

Abstract
Chronic stimulation of β-adrenoceptors with β-adrenoceptor agonists (β-agonists) can induce substantial skeletal muscle hypertrophy, but the mechanisms mediating this muscle growth have yet to be elucidated. We investigated whether chronic β-adrenoceptor stimulation in mice with the β-agonist formoterol alters the muscle anabolic response following β-adrenoceptor stimulation. Twelve-week-old C57BL/6 mice were treated for up to 28 days with a once-daily injection of either saline (control, n= 9) or formoterol (100 μg kg−1; n= 9). Rates of muscle protein synthesis were assessed at either 1, 7 or 28 days of treatment, 6 h after injection. Protein synthesis rates were higher in formoterol-treated mice at day 7 (∼1.5-fold, P < 0.05), but not at day 1 or 28. The increased muscle protein synthesis was associated with increased phosphorylation of S6K1 (r= 0.49, P < 0.01). Formoterol treatment acutely reduced maximal calpain activity by ∼25% (P < 0.05) but did not affect atrogin-1 protein levels and proteasome-mediated proteolytic activity, despite significantly enhanced phosphorylation of Akt (P < 0.05). Formoterol increased CREB phosphorylation by ∼30% (P < 0.05) and PPARγ coactivator-1α (PGC-1α) by 11-fold (P < 0.05) on day 1 only. These observations identify that formoterol treatment induces muscle anabolism, by reducing calpain activity and by enhancing protein synthesis via increased PI-3 kinase/Akt signalling.
Introduction
The biological importance of β-adrenergic signalling has been well documented in the heart but only very recently have we begun to understand the importance of this signalling pathway in skeletal muscle (Lynch & Ryall, 2008). Chronic stimulation of β-adrenoceptor signalling with β-adrenoceptor agonists (β-agonists) has been shown to induce skeletal muscle growth and could represent an effective treatment for skeletal muscle wasting conditions. In addition to promoting muscle hypertrophy in healthy animals (Maltin et al. 1987; Ryall et al. 2006), treatment with β-agonists can prevent or reverse the muscle wasting and weakness associated with numerous conditions and pathologies, including sarcopaenia (age-related muscle wasting) (Ryall et al. 2007), cancer cachexia (Busquets et al. 2004) and muscular dystrophies (Harcourt et al. 2007; Gehrig et al. 2010). Chronic treatment with formoterol, a highly selective (newer generation) β2-agonist, increased muscle mass, fibre cross-sectional area (CSA) and maximal force producing capacity in wild-type and mdx dystrophic mice (Harcourt et al. 2007; Gehrig et al. 2010) and in young, adult and aged rats (Ryall et al. 2004, 2006, 2007). However, the therapeutic potential of β-agonists for conditions associated with muscle wasting and weakness has so far been compromized by deleterious side effects, including hypertrophy and dysfunction of the heart (Ryall et al. 2008b). It is therefore important to identify the mechanisms underlying β-adrenoceptor mediated skeletal muscle hypertrophy so that targets can be identified that modulate β-adrenergic signalling to enhance muscle mass and function without side-effects. Such an outcome would have considerable clinical significance for the treatment of many muscle wasting disorders.
The hypertrophic response of skeletal muscle following chronic, high-dose β-agonist administration has been associated with an increase in muscle protein synthesis (MPS), a decrease in protein degradation, or a combination of both mechanisms (Ryall et al. 2008a). The data are equivocal as to the precise mechanisms that mediate β-agonist-induced skeletal muscle hypertrophy. Canonical β-agonist signalling has been well described and involves selective coupling to a heterotrimeric G-protein (Gαβγ) to initiate downstream signalling. Traditionally, β-agonist signalling is believed to occur via the stimulatory Gα subunit (Gαs) coupling to adenylate cyclase (AC), with production of cyclic AMP (cAMP) and subsequent activation of protein kinase A (PKA) and phosphorylation of cAMP response element binding (CREB) protein (Lynch & Ryall, 2008). The activation of this pathway has been linked to the inhibition of Ca2+-dependent proteolytic pathways, and possibly to increased mixed muscle and mitochondrial protein synthesis, via increases in transcription of genes encoding muscle regulatory factors such as MyoD and peroxisome proliferator-activated receptor γ (PPARγ) coactivator-1α (PGC-1α) (Pearen et al. 2008). The Gβγ subunits of the G-protein have also been suggested to play an active role in various cell signalling processes, which may have important roles in β-agonist-induced hypertrophy of skeletal muscle. Specifically, β-agonists initiate Gβγ-mediated activation of the phosphoinositol 3-kinase (PI3K)–protein kinase B (Akt) signalling pathway, which has been implicated as a regulator of both MPS and ubiquitin-proteasome-dependent protein degradation (Kline et al. 2007).
It has been shown previously that chronic administration of most β-agonists in rats activates MPS significantly (Maltin et al. 1989) but MPS does not appear to be increased in rats after acute (oral) administration of the (older generation) β2-agonist clenbuterol (Maltin et al. 1989), or infusion of isoproterenol, a non-specific β-agonist in humans (Robinson et al. 2010). In contrast, protein degradation is affected more rapidly (Maltin et al. 1987). Thus, the anabolic response to a single administration of a β-agonist might be different from that with chronic treatment because of differences in the activation of β-adrenoceptor signalling. To investigate whether there is a shift in the mechanism that primarily governs β-adrenergic signalling mediated muscle hypertrophy with chronic stimulation, muscle protein synthesis and degradation and the associated signalling events need to be measured at different times after β-agonist administration.
The aim of this study was to determine the pathways responsible for skeletal muscle hypertrophy after chronic β-adrenoceptor stimulation with administration of formoterol. We measured mitochondrial, myofibrillar and sarcoplasmic protein synthesis and degradation and associated signalling after 1, 7 and 28 days of β-adrenoceptor stimulation. We tested the hypothesis that changes in muscle protein degradation predominate in the initial stages of formoterol treatment which are followed by a shift to changes in MPS during the later stages of chronic β-adrenoceptor stimulation.
Methods
Animals
Twelve-week-old C57BL/6 mice (n= 72) were used in this study. All mice were obtained from the Animal Resources Centre (Canning Vale, Western Australia), and housed in the Biological Research Facility at The University of Melbourne under a 12 h light/dark cycle, with drinking water and standard chow provided ad libitum. All experiments were approved by the Animal Experimental Ethics Committee of The University of Melbourne and conducted in accordance with the Australian code of practice for the care and use of animals for scientific purposes as stipulated by the National Health and Medical Research Council (NHMRC Australia).
Mice were allocated into either control (3 groups, n= 12 per group) or formoterol treated groups (3 groups, n= 12 per group). Treated animals received 100 μg kg−1 of formoterol (Sigma-Aldrich Co., Castle Hill, NSW, Australia) in saline once daily via intraperitoneal (i.p.) injections for up to 28 days. Control mice received an identical volume of saline only. All animals were weighed daily throughout the study. We have shown previously that 4 weeks of treatment with this dose of formoterol increases muscle size and strength in mice by 20–30% (Gehrig et al. 2010).
Experimental procedures
We have shown previously that the increase in muscle fibre CSA with chronic administration of the β-agonist fenoterol was associated with a reduction in oxidative enzyme activity in selected muscles of rats (Ryall et al. 2004). As such, we hypothesized that the acute and chronic anabolic response to formoterol administration would be different in mitochondrial and myofibrillar protein fractions in skeletal muscle. Therefore, protein synthesis rates in myofibrillar, mitochondrial and sarcoplasmic fractions and muscle protein breakdown rates were determined after 1, 7 or 28 days of formoterol treatment. MPS was measured 6 h after the injection of either formoterol or saline, with food withdrawn during the last 5 h. MPS was assessed in the gastrocnemius/soleus/plantaris muscle complex using the flooding-dose method as described previously (Osowska et al. 2006). Briefly, 50 min before being killed, each mouse was injected subcutaneously with a flooding dose of l-[1-13C]valine (50%, 300 μmol per 100 g; Cambridge Isotope laboratories, Andover, MA, USA). Three mice per group did not receive the tracer and served as controls to measure baseline free and protein-bound l-[1-13C]valine enrichments. Thirty minutes after the tracer injection, mice were anaesthetized with sodium pentobarbitone (Nembutal, 60 mg kg−1, Sigma-Aldrich) via i.p. injection. While anaesthetized deeply, the right and left gastrocnemius/soleus/plantaris muscles were dissected as a unit, weighed and rapidly frozen in liquid N2 for later determination of free and protein-bound l-[1-13C]valine enrichment. The tibialis anterior (TA) and quadriceps muscles were carefully dissected and trimmed of tendon and any adhering non-muscle tissue. The right TA was then mounted in embedding medium and frozen in thawing isopentane, while the other muscles were frozen directly in liquid N2 and stored at −80°C for subsequent analyses. Mice were killed as a consequence of heart excision, while anaesthetized deeply.
Muscle analyses
Muscle protein synthesis
The two gastrocnemius/soleus/plantaris complexes (∼350 mg) were used for isolation of myofibrillar, sarcoplasmic and mitochondrial proteins as described previously (Rooyackers et al. 1996; Guillet et al. 2004; Zangarelli et al. 2004). Briefly, muscle samples were homogenized in a 5% ice-cold buffer containing 0.25 m sucrose, 2 mm EDTA and 10 mm Tris-HCl (pH 7.4) using a Potter-Elvehjem homogenizer. The homogenate was centrifuged at low speed (600 g) and the pellet containing myofibrillar protein was collected. From the supernatant centrifuged at 7000 g, the sarcoplasmic protein fraction was isolated after centrifugation at 100,000 g for 60 min. The pellet of the 7000 g supernatant, containing mitochondria, was washed twice using an ice-cold buffer containing 100 mm KCl, 5 mm EGTA, 5 mm MgSO4, 50 mm Tris-HCl (pH 7.4) and 1 mm ATP and spun at 7000 g. The final mitochondrial pellet was suspended in the sucrose/EDTA/Tris-HCl buffer for analysis (Guillet et al. 2004). The applied protein fractionation method separates subsarcolemmal mitochondria from myofibrillar and sarcoplasmic proteins (Rooyackers et al. 1996). The purity of the myofibrillar, mitochondrial and sarcoplasmic fractions was confirmed by Western blot analyses for myosin and COX-IV protein abundance and citrate synthase activity (data not shown). The myofibrillar protein fraction is enriched in contractile proteins, but also contains some intermyofibrillar mitochondria. Therefore, we refer to the three protein fractions as being enriched in myofibrillar, mitochondrial or sarcoplasmic protein.
Each protein fraction was hydrolysed using 6 m HCl (110°C for 24 h), and the constituent amino acids in the hydrolysate were purified by cation exchange chromatography (Dowex 50W 8X; Bio-Rad Laboratories, Hercules, CA, USA). Amino acids were eluted in 4 ml of 4 m NH4OH and dried in a SpeedVac (Savant Instruments Inc., USA). Amino acids were derivatized as their N-acetyl-propyl and isotopic enrichments measured by gas chromatography–combustion–isotope ratio mass spectrometry (GC-C-IRMS, mGas System, Fisons Instruments/VG Isotech, Middlewich, UK) (Osowska et al. 2006). Tissue fluid valine enrichments were determined using t-butyldimethylsilyl derivatives, as described previously (Osowska et al. 2006) and used as the precursor pool enrichment to calculate fractional synthesis rates (FSRs). Another set of three mice was used for the determination of basal isotopic abundance in skeletal muscle.
Calpain activity
An ∼15 mg portion of the TA muscle was homogenized in a Tris-based buffer containing EDTA, 0.1%β-mercaptoethanol and a protease inhibitor cocktail (Wei et al. 2005). Following centrifugation, supernatants were equalized to 1 mg protein per ml and calpain activity was determined using 4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene-3-propionic acid labelled casein (BODIPY-FL-casein, Invitrogen) as a calpain substrate. Muscle extracts (50 μg) and the labelled casein were added to a micro-titre plate and incubated at 25°C for 60 min. The reaction was stopped by adding 25 μl of 100 mm EDTA, and fluorescence was read at 485 nm excitation and 530 nm emission wavelengths. Duplicate assays were performed under identical conditions, except that calcium was omitted and 100 mm EDTA was added to the assay. Calpain activity was calculated as the difference between activity measured in the presence of 10 mm calcium and the activity measured in the absence of calcium and the presence of 100 mm EDTA. The assay measured calcium-dependent degradation of casein (Wei et al. 2005).
Proteasome activity
The 20S proteasome activities were determined in quadriceps muscles ex vivo as described in detail elsewhere (Hobler et al. 1999). Briefly, muscles were homogenized and proteasomes isolated by sequential (ultra)centrifugation steps. Protein content (determined with Bio-Rad DC Protein Assay; Bio-Rad, Gladesville, NSW, Australia) of the isolated proteasome fractionations were equalized and the peptidase activities of the 20S proteasome determined fluorometrically by measuring the hydrolysis of the fluorogenic substrates succinyl-Leu-Leu-Val-Tyr-7-amido-4-methylcoumarin (Suc-LLVY-AMC, Sapphire Bioscience, Redfern, NSW, Australia), t-butoxycarbonyl-Leu-Arg-Arg-7-amido-4-methylcoumarin (Boc-LRR-AMC, Sigma), and Z-Leu-Leu-Glu-amido-4-methylcoumarin (Z-LLE-AMC, Sapphire Bioscience). These substrates are hydrolysed preferentially by the chymotrypsin-, trypsin-like and the peptidyl glutamyl peptide hydrolase (PGPH) activities of the 20S proteasome, respectively (Minnaard et al. 2005). Peptidase activity was determined by measuring the accumulation of the fluorogenic cleavage product amido-4-methylcoumarin (AMC) using a luminescence spectrometer. The difference between arbitrary fluorescence units recorded with or without 40 μm of the proteasome inhibitor MG132 (Sigma) in the reaction medium was corrected for the amount of protein in the reaction and the time course for the accumulation of AMC after hydrolysis of the substrate was analysed by linear regression to calculate peptidase activities, as described previously (Minnaard et al. 2005).
Western blot
Thirty milligram portions of the TA muscle were homogenized in ice-cold buffer (10 mm Tris-HCl (pH 7.4), 100 mm NaCl, 1 mm EDTA, 1 mm EGTA, 1 mm NaF, 1% Triton, 10% glycerol, 0.1% SDS, 20 mm Na4P2O7, 0.5 mm Na3VO4, 0.5% sodium deoxycholate, 0.1 mm PMSF and protease and phosphatase inhibitors, all from Sigma). Samples were centrifuged at 10,000 g for 5 min at 4°C and the resulting supernatant analysed for total protein content (Bio-Rad DC Protein Assay), with BSA as the standard. Samples were normalized to 3 μg μl−1 in homogenizing buffer, resolved in SDS-buffer and heated for 5 min at 95°C. Primary phospho-specific antibodies (anti-phospho Akt (S473), anti-phospho S6K1 (T389), anti-phospho CREB (S133) and anti-Akt, anti-S6K1, anti-CREB and anti-calpastatin were purchased from Cell Signaling Technology (Beverly, MA, USA). Anti-atrogin-1 and anti-tubulin were purchased from ECM-Biosciences (Versailles, KY, USA) and Sigma, respectively.
Equal amounts of protein (60 μg per lane) were run on SDS-polyacrylamide gels and proteins were transferred to 0.45 mm PVDF. After Ponceau S staining and de-staining, membranes were blocked for 2 h at room temperature (RT) in Tris-buffered saline–Tween 20 (TBST) containing 5% BSA. Membranes were incubated overnight at 4°C in primary antibodies. The following day membranes were washed for 3 × 5 min in TBST and then incubated for 1 h at RT in horseradish peroxidase-conjugated secondary antibodies (mouse anti-rabbit or goat anti-mouse immunoglobulins) diluted in TBST containing 5% BSA. After 3 × 5 min washes in TBST, membranes were treated with enhanced chemiluminescence (ECL plus; GE Healthcare, Little Chalfont, UK). The signal was imaged using a ChemiDoc XRS machine (Bio-Rad) and blots were quantified using Quantity One software (Bio-Rad). Tubulin was used as the loading control.
Real-time RT-PCR
MuRF-1, atrogin-1, myostatin, calpastatin, MyoD, PGC-1α, Myf5, β-arrestin-2, β2-adrenoceptor, BNIP3, LC3, myosin heavy chain (MHC) IIa, MHCIIb, MHCIIx, calpain-1, calpain-2 and calpain-3 mRNA expression were determined by real-time RT-PCR as described in detail elsewhere (Murphy et al. 2010). Total RNA was extracted from 10–20 mg of TA muscle using a commercially available kit, according to the manufacturer's instructions (PureLink RNA Mini Kit, Invitrogen, Carlsbad, CA, USA). RNA was transcribed into cDNA using the Invitrogen SuperScript™ VILO cDNA Synthesis Kit, and the resulting cDNA was stored at −20°C for subsequent analysis. Real-time PCR was performed using the Bio-Rad iCycler Thermal Cycler. Primer sequences are shown in Table 1. Measurements included a no-template control as well as an RT (reverse transcription) negative control. The content of single stranded DNA (ssDNA) in each sample was determined using the Quanti-iT OliGreen ssDNA Assay Kit (Molecular Probes/Invitrogen, Eugene, OR, USA), as described previously (Murphy et al. 2010). Gene expression was quantified by normalizing to the cDNA content of each sample and expressed as arbitrary units (AU).
Table 1.
Primer sequences used
| Gene | GeneBank accession no. | Sense primer (5′–3′) | Antisense primer (5′–3′) |
|---|---|---|---|
| MuRF-1 | NM_009066 | AGGTGTCAGCGAAAAGCAGT | CCTCCTTTGTCCTCTTGCTG |
| Atrogin-1 | AF_441120 | GTTTTCAGCAGGCCAAGAAG | TTGCCAGAGAACACGCTATG |
| MyoD | NM_010866 | AGTGAATGAGGCCTTCGAGA | GCATCTGAGTCGCCACTGTA |
| Myf5 | NM_008656 | AACCAGAGACTCCCCAAGGT | AGCTGGACACGGAGCTTTTA |
| BNIP3 | NM_009760 | GGGTTTTCCCCAAAGGAATA | TGACCACCCAAGGTAATGGT |
| LC3 | NM_026160 | CGGCTTCCTGTACATGGTTT | ATGTGGGTGCCTACGTTCTC |
| β2-ADR | NM_007420 | GAGCACAAAGCCCTCAAGAC | GTTGACGTAGCCCAACCAGT |
| β-Arrestin-2 | NM_145429 | AAGTCGAGCCCTAACTGCAA | GGTGAGGGTCACGAACACTT |
| Myostatin | NM_010834 | GGTACAAGGTATACTGGAATCCGATC | CTTCACATCAATGCTCTGCCA |
| PGC-1α | NM_008904 | CCGAGAATTCATGGAGCAAT | TTTCTGTGGGTTTGGTGTGA |
| MHCIIa | NM_001039545 | GAACCCTCCCAAGTACGACA | TAAGGGTTGACGGTGACACA |
| MHCIIb | NM_030679 | CTTCAACCACCACATGTTCG | GAGCTTCAACCTTGCCTTTG |
| MHCIIx | NM_010855 | CCAGAGTCACCTTCCAGCTC | CTTCCCTTTGCTTTTGCTTG |
| Calpastatin | NM_009817 | TTGATGATGCCTTGGATGAA | TCCCAGTTTCTCGCTATGCT |
| Calpain-1 | NM_007600 | ACATTTTACGAGGGCACCTG | CTCCCGGTTGTCATAGTCGT |
| Calpain-2 | NM_009794 | TGACAATTTTGTGCGGTGTT | GAAAAACTCAGCCACGAAGC |
| Calpain-3 | NM_007601 | ACGGGAATAAGCAACACCTG | AAACGAAGATGATGGGCTTG |
Primers were designed using Invitrogen software from gene sequences obtained from GeneBank. Primer specificity was determined using a BLAST search.
Skeletal muscle histology
Serial sections were cut transversely through the TA muscles from mice killed after 28 days of treatment with formoterol or saline, using a refrigerated (−20°C) cryostat (CTI Cryostat, IEC, Needham Heights, MA, USA). Sections were reacted with caveolin-3 to assist in the determination of mean myofibre CSA and succinate dehydrogenase (SDH) to determine fibre oxidative capacity (Koopman et al. 2006). Digital images of stained sections were obtained using an upright microscope with camera (Axio Imager D1, Carl Zeiss MicroImaging GmbH, Jena, Germany), controlled by AxioVision AC software (AxioVision AC Rel. 4.7.1, Carl Zeiss). Images were quantified using AxioVision 4.7.1 software as described previously (Koopman et al. 2006).
Statistical analyses
All values are expressed as means ±s.e.m. unless stated otherwise. Saline- and formoterol-treated groups were compared using Student's unpaired t test or a two-way ANOVA (time, treatment), where appropriate. Bonferroni's post hoc test was used to determine significant differences between individual groups. The level of significance was set at P < 0.05 for all comparisons.
Results
Formoterol treatment induces skeletal muscle hypertrophy
Daily injections with 100 μg kg−1 formoterol for 28 days increased body mass in C57BL/6 mice (Fig. 1A, significant treatment main effect; P < 0.05). The higher body mass in formoterol treated animals was associated with greater TA muscle mass (Fig. 1B, significant treatment main effect; P < 0.05). The increase in muscle mass after 28 days of treatment was associated with a 15% increase in muscle fibre cross-sectional area (P < 0.01), without changes in muscle fibre oxidative enzyme activity (P= 0.40, Fig. 1C–H). Total muscle oxidative capacity (sum of all muscle fibre CSA × SDH density) was significantly higher (+27%) in the formoterol treated mice compared with saline treated controls (P < 0.05).
Figure 1. Effect of formoterol treatment on body and muscle mass, fibre cross-sectioned area and oxidative enzyme capacity.
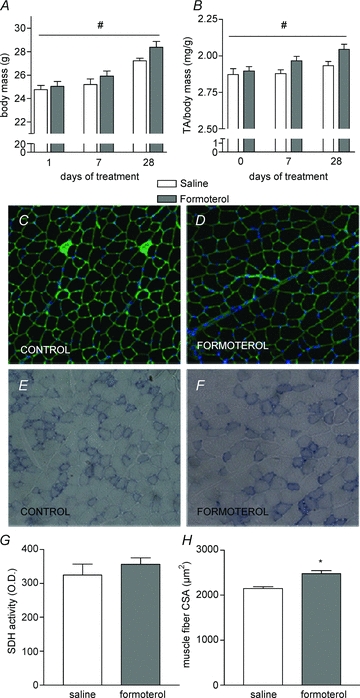
A and B, body mass (A), TA muscle mass to body mass rate (B) from C57BL/6 mice after 1, 7 or 28 days treatment with either saline or formoterol (100 μg kg−1 day−1). C–F, muscle cross-sections from mice treated for 28 days reacted for laminin and DAPI (C and D) and SDH activity (E and F). G and H, the average muscle fibre SDH activity (G) and cross-sectional area (CSA, H) were quantified using Axiovision 4.7 software. TA, tibialis anterior; SDH, succinate dehydrogenase. Data are means ±s.e.m.; n= 9 per group. *P < 0.05 versus saline, #P < 0.05 treatment main effect.
Formoterol treatment does not acutely increase muscle protein synthesis
MPS was measured 6 h after administration of 100 μg kg−1 formoterol for 1, 7 and 28 days of treatment in skeletal muscle mitochondrial, myofibrillar and sarcoplasmic enriched protein fractions (Fig. 2) to identify changes in the muscle anabolic response to formoterol over time. A single i.p. injection of formoterol (day 1) did not acutely increase protein synthesis in any of the muscle protein fractions (Fig. 2B–D). After 7 days of treatment (i.e. 6 h after the 7th daily injection), MPS was elevated significantly in the formoterol treated mice compared with control mice in the mitochondrial (2-fold), myofibrillar (1.6-fold), and sarcoplasmic protein enriched fractions (1.6-fold), as seen in Fig. 2B–D (P < 0.05). After 28 days of treatment (i.e. 6 h after the 28th daily injection), no significant differences in the rates of myofibrillar and sarcoplasmic protein synthesis were observed between treatments, although mitochondrial protein synthesis rates tended to be higher (∼20%, P= 0.10) in the formoterol treated group compared with the saline treated controls.
Figure 2. Effect of formoterol treatment on the acute protein synthetic response to β-agonist administration.
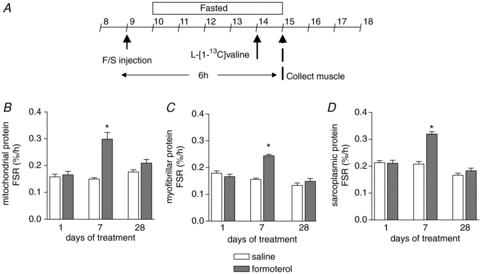
A, muscle protein synthetic response was determined 6 h after a single injection of formoterol (100 μg kg−1) or saline at 1, 7 and 28 days of treatment by injecting a large flooding dose of l-[1-13C]valine 50 min before muscle collection. B–D, protein synthetic rates were determined in mitochondrial (B), myofibrillar (C) and sarcoplasmic (D) protein enriched fractions. Data are means ±s.e.m.; n= 9. *P < 0.05 versus saline at same time point.
Formoterol treatment reduces maximum calpain activity but does not affect ubiquitin-proteasome-dependent proteasome activity
In addition to measuring MPS we also assessed muscle protein degradation, specifically the Ca2+-dependent (calpains) and ATP-dependent (proteasome) proteolytic activity ex vivo. Maximal calpain activity was reduced significantly with chronic administration of formoterol (Fig. 3A, treatment main effect, P < 0.05). In contrast, there was no acute or chronic effect of formoterol administration on ubiquitin-proteasome-dependent protein degradation. Maximal proteasome activities (i.e. trypsin-like, chymotrypsin-like and peptidyl glutamyl peptide hydrolase) were unchanged 6 h after formoterol administration on days 1, 7 and 28 of treatment (Fig. 3B–D).
Figure 3. Effect of formoterol treatment on Ca2+-dependent (A) and ubiquitin-proteasome dependent (B–D) protein degradation.
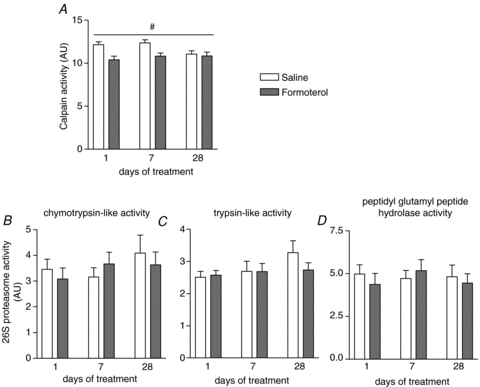
Maximal calpain activity (A) and proteasome activity (B–D, chymotrypsin-like, trypsin-like and peptidyl glutamyl peptide hydrolase activities) were determined ex vivo as described in Methods. Data are means ±s.e.m.; n= 9. #P < 0.05 treatment main effect.
Formoterol induces signalling through p70 S6K1 to increase muscle protein synthesis
To elucidate the signalling pathway regulating protein synthesis and breakdown with β-adrenoceptor stimulation, we performed Western blotting using phospho-specific antibodies and real-time RT-PCR. Formoterol treatment induced significant increases in Akt phosphorylation (Fig. 4A and B, main treatment effect, P < 0.05), without altering S6K1 phosphorylation at days 1 and 28 (Fig. 4C). Although we could not detect significant treatment, time and interaction effects of formoterol treatment on S6K1 phosphorylation, we observed significant correlations between the extent of S6K1 phosphorylation and the fractional synthetic rate of myofibrillar (Fig. 4F), mitochondrial (r= 0.49, P < 0.01) and sarcoplasmic protein (r= 0.42, P < 0.01). The increased Akt phosphorylation was associated with a reduction in atrogin-1 mRNA expression after 7 days of formoterol treatment, but there was no effect on atrogin-1 protein expression (Fig. 4D/E). We did not observe any changes in MuRF-1 mRNA levels following formoterol administration (Table 2). Interestingly, mRNA levels of BNIP3 and LC3, genes involved in the regulation of autophagy, tended to be reduced at day 7 of treatment (Table 2), but there were no significant correlations between Akt or S6K1 phosphorylation status and BNIP and LC3 mRNA levels.
Figure 4. Representative blots (A) and group data for Akt (S457) phosphorylation (B), S6K1 (T389) phosphorylation (C), atrogin-1 mRNA (D) and protein (E) levels in TA muscles of mice after 1, 7 or 28 days of treatment with either saline or formoterol, and the plotted relationship between S6K1 phosphorylation and myofibrillar protein synthesis rates (F).
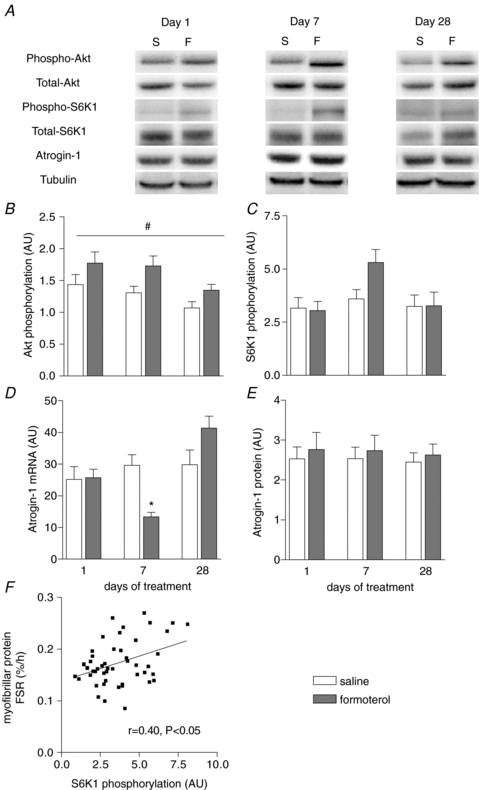
Data are means ±s.e.m.; n= 9. *P < 0.05 versus saline at same time point. #P < 0.05 treatment main effect.
Table 2.
mRNA expression in mouse TA muscles 6 h after formoterol administration at day 1, 7 or 28 of treatment
| Gene | Time (days) | Saline | Formoterol | P-values Time/treatment/interaction |
|---|---|---|---|---|
| MuRF-1 | 1 | 1.78 ± 0.11 | 1.80 ± 0.16 | 0.09/0.57/0.17 |
| 7 | 2.19 ± 0.36 | 1.93 ± 0.22 | ||
| 28 | 1.33 ± 0.12 | 1.86 ± 0.16 | ||
| Myf5 | 1 | 1.27 ± 0.34 | 1.37 ± 0.13 | <0.01/0.24/0.39 |
| 7 | 1.39 ± 0.23 | 1.36 ± 0.14 | ||
| 28 | 0.52 ± 0.04 | 1.04 ± 0.17 | ||
| BNIP3 | 1 | 1.92 ± 0.29 | 1.92 ± 0.24 | 0.13/0.70/<0.05 |
| 7 | 2.65 ± 0.24 | 1.81 ± 0.25‡ | ||
| 28 | 2.21 ± 0.20 | 2.79 ± 0.38 | ||
| LC3 | 1 | 1.58 ± 0.13 | 1.53 ± 0.14 | 0.11/0.14/0.07 |
| 7 | 1.97 ± 0.12 | 1.38 ± 0.13 | ||
| 28 | 1.84 ± 0.13 | 1.92 ± 0.21 | ||
| Myostatin | 1 | 10.24 ± 1.69 | 14.43 ± 1.32 | 0.05/0.74/<0.05 |
| 7 | 20.32 ± 3.83 | 14.49 ± 1.82 | ||
| 28 | 11.98 ± 0.75 | 15.36 ± 1.76 | ||
| β2-ADR | 1 | 1.32 ± 0.21 | 0.83 ± 0.12 | 0.25/0.19/0.20 |
| 7 | 1.56 ± 0.23 | 1.18 ± 0.28 | ||
| 28 | 1.28 ± 0.14 | 1.48 ± 0.20 | ||
| β-Arrestin-2 | 1 | 0.63 ± 0.11 | 0.71 ± 0.07 | 0.12/0.09/0.65 |
| 7 | 0.62 ± 0.11 | 0.70 ± 0.09 | ||
| 28 | 0.39 ± 0.06 | 0.61 ± 0.07 | ||
| MHCIIa | 1 | 26.70 ± 4.29 | 36.02 ± 3.51 | <0.01/0.12/0.16 |
| 7 | 49.90 ± 6.74 | 45.07 ± 4.06 | ||
| 28 | 56.68 ± 9.19 | 81.71 ± 12.33 | ||
| MHCIIb | 1 | 5.50 ± 1.20 | 7.11 ± 1.05 | <0.01/0.68/0.45 |
| 7 | 8.80 ± 1.32 | 6.72 ± 0.43 | ||
| 28 | 14.50 ± 2.63 | 13.45 ± 1.65 | ||
| MHCIIx | 1 | 3.51 ± 0.64 | 4.43 ± 0.51 | 0.10/0.16/0.54 |
| 7 | 4.01 ± 0.54 | 3.96 ± 0.36 | ||
| 28 | 4.52 ± 1.09 | 5.93 ± 0.66 | ||
| Calpain-1 | 1 | 3.94 ± 0.42 | 3.65 ± 0.33 | <0.05/0.77/0.15 |
| 7 | 4.77 ± 0.33 | 4.22 ± 0.37 | ||
| 28 | 4.46 ± 0.60 | 5.64 ± 0.63 | ||
| Calpain-2 | 1 | 3.85 ± 0.33 | 4.13 ± 0.28 | <0.01/0.57/0.44 |
| 7 | 4.50 ± 0.29 | 4.90 ± 0.30 | ||
| 28 | 4.92 ± 0.60 | 6.26 ± 0.68 | ||
| Calpain-3 | 1 | 1.54 ± 0.15 | 1.30 ± 0.14 | 0.56/0.25/0.21 |
| 7 | 1.51 ± 0.12 | 1.14 ± 0.11 | ||
| 28 | 1.41 ± 0.15 | 1.57 ± 0.22 |
Data are in means ±s.e.m.; n= 9 per group. ‡P= 0.09 versus saline.
Chronic β-adrenoceptor stimulation alters the change in CREB phosphorylation after acute formoterol administration
In addition to studying the PI3 kinase/Akt/mTOR signalling pathway, we performed measurements of the proteins involved in β-adrenergic signalling. A single injection of formoterol increased the phosphorylation status of CREB by 30% (P < 0.05, Fig. 5A and B), but with repeated injections (i.e. after 7 and 28 days of treatment) CREB phosphorylation was not different from the saline control. MyoD mRNA expression was significantly higher (Fig. 5C, treatment main effect, P < 0.05) in the formoterol treated group compared with controls. Formoterol injections (acute and chronic) did not affect mRNA expression of the β2-adrenoceptor, β-arrestin-2, myostatin, calpastatin (Fig. 5D), calpain-1, calpain-2, calpain-3, Myf-5, or myosin heavy chain (MHC) IIa, IIb and IIx isoforms (Table 2). In contrast, muscle calpastatin protein levels were increased at day 7 in the formoterol treated mice compared with the saline treated controls (Fig. 5E, P < 0.05). No significant correlations were observed between calpastatin protein levels, calpain mRNA levels and the measured calpain activity (data not shown). Formoterol injections acutely increased PGC-1α mRNA expression (Fig. 5F, P < 0.01), but following repeated injections (i.e. after 7 and 28 days of treatment) PGC-1α mRNA was not different from the saline control (Fig. 5F). Formoterol treatment did not alter PGC-1α protein levels (Fig. 5G).
Figure 5. Representative blots (A) and group data for CREB (S133) phosphorylation (B), MyoD (C) and calpastatin (D) mRNA expression, calpastatin protein levels (E), PGC-1α mRNA expression (F), and PGC-1α protein levels (G) in TA muscle of mice after 1, 7 or 28 days of treatment with either saline or formoterol.
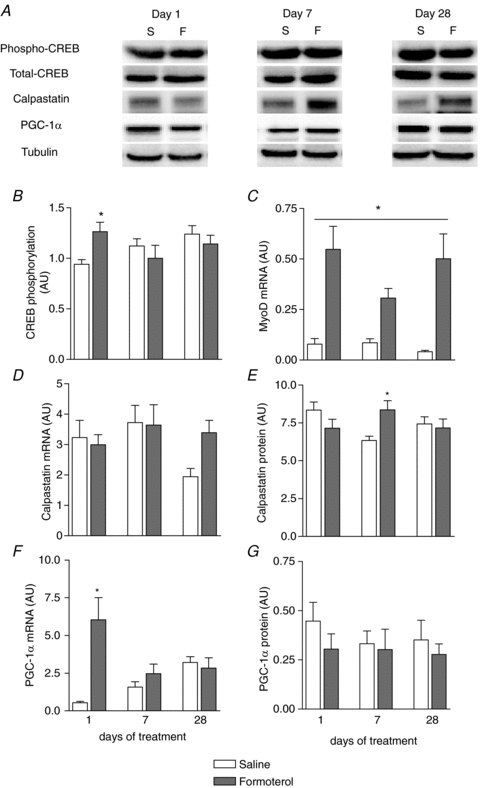
Data are means ±s.e.m.; n= 9. *P < 0.05 versus saline at same time point. #P < 0.05 treatment main effect.
Discussion
The importance of β-adrenergic signalling in skeletal muscle is only beginning to emerge, especially the potential therapeutic applications of modulating β-adrenoceptor mediated pathways that regulate muscle growth. In this study, we found that muscle protein synthesis was increased only after chronic (7 days) β-adrenoceptor stimulation with formoterol administration and that treatment induced muscle anabolism, by reducing calpain activity, and by enhancing muscle protein synthesis via increased PI-3 kinase/Akt signalling. This knowledge enhances our understanding of the pathways that maintain skeletal muscle homeostasis with implications for treating muscle wasting conditions.
A single injection of formoterol did not increase the rates of mitochondrial, myofibrillar and sarcoplasmic protein synthesis within 6 h of administration (Fig. 2). Since it has been shown previously, at least for the β-agonist clenbuterol, that maximum muscle concentrations of the drug are attained 30–45 min after administration (Chang et al. 2009), and the elimination half-life from muscle is about 6 h (Chang et al. 2009), we expected that formoterol administration would acutely affect muscle protein synthesis. In fact, MPS in all measured protein fractions were elevated significantly 6 h after formoterol injection at day 7, but protein synthesis rates were not increased at day 28 (Fig. 2). These findings are consistent with data from studies performed in rats (Maltin et al. 1992) and humans (Robinson et al. 2010), using similar methods (i.e. injection of labelled phenylalanine) to measure MPS in vivo, showing that MPS rates are not increased immediately after β-agonist administration.
We observed increased phosphorylation of CREB, combined with increased mRNA levels of MyoD and PGC-1α, 6 h after the first injection of formoterol, indicating that stimulation of the β-adrenoceptor initially signals through Gαs coupling to AC, production of cAMP with subsequent activation of PKA, and phosphorylation of CREB protein. Interestingly, the increased PGC-1α mRNA expression in the formoterol treated mice was not associated with altered PGC-1α protein levels or mitochondrial FSR indicating that subsequent changes in protein levels (if any) occur after 6 h post-treatment. We did not observe any increase in CREB phosphorylation, PGC-1α mRNA or protein expression at days 7 and 28, suggesting that other signalling pathways become more important in the regulation of MPS with chronic β-adrenoceptor stimulation. By using rapamycin to inhibit mTOR activity, it has been established that PI-3 kinase/Akt signalling is implicated in β-agonist mediated hypertrophy (Kline et al. 2007). Our new findings reveal an increase in Akt phosphorylation after β-adrenoceptor stimulation with formoterol treatment and significant correlations between the extent of S6K1 phosphorylation and MPS in mitochondrial, myofibrillar and sarcoplasmic protein enriched fractions after formoterol treatment. These data indicate that the increase in overall protein synthesis during formoterol treatment is driven mainly by signalling through the PI-3 kinase/Akt pathway.
In addition to our measurements of MPS, we examined potential changes in muscle protein breakdown by assessing maximal Ca2+-dependent (calpains) and ubiquitin-proteasome-dependent proteolytic activity ex vivo. Calpain activity was acutely and chronically reduced after formoterol administration (Fig. 3A), consistent with data from in vitro studies in rats that showed reduced Ca2+-dependent protein degradation with clenbuterol (Navegantes et al. 2001). We found that the reduction in calpain activity was not associated with changes in calpain-1–3 and calpastatin mRNA expression with formoterol treatment, indicating that β-adrenergic signalling does not control the transcription of these factors. These observations are consistent with previous studies showing that mRNA expression of calpain-1, -2 or -3 and calpastatin are either increased (Parr et al. 1992) or not changed (Busquets et al. 2004) after β-agonist administration. Calpastatin protein levels were increased on day 7 of formoterol treatment suggesting that part of the reduction in calpain activity with formoterol treatment was due to increased expression of calpastatin protein. Further work is needed to investigate the interaction between β-adrenergic signalling and calpain activity and the regulation of calpain activity by calpastatin in response to β2-agonist administration.
We did not observe any acute or chronic effects of formoterol treatment on proteasome-dependent protein breakdown, which is different from that reported by Busquets et al. (2004), who found reduced proteasome-dependent protein breakdown after chronic formoterol treatment in control and cachectic mice, without substantial changes in mRNA expression in proteasome subunits (Busquets et al. 2004). Although we observed a reduction in atrogin-1 mRNA expression with formoterol administration on day 7 (but not on days 1 and 28), consistent with the increased Akt signalling, altered mRNA expression did not translate to changes in atrogin-1 protein expression. In addition, MuRF-1 mRNA expression was not affected by formoterol treatment. Our results for MuRF-1 mRNA and atrogin-1 mRNA and protein expression data are consistent with our observation that proteasome activity is unchanged with acute and chronic formoterol treatment in C57BL/6 mice.
Taken together, our data show that acute stimulation of the β-adrenergic signalling pathway can reduce muscle protein breakdown, while prolonged stimulation promotes protein synthesis, findings that highlight how modulating the β-adrenergic signalling pathway is an appropriate therapeutic target for skeletal muscle wasting and weakness due to its dual roles in the mechanisms regulating protein synthesis and degradation. This may be particularly important for muscle wasting conditions that result from activation of Ca2+-dependent protein breakdown, as occurs during the later stages of sepsis (Voisin et al. 1996), or for wasting disorders that result from reduced (basal) MPS, such as immobilization (de Boer et al. 2007). It has been suggested that a reduced anabolic response (e.g. S6K1 phosphorylation and MPS) to food intake, insulin and exercise in the elderly plays a key role in the aetiology of sarcopaenia (Cuthbertson et al. 2005; Rasmussen et al. 2006; Kumar et al. 2009) and as such, our findings that acute and chronic formoterol treatment effectively stimulates PI3-kinase/Akt signalling supports the contention that stimulation of the β-adrenergic signalling pathway has therapeutic potential for age-related muscle wasting and weakness.
Acknowledgments
This work was supported by grants from the National Health & Medical Research Council (NHMRC, Australia, Project grant 509313) and the Association Française contre les Myopathies (France). R.K. is supported by a C. R. Roper Research Fellowship from the Faculty of Medicine, Dentistry and Health Sciences of The University of Melbourne. B.L. is supported by a Fellowship from the Swiss National Funds. K.T.M. is supported by a Biomedical Australian Fellowship from the National Health and Medical Research Council (NHMRC). S.M.G. was supported by a Postgraduate Scholarship from the National Heart Foundation (Australia). We thank Benjamin Gleeson for expert technical assistance.
Glossary
Abbreviations
- AC
adenylate cyclase
- FSR
fractional synthetic rate
- MHC
myosin heavy chain
- MPS
muscle protein synthesis
- PGC-1α
peroxisome proliferator-activated receptor γ coactivator-1α
- PI3K
phosphoinositol 3-kinase
- PKA
protein kinase A
- SDH
succinate dehydrogenase
- TA
tibialis anterior
Author contributions
R.K., S.M.G. and G.S.L. designed the study. R.K. organized and carried out the animal experiments together with S.M.G. S.W. performed the stable isotope analyses. R.K., B.L., J.T. and K.T.M. performed PCR, Western and enzyme activity analyses. R.K. and K.T.M. performed the statistical analyses of the data. R.K., K.T.M. and G.S.L. wrote the manuscript. No author had any financial or personal conflicts of interest.
References
- Busquets S, Figueras MT, Fuster G, Almendro V, Moore-Carrasco R, Ametller E, Argilés JM, Lopez-Soriano FJ. Anticachectic effects of formoterol: a drug for potential treatment of muscle wasting. Cancer Res. 2004;64:6725–6731. doi: 10.1158/0008-5472.CAN-04-0425. [DOI] [PubMed] [Google Scholar]
- Chang JC, Lee WC, Wu YT, Tsai TH. Distribution of blood-muscle for clenbuterol in rat using microdialysis. Int J Pharm. 2009;372:91–96. doi: 10.1016/j.ijpharm.2009.01.015. [DOI] [PubMed] [Google Scholar]
- Cuthbertson D, Smith K, Babraj J, Leese G, Waddell T, Atherton P, Wackerhage H, Taylor PM, Rennie MJ. Anabolic signaling deficits underlie amino acid resistance of wasting, aging muscle. FASEB J. 2005;19:422–424. doi: 10.1096/fj.04-2640fje. [DOI] [PubMed] [Google Scholar]
- de Boer MD, Selby A, Atherton P, Smith K, Seynnes OR, Maganaris CN, Maffulli N, Movin T, Narici MV, Rennie MJ. The temporal responses of protein synthesis, gene expression and cell signalling in human quadriceps muscle and patellar tendon to disuse. J Physiol. 2007;585:241–251. doi: 10.1113/jphysiol.2007.142828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehrig SM, Koopman R, Naim T, Tjoakarfa C, Lynch GS. Making fast-twitch dystrophic muscles bigger protects them from contraction injury and attenuates the dystrophic pathology. Am J Pathol. 2010;176:29–33. doi: 10.2353/ajpath.2010.090760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillet C, Zangarelli A, Mishellany A, Rousset P, Sornet C, Dardevet D, Boirie Y. Mitochondrial and sarcoplasmic proteins, but not myosin heavy chain, are sensitive to leucine supplementation in old rat skeletal muscle. Exp Gerontol. 2004;39:745–751. doi: 10.1016/j.exger.2004.02.011. [DOI] [PubMed] [Google Scholar]
- Harcourt LJ, Schertzer JD, Ryall JG, Lynch GS. Low dose formoterol administration improves muscle function in dystrophic mdx mice without increasing fatigue. Neuromuscul Disord. 2007;17:47–55. doi: 10.1016/j.nmd.2006.08.012. [DOI] [PubMed] [Google Scholar]
- Hobler SC, Williams A, Fischer D, Wang JJ, Sun X, Fischer JE, Monaco JJ, Hasselgren PO. Activity and expression of the 20S proteasome are increased in skeletal muscle during sepsis. Am J Physiol Regul Integr Comp Physiol. 1999;277:R434–440. doi: 10.1152/ajpregu.1999.277.2.R434. [DOI] [PubMed] [Google Scholar]
- Kline WO, Panaro FJ, Yang H, Bodine SC. Rapamycin inhibits the growth and muscle-sparing effects of clenbuterol. J Appl Physiol. 2007;102:740–747. doi: 10.1152/japplphysiol.00873.2006. [DOI] [PubMed] [Google Scholar]
- Koopman R, Manders RJ, Jonkers RA, Hul GB, Kuipers H, van Loon LJ. Intramyocellular lipid and glycogen content are reduced following resistance exercise in untrained healthy males. Eur J Appl Physiol. 2006;96:525–534. doi: 10.1007/s00421-005-0118-0. [DOI] [PubMed] [Google Scholar]
- Kumar V, Selby A, Rankin D, Patel R, Atherton P, Hildebrandt W, Williams J, Smith K, Seynnes O, Hiscock N, Rennie MJ. Age-related differences in the dose–response relationship of muscle protein synthesis to resistance exercise in young and old men. J Physiol. 2009;587:211–217. doi: 10.1113/jphysiol.2008.164483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch GS, Ryall JG. Role of β-adrenoceptor signaling in skeletal muscle: implications for muscle wasting and disease. Physiol Rev. 2008;88:729–767. doi: 10.1152/physrev.00028.2007. [DOI] [PubMed] [Google Scholar]
- Maltin CA, Hay SM, Delday MI, Lobley GE, Reeds PJ. The action of the β-agonist clenbuterol on protein metabolism in innervated and denervated phasic muscles. Biochem J. 1989;261:965–971. doi: 10.1042/bj2610965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltin CA, Hay SM, Delday MI, Smith FG, Lobley GE, Reeds PJ. Clenbuterol, a βagonist, induces growth in innervated and denervated rat soleus muscle via apparently different mechanisms. Biosci Rep. 1987;7:525–532. doi: 10.1007/BF01116510. [DOI] [PubMed] [Google Scholar]
- Maltin CA, Hay SM, McMillan DN, Delday MI. Tissue specific responses to clenbuterol; temporal changes in protein metabolism of striated muscle and visceral tissues from rats. Growth Regul. 1992;2:161–166. [PubMed] [Google Scholar]
- Minnaard R, Wagenmakers AJ, Combaret L, Attaix D, Drost MR, van Kranenburg GP, Schaart G, Hesselink MK. Ubiquitin-proteasome-dependent proteolytic activity remains elevated after zymosan-induced sepsis in rats while muscle mass recovers. Int J Biochem Cell Biol. 2005;37:2217–2225. doi: 10.1016/j.biocel.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Murphy KT, Koopman R, Naim T, Leger B, Trieu J, Ibebunjo C, Lynch GS. Antibody-directed myostatin inhibition in 21-mo-old mice reveals novel roles for myostatin signaling in skeletal muscle structure and function. FASEB J. 2010 doi: 10.1096/fj.10-159608. (in press) [DOI] [PubMed] [Google Scholar]
- Navegantes LC, Resano NM, Migliorini RH, Kettelhut IC. Catecholamines inhibit Ca2+-dependent proteolysis in rat skeletal muscle through β2-adrenoceptors and cAMP. Am J Physiol Endocrinol Metab. 2001;281:E449–454. doi: 10.1152/ajpendo.2001.281.3.E449. [DOI] [PubMed] [Google Scholar]
- Osowska S, Duchemann T, Walrand S, Paillard A, Boirie Y, Cynober L, Moinard C. Citrulline modulates muscle protein metabolism in old malnourished rats. Am J Physiol Endocrinol Metab. 2006;291:E582–586. doi: 10.1152/ajpendo.00398.2005. [DOI] [PubMed] [Google Scholar]
- Parr T, Bardsley RG, Gilmour RS, Buttery PJ. Changes in calpain and calpastatin mRNA induced by beta-adrenergic stimulation of bovine skeletal muscle. Eur J Biochem. 1992;208:333–339. doi: 10.1111/j.1432-1033.1992.tb17191.x. [DOI] [PubMed] [Google Scholar]
- Pearen MA, Myers SA, Raichur S, Ryall JG, Lynch GS, Muscat GE. The orphan nuclear receptor, NOR-1, a target of β-adrenergic signaling, regulates gene expression that controls oxidative metabolism in skeletal muscle. Endocrinology. 2008;149:2853–2865. doi: 10.1210/en.2007-1202. [DOI] [PubMed] [Google Scholar]
- Rasmussen BB, Fujita S, Wolfe RR, Mittendorfer B, Roy M, Rowe VL, Volpi E. Insulin resistance of muscle protein metabolism in aging. FASEB J. 2006;20:768–769. doi: 10.1096/fj.05-4607fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MM, Richards JC, Hickey MS, Moore DR, Phillips SM, Bell C, Miller BF. Acute β-adrenergic stimulation does not alter mitochondrial protein synthesis or markers of mitochondrial biogenesis in adult men. Am J Physiol Regul Integr Comp Physiol. 2010;298:R25–33. doi: 10.1152/ajpregu.00524.2009. [DOI] [PubMed] [Google Scholar]
- Rooyackers OE, Adey DB, Ades PA, Nair KS. Effect of age on in vivo rates of mitochondrial protein synthesis in human skeletal muscle. Proc Natl Acad Sci U S A. 1996;93:15364–15369. doi: 10.1073/pnas.93.26.15364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryall JG, Plant DR, Gregorevic P, Sillence MN, Lynch GS. β2-Agonist administration reverses muscle wasting and improves muscle function in aged rats. J Physiol. 2004;555:175–188. doi: 10.1113/jphysiol.2003.056770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryall JG, Schertzer JD, Lynch GS. Attenuation of age-related muscle wasting and weakness in rats after formoterol treatment: therapeutic implications for sarcopenia. J Gerontol A Biol Sci Med Sci. 2007;62:813–823. doi: 10.1093/gerona/62.8.813. [DOI] [PubMed] [Google Scholar]
- Ryall JG, Schertzer JD, Lynch GS. Cellular and molecular mechanisms underlying age-related skeletal muscle wasting and weakness. Biogerontology. 2008a;9:213–228. doi: 10.1007/s10522-008-9131-0. [DOI] [PubMed] [Google Scholar]
- Ryall JG, Schertzer JD, Murphy KT, Allen AM, Lynch GS. Chronic β2-adrenoceptor stimulation impairs cardiac relaxation via reduced SR Ca2+-ATPase protein and activity. Am J Physiol Heart Circ Physiol. 2008b;294:H2587–2595. doi: 10.1152/ajpheart.00985.2007. [DOI] [PubMed] [Google Scholar]
- Ryall JG, Sillence MN, Lynch GS. Systemic administration of β2-adrenoceptor agonists, formoterol and salmeterol, elicit skeletal muscle hypertrophy in rats at micromolar doses. Br J Pharmacol. 2006;147:587–595. doi: 10.1038/sj.bjp.0706669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voisin L, Breuille D, Combaret L, Pouyet C, Taillandier D, Aurousseau E, Obled C, Attaix D. Muscle wasting in a rat model of long-lasting sepsis results from the activation of lysosomal, Ca2+-activated, and ubiquitin-proteasome proteolytic pathways. J Clin Invest. 1996;97:1610–1617. doi: 10.1172/JCI118586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Fareed MU, Evenson A, Menconi MJ, Yang H, Petkova V, Hasselgren PO. Sepsis stimulates calpain activity in skeletal muscle by decreasing calpastatin activity but does not activate caspase-3. Am J Physiol Regul Integr Comp Physiol. 2005;288:R580–590. doi: 10.1152/ajpregu.00341.2004. [DOI] [PubMed] [Google Scholar]
- Zangarelli A, Walrand S, Guillet C, Gachon P, Rousset P, Giraudet C, Picard B, Boirie Y. Centrifugation-based isolation of myosin for measurement of its synthesis rate in small muscle samples. Anal Biochem. 2004;327:55–60. doi: 10.1016/j.ab.2003.11.024. [DOI] [PubMed] [Google Scholar]