time points during training. Transient responses in certain genes may occur early and be missed with an end-point study design.
Temporal relationship between transcription factors and mitochondrial biogenesis
The PPAR β/δ, α and γ isoforms regulate the expression of nuclear genes involved in fatty acid metabolism, are co-activated by PGC-1α (Gilde & Van Bilsen, 2003) and possess considerable influence on either lipid catabolic capacities (Gilde & Van Bilsen, 2003; Seedorf & Aberle, 2007) or skeletal muscle insulin sensitivity (Hevener et al. 2003; Norris et al. 2003). We found that acute high-intensity exercise increased PPARα and γ protein whereas multiple bouts were required for PPAR β/δ protein expression. Similar to the arguments presented for PGC-1α, the initial increases in PPARα and γ protein were likely not to be important for the initial increases in CS and β-HAD mRNA since all of these increases were observed after one training session. However, the initial increases in PPARα and γ protein may have contributed to the relatively high rate of increase in β-HAD maximal activity by the 3rd session given that the β-HAD gene contains a PPAR response element (Zhang et al. 1992). Conversely, the delayed increase in PPARβ/δ suggests an important role near the end of the 2 week training period although its contribution to initial phases of mitochondrial biogenesis also seems likely. Finally, increased PPARα and γ protein during training were not related to greater mRNA of these genes at the measured time points. This suggests these mRNA transcripts may appear at points other than 4 and 24 h. In support of this, increased PPARα mRNA has been reported 6 h after exercise but not 2 or 24 h into recovery in humans (Pilegaard et al. 2003).
Our data suggests that Tfam protein experienced dynamic turnover early during training but essentially did not change after 2 weeks. Furthermore, the increase in mtDNA with no change in Tfam protein suggests that activation of existing Tfam may contribute to mitochondrial biogenesis during training, which is similar to previous findings (Gordon et al. 2001). The erratic increases in NRF-2 protein are difficult to interpret but considerable variability in the mRNA of NRF-2 and Tfam in human skeletal muscle following exercise have also been reported previously (Pilegaard et al. 2003).
Speculations on the transcriptional regulation of mitochondrial biogenesis during training
Interestingly, certain genes for transcription proteins (PGC-1α, PGC-1β, PPARβ/δ and Tfam) responded with greater magnitudes than any of the genes for mitochondrial proteins. This suggests that a considerable effort is made to quickly boost the transcriptional capacity of the cell, which indeed happens early for the PGC-1 and PPAR isoforms. Importantly, these rapid increases in transcription proteins are likely to contribute to the very early and modest increases in mitochondrial enzymes. The greater up-regulation of these transcription protein contents relative to the genes for the oxidative enzymes CS and COX-IV has also been proposed previously (Williams & Neufer, 1996; Booth & Neufer, 2006). Nevertheless, many protein increases appeared to occur in response to the cumulative effects of transient increases in their respective mRNA transcripts following multiple exercise sessions. A stark example is PGC-1α whereby very large increases in mRNA contributed to gradual increases in PGC-1α protein that preceded the increase in CS maximal activity (shown schematically in Fig. 7) as well as β-HAD maximal activity and COX-IV protein. Overall, these data suggest a complexity in the transcriptional regulation of exercise-induced mitochondrial biogenesis that has not been apparent in previous investigations. While activation of these and other transcription proteins is surely critical for increasing the expression of metabolic genes during exercise, increases in the content of many of these proteins between only 1–5 exercise sessions (PGC-1α, PGC-1β, PPARα, β/δ, γ and NRF-2) likely increases the ‘transcriptional capacity’ of the cell such that the rate and magnitude of mitochondrial biogenesis is amplified early in training (Wright et al. 2007).
Figure 7.
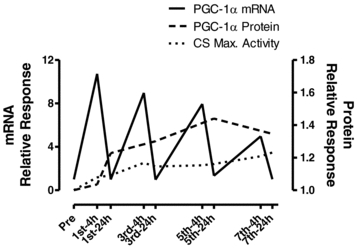
Representative figure demonstrating temporal responses and relationship of PGC-1α mRNA, PGC-1α protein and CS maximal activity throughout 2 weeks of high-intensity interval training
Temporal relationship between mitochondrial biogenesis and fusion and fission proteins
A fused mitochondrial reticulum possesses a greater capacity to oxidize carbohydrate and fatty acid (Bach et al. 2003; Pich et al. 2005). Hence, the greater mitochondrial oxidative capacity following training may be due in part to greater fusion and elongation of the reticular network (Kirkwood et al. 1987). We hypothesized that the fusion proteins MFN-1 and -2 would increase and the fission proteins, Fis-1 and DRP-1 would decrease resulting in a greater ratio of fusion to fission proteins. Unexpectedly, training stimulated a delayed increase in MFN-1 protein with no change in MFN-2 but considerable increases in both Fis-1 and DRP-1. In support of these findings, a recent report showed decreased MFN-1 mRNA and protein and MFN-2 mRNA during and following 150 min of exercise in rat skeletal muscle whereas Fis-1 mRNA and protein increased considerably after exercise (Ding et al. 2009). Conceivably, fission may be required to remodel and/or expand the reticulum during proliferation or to disassemble parts of the reticulum into fragments for re-location (Skulachev, 2001) and fusion into other regions of the network. However, in the absence of visual confirmation of morphology, we are left to speculate that the previous demonstration of increased reticular formation following training (Kirkwood et al. 1987) occurred in the present study through activation of existing fusion proteins.
Measurement of PGC-1α protein
Within the scientific community concerns have been raised about the specificity of some PGC-1α antibodies and, therefore, the proper detection of the PGC-1α protein band on Western blots. We, therefore, in the next paragraphs summarize the objective evidence that the antibody we have used in this and earlier publications is specific.
At first glance testing of the PGC-1α antibody specificity in tissues of PGC-1α KO mice would seem to be the ‘gold standard’ for determining the specificity of antibodies against PGC-1α. Some reports appear to show that there are multiple PGC-1α bands in tissues of PGC-1α KO mice (Lin et al. 2004; Handschin et al. 2007b; Estall et al. 2009). However, one study (Lin et al. 2004) used a polyclonal antibody in PGC-1α KO mice, which would of course yield multiple bands. In two other reports with PGC-1α KO mice it is not clear whether polyclonal or monoclonal antibodies were used (Handschin et al. 2007b; Estall et al. 2009). However, a recent published report employing a monoclonal antibody against PGC-1α demonstrates that, as expected, PGC-1α protein is found in muscle of WT mice (∼100 kDa), but it is not detected in muscle of PGC-1α KO mice (Little et al. 2010). As far as we can determine this appears to be the only published investigation in which the specificity of a monoclonal antibody against PGC-1α has been tested explicitly. These very recent data (Little et al. 2010) demonstrate that PGC-1α protein can be reliably detected, and these observations contrast with difficult to substantiate, unpublished, informal concerns that have been raised about detecting PGC-1α in PGC-1α KO mice.
We (Benton et al. 2008a,b, 2010; Holloway et al. 2010; Buddo & Bonen unpublished data) and other groups (Aquilano et al. 2010; Consitt et al. 2010; Xiong et al. 2010) have utilized alternative methods to validate PGC-1α antibodies, namely overexpression or knock down of PGC-1α in mammalian muscle and selected cell lines. In all of these studies that have employed genetic approaches, the polyclonal antibodies against PGC-1α detected the expected increases and decreases in this protein, as well as the expected PGC-1α-induced changes in mitochondrial content. Specifically, in the work from our laboratory we have overexpressed PGC-1α modestly in muscle to be within physiologically realistic limits (Benton et al. 2008a,b, 2010; Holloway et al. 2010; Buddo & Bonen unpublished data). In these PGC-1α transfection studies we routinely observe an increase of 25–40% in PGC-1α protein, a change that closely reflects the increase observed in PGC-1α mRNA (Benton et al. 2008a,b, 2010; Holloway et al. 2010; Buddo & Bonen unpublished data). Importantly, in these experiments inducing PGC-1α upregulation (25–40%) fails to alter the content of the non-specific bands that are detected when using a polyclonal antibody. Specifically, in 17 randomly selected experiments from among ∼50 experiments performed by four different people in our group (Benton et al. 2008a,b, 2010; Buddo & Bonen unpublished data), non-specific bands in sham-transfected muscle are 100 ± 4% while non-specific bands in the contralateral PGC-1α transfected muscles are 102 ± 5.9% (P > 0.05). Furthermore, the excellent correlations that we obtain among PGC-1α protein and many proteins regulated by PGC-1α (range of correlation coefficients: r= 0.87–0.99), but not with non-PGC-1α-regulated proteins (r < 0.30) (Benton et al. 2008a,b, 2010), also strongly suggest that we are measuring PGC-1α protein. Comparable observations have been published recently by others in selected cell lines transfected with PGC-1α (Aquilano et al. 2010; Consitt et al. 2010; Xiong et al. 2010). All of these very recent genetic approaches by us and others suggest strongly that PGC-1α protein can be detected appropriately by Western blotting with polyclonal antibodies. Despite this we do recognize that these experiments may require further confirmation and that additional work is required to establish with certainty that PGC-1α is being measured correctly.
We do recognize that there have been ongoing concerns with the detection of PGC-1α protein for some years. However, on balance, it would seem that in the past several years many groups have independently amassed considerable evidence, using genetic manipulations, to allay, in part, the earlier concerns about the detection of PGC-1α protein. This work has been performed in animal and selected cell lines. We presume that by using the same antibody in human muscle as has been used in our published animal studies in which we overexpressed PGC-1α (Benton et al. 2008a,b, 2010; Holloway et al. 2010; Buddo & Bonen unpublished data) that changes in human muscle PGC-1α protein are therefore also appropriately reflected. Similar assumptions have been made by others when performing experimental work in humans (Russell et al. 2003; Kuhl et al. 2006; Burgomaster et al. 2008; Little et al. 2010). Note, however, that care is required with commercial antibodies and we have found it necessary to routinely check each lot of PGC-1α antibody obtained as to its suitability for our work.
Summary
This investigation revealed numerous rapid mRNA and protein expression responses to repeated high-intensity interval training sessions with a temporal precision that has not been reported previously. Our findings demonstrated the occurrence of repeated, transient increases in mRNA to produce sustained increases in the content of transcription and metabolic proteins as previously proposed (Williams & Neufer, 1996; Booth & Neufer, 2006). The mRNA response to exercise was attenuated as the muscle adapted to the exercise challenge, even as the training intensity increased. Nevertheless, we found that increases in mitochondrial proteins occurred within five days and following three sessions of high-intensity interval training. Furthermore, the rapid increase in β-HAD mRNA (4 h post-session 1) suggests the initial increases in mRNA for certain mitochondrial proteins can occur without an initial increase in the protein content of the PGC-1 and PPAR isoforms, and NRF-2 and Tfam. However, the rapid increase in PGC-1 and PPAR isoform protein contents and NRF-2 within three exercise sessions may accelerate the adaptive response to subsequent exercise bouts. This may be especially true for PGC-1α and PPARα, which increased after one session but before the initial increase in mitochondrial proteins. Finally, the increase in MFN-1 supported the greater degree of fusion that occurs during training whereas the increases in Fis-1 and DRP-1 suggested fission was also required for expanding and/or re-modelling the morphology of the mitochondrial reticulum. These observations reveal a dynamic temporal sequence of molecular (mRNA and protein expression) and potential morphological events during the early periods of exercise training in human skeletal muscle that has not been apparent previously. These unique responses raise the intriguing possibility that the contribution of each transcription protein in mediating mitochondrial biogenesis may be time dependent.
Acknowledgments
We thank the subjects for their extraordinary effort and dedication. We also thank Dr P. Darrell Neufer for his expertise and valuable insight during the preparation of this manuscript. This study was supported by the Natural Sciences and Engineering Research Council of Canada (L.L.S. and A.B.) and the Canadian Institutes of Health Research (L.L.S., G.J.F.H. and A.B.). A.B. is the Canada Research Chair in Metabolism and Health. C.G.R.P. was supported by an Ontario Graduate Scholarship.
Author contributions
All experimental trials including human testing and training as well as laboratory analyses were performed in the Department of Human Health and Nutritional Sciences at the University of Guelph and the Department of Medicine at McMaster University. The authors contributions to the completion of this investigation were as follows: conception and design of the experiments (all authors); collection (C.G.R.P., J.L., G.P.H., G.J.F.H.), analysis (all authors) and interpretation of data (all authors); drafting the article or revising it critically for important intellectual content (all authors).
Author's present address
C. G. R. Perry: Department of Physiology, East Carolina University, Greenville, NC 27834, USA (perrych@ecu.edu).
References
- Aquilano K, Vigilanza P, Baldelli S, Pagliei B, Rotilio G, Ciriolo MR. Peroxisome proliferator-activated receptor γ co-activator 1α (PGC-1α) and sirtuin 1 (SIRT1) reside in mitochondria: possible direct function in mitochondrial biogenesis. J Biol Chem. 2010;285:21590–21599. doi: 10.1074/jbc.M109.070169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arany Z, Lebrasseur N, Morris C, Smith E, Yang W, Ma Y, Chin S, Spiegelman BM. The transcriptional coactivator PGC-1β drives the formation of oxidative type IIX fibers in skeletal muscle. Cell Metab. 2007;5:35–46. doi: 10.1016/j.cmet.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Bach D, Pich S, Soriano FX, Vega N, Baumgartner B, Oriola J, Daugaard JR, Lloberas J, Camps M, Zierath JR, Rabasa-Lhoret R, Wallberg-Henriksson H, Laville M, Palacin M, Vidal H, Rivera F, Brand M, Zorzano A. Mitofusin-2 determines mitochondrial network architecture and mitochondrial metabolismA novel regulatory mechanism altered in obesity. J Biol Chem. 2003;278:17190–17197. doi: 10.1074/jbc.M212754200. [DOI] [PubMed] [Google Scholar]
- Bengtsson J, Gustafsson T, Widegren U, Jansson E, Sundberg CJ. Mitochondrial transcription factor A and respiratory complex IV increase in response to exercise training in humans. Pflugers Arch. 2001;443:61–66. doi: 10.1007/s004240100628. [DOI] [PubMed] [Google Scholar]
- Benton CR, Holloway GP, Han XX, Yoshida Y, Snook LA, Lally J, Glatz JF, Luiken JJ, Chabowski A, Bonen A. Increased levels of peroxisome proliferator-activated receptor γ, coactivator 1α (PGC-1α) improve lipid utilisation, insulin signalling and glucose transport in skeletal muscle of lean and insulin-resistant obese Zucker rats. Diabetologia. 2010;53:2008–2019. doi: 10.1007/s00125-010-1773-1. [DOI] [PubMed] [Google Scholar]
- Benton CR, Nickerson JG, Lally J, Han XX, Holloway GP, Glatz JF, Luiken JJ, Graham TE, Heikkila JJ, Bonen A. Modest PGC-1α overexpression in muscle in vivo is sufficient to increase insulin sensitivity and palmitate oxidation in subsarcolemmal, not intermyofibrillar, mitochondria. J Biol Chem. 2008a;283:4228–4240. doi: 10.1074/jbc.M704332200. [DOI] [PubMed] [Google Scholar]
- Benton CR, Yoshida Y, Lally J, Han XX, Hatta H, Bonen A. PGC-1α increases skeletal muscle lactate uptake by increasing the expression of MCT1 but not MCT2 or MCT4. Physiol Genom. 2008b;35:45–54. doi: 10.1152/physiolgenomics.90217.2008. [DOI] [PubMed] [Google Scholar]
- Bergmeyer HU. Methods in Enzymatic Analyses. New York: Academic; 1974. [Google Scholar]
- Booth F, Neufer PD. Exercise Genomics and Proteomics. In: Tipton CM, editor. ACSM's Advanced Exercise Physiology. Baltimore, MD: Lippincott Williams & Wilkins; 2006. pp. 623–651. [Google Scholar]
- Burgomaster KA, Howarth KR, Phillips SM, Rakobowchuk M, Macdonald MJ, McGee SL, Gibala MJ. Similar metabolic adaptations during exercise after low volume sprint interval and traditional endurance training in humans. J Physiol. 2008;586:151–160. doi: 10.1113/jphysiol.2007.142109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgomaster KA, Hughes SC, Heigenhauser GJ, Bradwell SN, Gibala MJ. Six sessions of sprint interval training increases muscle oxidative potential and cycle endurance capacity in humans. J Appl Physiol. 2005;98:1985–1990. doi: 10.1152/japplphysiol.01095.2004. [DOI] [PubMed] [Google Scholar]
- Cartoni R, Leger B, Hock MB, Praz M, Crettenand A, Pich S, Ziltener JL, Luthi F, Deriaz O, Zorzano A, Gobelet C, Kralli A, Russell AP. Mitofusins 1/2 and ERRα expression are increased in human skeletal muscle after physical exercise. J Physiol. 2005;567:349–358. doi: 10.1113/jphysiol.2005.092031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerveny KL, Tamura Y, Zhang Z, Jensen RE, Sesaki H. Regulation of mitochondrial fusion and division. Trends Cell Biol. 2007;17:563–569. doi: 10.1016/j.tcb.2007.08.006. [DOI] [PubMed] [Google Scholar]
- Chesley A, Heigenhauser GJ, Spriet LL. Regulation of muscle glycogen phosphorylase activity following short-term endurance training. Am J Physiol Endocrinol Metab. 1996;270:E328–335. doi: 10.1152/ajpendo.1996.270.2.E328. [DOI] [PubMed] [Google Scholar]
- Consitt LA, Bell JA, Koves TR, Muoio DM, Hulver MW, Haynie KR, Dohm GL, Houmard JA. Peroxisome proliferator-activated receptor-gamma coactivator-1alpha overexpression increases lipid oxidation in myocytes from extremely obese individuals. Diabetes. 2010;59:1407–1415. doi: 10.2337/db09-1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding H, Jiang N, Liu H, Liu X, Liu D, Zhao F, Wen L, Liu S, Ji LL, Zhang Y. Response of mitochondrial fusion and fission protein gene expression to exercise in rat skeletal muscle. Biochim Biophys Acta. 2009;1800:250–256. doi: 10.1016/j.bbagen.2009.08.007. [DOI] [PubMed] [Google Scholar]
- Estall JL, Kahn M, Cooper MP, Fisher FM, Wu MK, Laznik D, Qu L, Cohen DE, Shulman GI, Spiegelman BM. Sensitivity of lipid metabolism and insulin signaling to genetic alterations in hepatic peroxisome proliferator-activated receptor-γ coactivator-1α expression. Diabetes. 2009;58:1499–1508. doi: 10.2337/db08-1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fluck M, Hoppeler H. Molecular basis of skeletal muscle plasticity: from gene to form and function. Rev Physiol Biochem Pharmacol. 2003;146:159–216. doi: 10.1007/s10254-002-0004-7. [DOI] [PubMed] [Google Scholar]
- Garcia-Roves PM, Huss J, Holloszy JO. Role of calcineurin in exercise-induced mitochondrial biogenesis. Am J Physiol Endocrinol Metab. 2006;290:E1172–1179. doi: 10.1152/ajpendo.00633.2005. [DOI] [PubMed] [Google Scholar]
- Gibala MJ, Little JP, van Essen M, Wilkin GP, Burgomaster KA, Safdar A, Raha S, Tarnopolsky MA. Short-term sprint interval versus traditional endurance training: similar initial adaptations in human skeletal muscle and exercise performance. J Physiol. 2006;575:901–911. doi: 10.1113/jphysiol.2006.112094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilde AJ, Van Bilsen M. Peroxisome proliferator-activated receptors (PPARS): regulators of gene expression in heart and skeletal muscle. Acta Physiol Scand. 2003;178:425–434. doi: 10.1046/j.1365-201X.2003.01161.x. [DOI] [PubMed] [Google Scholar]
- Gleyzer N, Vercauteren K, Scarpulla RC. Control of mitochondrial transcription specificity factors (TFB1M and TFB2M) by nuclear respiratory factors (NRF-1 and NRF-2) and PGC-1 family coactivators. Mol Cell Biol. 2005;25:1354–1366. doi: 10.1128/MCB.25.4.1354-1366.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodyear LJ, Kahn BB. Exercise, glucose transport, and insulin sensitivity. Annu Rev Med. 1998;49:235–261. doi: 10.1146/annurev.med.49.1.235. [DOI] [PubMed] [Google Scholar]
- Gordon JW, Rungi AA, Inagaki H, Hood DA. Effects of contractile activity on mitochondrial transcription factor A expression in skeletal muscle. J Appl Physiol. 2001;90:389–396. doi: 10.1152/jappl.2001.90.1.389. [DOI] [PubMed] [Google Scholar]
- Gulve EA, Spina RJ. Effect of 7–10 days of cycle ergometer exercise on skeletal muscle GLUT-4 protein content. J Appl Physiol. 1995;79:1562–1566. doi: 10.1152/jappl.1995.79.5.1562. [DOI] [PubMed] [Google Scholar]
- Gurd BJ, Yoshida Y, Lally J, Holloway GP, Bonen A. The deacetylase enzyme SIRT1 is not associated with oxidative capacity in rat heart and skeletal muscle and its overexpression reduces mitochondrial biogenesis. J Physiol. 2009;587:1817–1828. doi: 10.1113/jphysiol.2008.168096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handschin C, Chin S, Li P, Liu F, Maratos-Flier E, Lebrasseur NK, Yan Z, Spiegelman BM. Skeletal muscle fiber-type switching, exercise intolerance, and myopathy in PGC-1α muscle-specific knock-out animals. J Biol Chem. 2007a;282:30014–30021. doi: 10.1074/jbc.M704817200. [DOI] [PubMed] [Google Scholar]
- Handschin C, Choi CS, Chin S, Kim S, Kawamori D, Kurpad AJ, Neubauer N, Hu J, Mootha VK, Kim YB, Kulkarni RN, Shulman GI, Spiegelman BM. Abnormal glucose homeostasis in skeletal muscle-specific PGC-1α knockout mice reveals skeletal muscle-pancreatic β cell crosstalk. J Clin Invest. 2007b;117:3463–3474. doi: 10.1172/JCI31785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellsten Y, Nielsen JJ, Lykkesfeldt J, Bruhn M, Silveira L, Pilegaard H, Bangsbo J. Antioxidant supplementation enhances the exercise-induced increase in mitochondrial uncoupling protein 3 and endothelial nitric oxide synthase mRNA content in human skeletal muscle. Free Radic Biol Med. 2007;43:353–361. doi: 10.1016/j.freeradbiomed.2007.02.029. [DOI] [PubMed] [Google Scholar]
- Hevener AL, He W, Barak Y, Le J, Bandyopadhyay G, Olson P, Wilkes J, Evans RM, Olefsky J. Muscle-specific Pparg deletion causes insulin resistance. Nat Med. 2003;9:1491–1497. doi: 10.1038/nm956. [DOI] [PubMed] [Google Scholar]
- Holloszy JO, Booth FW. Biochemical adaptations to endurance exercise in muscle. Annu Rev Physiol. 1976;38:273–291. doi: 10.1146/annurev.ph.38.030176.001421. [DOI] [PubMed] [Google Scholar]
- Holloszy JO, Coyle EF. Adaptations of skeletal muscle to endurance exercise and their metabolic consequences. J Appl Physiol. 1984;56:831–838. doi: 10.1152/jappl.1984.56.4.831. [DOI] [PubMed] [Google Scholar]
- Holloway GP, Gurd BJ, Snook LA, Lally J, Bonen A. Compensatory increases in nuclear PGC1α protein are primarily associated with subsarcolemmal mitochondrial adaptations in ZDF rats. Diabetes. 2010;59:819–828. doi: 10.2337/db09-1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holloway GP, Perry CG, Thrush AB, Heigenhauser GJ, Dyck DJ, Bonen A, Spriet LL. PGC-1α's relationship with skeletal muscle palmitate oxidation is not present with obesity despite maintained PGC-1α and PGC-1β protein. Am J Physiol Endocrinol Metab. 2008;294:E1060–1069. doi: 10.1152/ajpendo.00726.2007. [DOI] [PubMed] [Google Scholar]
- Hood DA. Invited Review: contractile activity-induced mitochondrial biogenesis in skeletal muscle. J Appl Physiol. 2001;90:1137–1157. doi: 10.1152/jappl.2001.90.3.1137. [DOI] [PubMed] [Google Scholar]
- Hoppeler H. Exercise-induced ultrastructural changes in skeletal muscle. Int J Sports Med. 1986;7:187–204. doi: 10.1055/s-2008-1025758. [DOI] [PubMed] [Google Scholar]
- Hoppeler H, Howald H, Conley K, Lindstedt SL, Claassen H, Vock P, Weibel ER. Endurance training in humans: aerobic capacity and structure of skeletal muscle. J Appl Physiol. 1985;59:320–327. doi: 10.1152/jappl.1985.59.2.320. [DOI] [PubMed] [Google Scholar]
- Hoppeler H, Luthi P, Claassen H, Weibel ER, Howald H. The ultrastructure of the normal human skeletal muscle. A morphometric analysis on untrained men, women and well-trained orienteers. Pflugers Arch. 1973;344:217–232. doi: 10.1007/BF00588462. [DOI] [PubMed] [Google Scholar]
- Horowitz JF, Leone TC, Feng W, Kelly DP, Klein S. Effect of endurance training on lipid metabolism in women: a potential role for PPARα in the metabolic response to training. Am J Physiol Endocrinol Metab. 2000;279:E348–355. doi: 10.1152/ajpendo.2000.279.2.E348. [DOI] [PubMed] [Google Scholar]
- Huss JM, Kelly DP. Nuclear receptor signaling and cardiac energetics. Circ Res. 2004;95:568–578. doi: 10.1161/01.RES.0000141774.29937.e3. [DOI] [PubMed] [Google Scholar]
- Joseph AM, Pilegaard H, Litvintsev A, Leick L, Hood DA. Control of gene expression and mitochondrial biogenesis in the muscular adaptation to endurance exercise. Essays Biochem. 2006;42:13–29. doi: 10.1042/bse0420013. [DOI] [PubMed] [Google Scholar]
- Kelly DP, Scarpulla RC. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. 2004;18:357–368. doi: 10.1101/gad.1177604. [DOI] [PubMed] [Google Scholar]
- Kirkwood SP, Munn EA, Brooks GA. Mitochondrial reticulum in limb skeletal muscle. Am J Physiol Cell Physiol. 1986;251:C395–402. doi: 10.1152/ajpcell.1986.251.3.C395. [DOI] [PubMed] [Google Scholar]
- Kirkwood SP, Packer L, Brooks GA. Effects of endurance training on a mitochondrial reticulum in limb skeletal muscle. Arch Biochem Biophys. 1987;255:80–88. doi: 10.1016/0003-9861(87)90296-7. [DOI] [PubMed] [Google Scholar]
- Koves TR, Li P, An J, Akimoto T, Slentz D, Ilkayeva O, Dohm GL, Yan Z, Newgard CB, Muoio DM. Peroxisome proliferator-activated receptor-γ co-activator 1α-mediated metabolic remodeling of skeletal myocytes mimics exercise training and reverses lipid-induced mitochondrial inefficiency. J Biol Chem. 2005;280:33588–33598. doi: 10.1074/jbc.M507621200. [DOI] [PubMed] [Google Scholar]
- Kuhl JE, Ruderman NB, Musi N, Goodyear LJ, Patti ME, Crunkhorn S, Dronamraju D, Thorell A, Nygren J, Ljungkvist O, Degerblad M, Stahle A, Brismar TB, Andersen KL, Saha AK, Efendic S, Bavenholm PN. Exercise training decreases the concentration of malonyl-CoA and increases the expression and activity of malonyl-CoA decarboxylase in human muscle. Am J Physiol Endocrinol Metab. 2006;290:E1296–1303. doi: 10.1152/ajpendo.00341.2005. [DOI] [PubMed] [Google Scholar]
- Leick L, Plomgaard P, Gronlokke L, Al-Abaiji F, Wojtaszewski JF, Pilegaard H. Endurance exercise induces mRNA expression of oxidative enzymes in human skeletal muscle late in recovery. Scand J Med Sci Sports. 2009;20:593–599. doi: 10.1111/j.1600-0838.2009.00988.x. [DOI] [PubMed] [Google Scholar]
- Leick L, Wojtaszewski JF, Johansen ST, Kiilerich K, Comes G, Hellsten Y, Hidalgo J, Pilegaard H. PGC-1α is not mandatory for exercise- and training-induced adaptive gene responses in mouse skeletal muscle. Am J Physiol Endocrinol Metab. 2008;294:E463–474. doi: 10.1152/ajpendo.00666.2007. [DOI] [PubMed] [Google Scholar]
- Lin J, Wu PH, Tarr PT, Lindenberg KS, St-Pierre J, Zhang CY, Mootha VK, Jager S, Vianna CR, Reznick RM, Cui L, Manieri M, Donovan MX, Wu Z, Cooper MP, Fan MC, Rohas LM, Zavacki AM, Cinti S, Shulman GI, Lowell BB, Krainc D, Spiegelman BM. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1α null mice. Cell. 2004;119:121–135. doi: 10.1016/j.cell.2004.09.013. [DOI] [PubMed] [Google Scholar]
- Little JP, Safdar A, Cermak N, Tarnopolsky MA, Gibala MJ. Acute endurance exercise increases the nuclear abundance of PGC-1α in trained human skeletal muscle. Am J Physiol Regul Integr Comp Physiol. 2010;298:R912–917. doi: 10.1152/ajpregu.00409.2009. [DOI] [PubMed] [Google Scholar]
- Mathai AS, Bonen A, Benton CR, Robinson DL, Graham TE. Rapid exercise-induced changes in PGC-1α mRNA and protein in human skeletal muscle. J Appl Physiol. 2008;105:1098–1105. doi: 10.1152/japplphysiol.00847.2007. [DOI] [PubMed] [Google Scholar]
- McGee SL, Hargreaves M. Exercise and myocyte enhancer factor 2 regulation in human skeletal muscle. Diabetes. 2004;53:1208–1214. doi: 10.2337/diabetes.53.5.1208. [DOI] [PubMed] [Google Scholar]
- Meirhaeghe A, Crowley V, Lenaghan C, Lelliott C, Green K, Stewart A, Hart K, Schinner S, Sethi JK, Yeo G, Brand MD, Cortright RN, O’Rahilly S, Montague C, Vidal-Puig AJ. Characterization of the human, mouse and rat PGC1β (peroxisome-proliferator-activated receptor-γ co-activator 1β) gene in vitro and in vivo. Biochem J. 2003;373:155–165. doi: 10.1042/BJ20030200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen OH, Plomgaard P, Fischer CP, Hansen AK, Pilegaard H, Pedersen BK. PGC-1β is downregulated by training in human skeletal muscle: no effect of training twice every second day vsonce daily on expression of the PGC-1 family. J Appl Physiol. 2007;103:1536–1542. doi: 10.1152/japplphysiol.00575.2007. [DOI] [PubMed] [Google Scholar]
- Neufer PD, Dohm GL. Exercise induces a transient increase in transcription of the GLUT-4 gene in skeletal muscle. Am J Physiol Cell Physiol. 1993;265:C1597–1603. doi: 10.1152/ajpcell.1993.265.6.C1597. [DOI] [PubMed] [Google Scholar]
- Norrbom J, Sundberg CJ, Ameln H, Kraus WE, Jansson E, Gustafsson T. PGC-1α mRNA expression is influenced by metabolic perturbation in exercising human skeletal muscle. J Appl Physiol. 2004;96:189–194. doi: 10.1152/japplphysiol.00765.2003. [DOI] [PubMed] [Google Scholar]
- Norrbom J, Wallman SE, Gustafsson T, Rundqvist H, Jansson E, Sundberg CJ. Training response of mitochondrial transcription factors in human skeletal muscle. Acta physiologica (Oxf) 2010;198:71–79. doi: 10.1111/j.1748-1716.2009.02030.x. [DOI] [PubMed] [Google Scholar]
- Norris AW, Chen L, Fisher SJ, Szanto I, Ristow M, Jozsi AC, Hirshman MF, Rosen ED, Goodyear LJ, Gonzalez FJ, Spiegelman BM, Kahn CR. Muscle-specific PPARγ-deficient mice develop increased adiposity and insulin resistance but respond to thiazolidinediones. J Clin Invest. 2003;112:608–618. doi: 10.1172/JCI17305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Doherty RM, Bracy DP, Granner DK, Wasserman DH. Transcription of the rat skeletal muscle hexokinase II gene is increased by acute exercise. J Appl Physiol. 1996;81:789–793. doi: 10.1152/jappl.1996.81.2.789. [DOI] [PubMed] [Google Scholar]
- Ogata T, Yamasaki Y. Ultra-high-resolution scanning electron microscopy of mitochondria and sarcoplasmic reticulum arrangement in human red, white, and intermediate muscle fibers. Anat Rec. 1997;248:214–223. doi: 10.1002/(SICI)1097-0185(199706)248:2<214::AID-AR8>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Perry CG, Heigenhauser GJ, Bonen A, Spriet LL. High-intensity aerobic interval training increases fat and carbohydrate metabolic capacities in human skeletal muscle. Appl Physiol Nutr Metab. 2008;33:1112–1123. doi: 10.1139/H08-097. [DOI] [PubMed] [Google Scholar]
- Pich S, Bach D, Briones P, Liesa M, Camps M, Testar X, Palacin M, Zorzano A. The Charcot-Marie-Tooth type 2A gene product, Mfn2, up-regulates fuel oxidation through expression of OXPHOS system. Hum Mol Genet. 2005;14:1405–1415. doi: 10.1093/hmg/ddi149. [DOI] [PubMed] [Google Scholar]
- Pilegaard H, Ordway GA, Saltin B, Neufer PD. Transcriptional regulation of gene expression in human skeletal muscle during recovery from exercise. Am J Physiol Endocrinol Metab. 2000;279:E806–814. doi: 10.1152/ajpendo.2000.279.4.E806. [DOI] [PubMed] [Google Scholar]
- Pilegaard H, Saltin B, Neufer PD. Exercise induces transient transcriptional activation of the PGC-1α gene in human skeletal muscle. J Physiol. 2003;546:851–858. doi: 10.1113/jphysiol.2002.034850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- Russell AP, Feilchenfeldt J, Schreiber S, Praz M, Crettenand A, Gobelet C, Meier CA, Bell DR, Kralli A, Giacobino JP, Deriaz O. Endurance training in humans leads to fiber type-specific increases in levels of peroxisome proliferator-activated receptor-γ coactivator-1 and peroxisome proliferator-activated receptor-α in skeletal muscle. Diabetes. 2003;52:2874–2881. doi: 10.2337/diabetes.52.12.2874. [DOI] [PubMed] [Google Scholar]
- Russell AP, Hesselink MK, Lo SK, Schrauwen P. Regulation of metabolic transcriptional co-activators and transcription factors with acute exercise. FASEB J. 2005;19:986–988. doi: 10.1096/fj.04-3168fje. [DOI] [PubMed] [Google Scholar]
- Scarpulla RC. Nuclear activators and coactivators in mammalian mitochondrial biogenesis. Biochim Biophys Acta. 2002;1576:1–14. doi: 10.1016/s0167-4781(02)00343-3. [DOI] [PubMed] [Google Scholar]
- Scarpulla RC. Nuclear control of respiratory gene expression in mammalian cells. J Cell Biochem. 2006;97:673–683. doi: 10.1002/jcb.20743. [DOI] [PubMed] [Google Scholar]
- Seedorf U, Aberle J. Emerging roles of PPARδ in metabolism. Biochim Biophys Acta. 2007;1771:1125–1131. doi: 10.1016/j.bbalip.2007.04.017. [DOI] [PubMed] [Google Scholar]
- Short KR, Vittone JL, Bigelow ML, Proctor DN, Rizza RA, Coenen-Schimke JM, Nair KS. Impact of aerobic exercise training on age-related changes in insulin sensitivity and muscle oxidative capacity. Diabetes. 2003;52:1888–1896. doi: 10.2337/diabetes.52.8.1888. [DOI] [PubMed] [Google Scholar]
- Sirikul B, Gower BA, Hunter GR, Larson-Meyer DE, Newcomer BR. Relationship between insulin sensitivity and in vivo mitochondrial function in skeletal muscle. Am J Physiol Endocrinol Metab. 2006;291:E724–728. doi: 10.1152/ajpendo.00364.2005. [DOI] [PubMed] [Google Scholar]
- Skulachev VP. Mitochondrial filaments and clusters as intracellular power-transmitting cables. Trends Biochem Sci. 2001;26:23–29. doi: 10.1016/s0968-0004(00)01735-7. [DOI] [PubMed] [Google Scholar]
- Spina RJ, Chi MM, Hopkins MG, Nemeth PM, Lowry OH, Holloszy JO. Mitochondrial enzymes increase in muscle in response to 7–10 days of cycle exercise. J Appl Physiol. 1996;80:2250–2254. doi: 10.1152/jappl.1996.80.6.2250. [DOI] [PubMed] [Google Scholar]
- Srere P. Citrate synthase. In: Lowenstein J, editor. Methods in Enzymology. New York: Academic Press; 1969. pp. 3–11. [Google Scholar]
- Talanian JL, Galloway SD, Heigenhauser GJ, Bonen A, Spriet LL. Two weeks of high-intensity aerobic interval training increases the capacity for fat oxidation during exercise in women. J Appl Physiol. 2007;102:1439–1447. doi: 10.1152/japplphysiol.01098.2006. [DOI] [PubMed] [Google Scholar]
- Watt MJ, Southgate RJ, Holmes AG, Febbraio MA. Suppression of plasma free fatty acids upregulates peroxisome proliferator-activated receptor (PPAR) α and δ and PPAR coactivator 1α in human skeletal muscle, but not lipid regulatory genes. J Mol Endocrinol. 2004;33:533–544. doi: 10.1677/jme.1.01499. [DOI] [PubMed] [Google Scholar]
- Westermann B. Molecular machinery of mitochondrial fusion and fission. J Biol Chem. 2008;283:13501–13505. doi: 10.1074/jbc.R800011200. [DOI] [PubMed] [Google Scholar]
- Williams RS, Neufer PD. Regulation of gene expression in skeletal muscle by contractile activity. In: Rowell LB, Shepherd JT, editors. The Handbook of Physiology, section 12, Exercise: Regulation and Integration of Multiple Systems. New York: Oxford University Press; 1996. pp. 1124–1150. [Google Scholar]
- Wright DC, Han DH, Garcia-Roves PM, Geiger PC, Jones TE, Holloszy JO. Exercise-induced mitochondrial biogenesis begins before the increase in muscle PGC-1α expression. J Biol Chem. 2007;282:194–199. doi: 10.1074/jbc.M606116200. [DOI] [PubMed] [Google Scholar]
- Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, Spiegelman BM. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- Xiong S, Salazar G, San Martin A, Ahmad M, Patrushev N, Hilenski L, Nazarewicz RR, Ma M, Ushio-Fukai M, Alexander RW. PGC-1α serine 570 phosphorylation and GCN5-mediated acetylation by angiotensin II drive catalase down-regulation and vascular hypertrophy. J Biol Chem. 2010;285:2474–2487. doi: 10.1074/jbc.M109.065235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Marcus SL, Sajjadi FG, Alvares K, Reddy JK, Subramani S, Rachubinski RA, Capone JP. Identification of a peroxisome proliferator-responsive element upstream of the gene encoding rat peroxisomal enoyl-CoA hydratase/3-hydroxyacyl-CoA dehydrogenase. Proc Natl Acad Sci U S A. 1992;89:7541–7545. doi: 10.1073/pnas.89.16.7541. [DOI] [PMC free article] [PubMed] [Google Scholar]