

Abstract
High altitude (HA)-induced pulmonary hypertension may be due to a free radical-mediated reduction in pulmonary nitric oxide (NO) bioavailability. We hypothesised that the increase in pulmonary artery systolic pressure (PASP) at HA would be associated with a net transpulmonary output of free radicals and corresponding loss of bioactive NO metabolites. Twenty-six mountaineers provided central venous and radial arterial samples at low altitude (LA) and following active ascent to 4559 m (HA). PASP was determined by Doppler echocardiography, pulmonary blood flow by inert gas re-breathing, and vasoactive exchange via the Fick principle. Acute mountain sickness (AMS) and high-altitude pulmonary oedema (HAPE) were diagnosed using clinical questionnaires and chest radiography. Electron paramagnetic resonance spectroscopy, ozone-based chemiluminescence and ELISA were employed for plasma detection of the ascorbate free radical (A·−), NO metabolites and 3-nitrotyrosine (3-NT). Fourteen subjects were diagnosed with AMS and three of four HAPE-susceptible subjects developed HAPE. Ascent decreased the arterio-central venous concentration difference (a-cvD) resulting in a net transpulmonary loss of ascorbate, α-tocopherol and bioactive NO metabolites (P < 0.05 vs. LA). This was accompanied by an increased a-cvD and net output of A·− and lipid hydroperoxides (P < 0.05 vs. sea level, SL) that correlated against the rise in PASP (r= 0.56–0.62, P < 0.05) and arterial 3-NT (r= 0.48–0.63, P < 0.05) that was more pronounced in HAPE. These findings suggest that increased PASP and vascular resistance observed at HA are associated with a free radical-mediated reduction in pulmonary NO bioavailability.
Introduction
Hypoxic pulmonary vasoconstriction (HPV) is a highly conserved adaptive response to alveolar hypoxia that serves to optimise ventilation–perfusion matching and gas exchange (Von Euler & Liljestrand, 1946). However, the prolonged systemic and alveolar hypoxia associated with high-altitude (HA) exposure can result in a potentially detrimental increase in pulmonary artery pressure and vascular resistance. When excessive, this can lead to high-altitude pulmonary oedema (HAPE), with potentially fatal consequences (Bartsch et al. 1991; Swenson et al. 2002).
Despite extensive research, the precise mechanisms underlying HA-induced pulmonary hypertension are not completely understood. Human studies suggest that this may be due to a functional imbalance favouring vasoconstriction through increased sympathetic activation of α-adrenergic efferent pathways (Duplain et al. 1999) with additional contributions from endothelin (Berger et al. 2009) and angiotensin II (Bartsch et al. 1988) in combination with reduced bioavailability of the endogenous vasodilator nitric oxide (NO) (Scherrer et al. 1996; Busch et al. 2001; Swenson et al. 2002; Berger et al. 2005; Ingram et al. 2010). At the cellular level, emerging evidence implicates the mitochondrial formation of reactive oxygen species (ROS) as the primary stimulus underlying HPV (Guzy & Schumacker, 2006; Waypa & Schumacker, 2008).
In support, there is now compelling evidence to suggest that hypoxia promotes systemic (Bailey et al. 2001, 2004, 2006b, 2009a) and focal cerebral (Bailey et al. 2009b) oxidative–nitrosative stress in humans. However, to what extent if indeed at all the pulmonary circulation through which the entire cardiac output must pass contributes to the intravascular accumulation of free radicals and subsequent implications for high-altitude pulmonary hypertension remains to be examined.
Therefore, our primary aim was to determine the temporal effects of hypoxia on the systemic and transpulmonary exchange of oxidative–nitrosative–inflammatory stress biomarkers during ascent to HA. The secondary aim was to examine if any subsequent relationships existed between exchange across the lungs and pulmonary hypertension. We hypothesised that compared to the sea level (SL) control condition, HA would promote the pulmonary formation and subsequent net output of free radicals (oxidative stress) and associated reactants (inflammatory stress) as indicated by a positive arterial–central venous concentration difference (a-cvD). This would be accompanied by a net loss of bioactive NO metabolites and output of 3-nitrotyrosine (3-NT), reflecting oxidative inactivation of NO (Pacher et al. 2007) (nitrosative stress). Finally, we hypothesised that exchange would be directly proportional to both the absolute values and the increase in pulmonary artery systolic pressure (PASP) and subsequently more marked in those subjects who developed HAPE.
Methods
Subjects
Thirty-eight (32 male, 6 female) healthy non-acclimatised native lowlanders aged 37 (mean) ± 10 (s.d.) years old volunteered for the study, which had been approved by the Medical Faculty of the University of Heidelberg. Thirty subjects had previously climbed to altitudes above 3000 m. Of these, eight had previously developed moderate to severe acute mountain sickness (AMS) and eight had prior radiographic evidence of HAPE (classified as HAPE susceptible; HAPE-S). No subject spent more than four nights above 2500 m within 30 days before the study and there was no history of chronic daily headache, habitual antioxidant vitamin or anti-inflammatory supplementation. All subjects were encouraged to follow a low nitrate/nitrite diet prior to and throughout the duration of the study with specific instructions to avoid fruits, salads and cured meats (Wang et al. 1997).
Compliance
Four subjects withdrew following baseline control measurements, two of whom were HAPE-S. A further eight subjects were excluded (two HAPE-S, one of whom developed HAPE) due to failure to measure pulmonary blood flow (PBF) and/or obtain blood samples at all time points. Thus, the current data are based on full data sets for 26 subjects (aged 36 ± 10 years) with four classified as HAPE-S.
Experimental design
Baseline control measurements were conducted at sea level (SL) in Heidelberg, Germany (110 m). Between 2 and 4 weeks later, subjects travelled to Alagna, Valsesia, Italy. They were transported by cable car to 3200 m before trekking to 3611 m where they spent the night (the Gnifetti Hut). In the morning they were accompanied by a certified mountain guide while they ascended to 4559 m (Capanna Regina Margherita) within 4–6 h where they remained for the following 2 days.
Blood sampling
At SL subjects attended the laboratory following a 12 h overnight fast. A radial arterial catheter (RA 04020, Arrow International, Inc., Reading, PA, USA) was placed under local anaesthesia (2% lidocaine) and a central venous catheter (Cavafix Certodyn 375, B. Braun Melsungen AG, Melsungen, Germany) was inserted via an antecubital vein and advanced under electrocardiographic guidance into the superior vena cava (Corsten et al. 1994). Mixed venous samples would have been preferred, though this was not an option in light of the risks associated with pulmonary artery catheterisation. At HA, catheters were placed within 1 h of arrival to the Margherita Hut. Samples were obtained 3 h thereafter (HA-3h) to ensure that the baseline blood samples were not contaminated by the combined stresses of catheterisation, exercise and/or post-prandial elevations in blood-borne biomarkers of oxidative–nitrosative–inflammatory stress. We were confident that given the short half-lives of the biomarkers employed that this ‘recovery period’ was sufficient to ensure an accurate baseline at HA. Catheters were kept patent overnight and the final sample was obtained within 1 h of waking at 07.00 h which equated to a 20 h exposure to 4559 m (HA-20h). With the exception of blood gas measurements, samples were centrifuged at 600 g (4°C) for 10 min and serum/plasma/red blood cells (RBC) immediately snap-frozen and stored in liquid nitrogen prior to analysis. Subjects remained at HA for a further 24 h having received medical treatment when necessary prior to guided descent.
Rescue medication
Subjects with clinical and radiographic signs of HAPE were treated with oral nifedipine (2 × 8 mg day−1) and supplemental oxygen. Those subjects who developed AMS received symptomatic treatment with acetaminophen (500 mg) and/or ibuprofen (400 mg). All measurements were performed prior to pharmacological intervention and no further assessments were made thereafter. A more detailed description of the safety aspects and clinical management of subjects has been described by Swenson et al. (2002) and more recently in two separate publications that report further data from the same study described here (Berger et al. 2009; Dehnert et al. 2010).
Pulmonary haemodynamics
Echocardiography
Doppler echocardiography (Accuson Cypress, Siemens, Germany) was employed for the measurement of PASP according to the modified Bernoulli equation (Allemann et al. 2000). Measurements were performed on three cardiac cycles (mean recorded) by an experienced sonographer and subsequently evaluated off-line in random order by a separate sonographer blinded to the study. The inter- and intra-observer co-efficients of variation (c.v.) were both <15%.
Pulmonary flow
Pulmonary blood flow (PBF) was determined non-invasively (Innocor, Innovision A/S, Odense, Denmark) using an oxygen-enriched mixture of 0.5% nitrous oxide and 0.1% sulfur hexafluoride measured with photo-acoustic analysers over a 5-breath interval (Lang et al. 2007). Pulmonary plasma flow (PPF) was calculated as PBF × (1 – Hct). The potential limitations of this technique have been discussed by Berger et al. (2009) who performed identical measurements in a sub-set of subjects enrolled in the same study. The inter and intra-observer c.v. were both <5%.
Blood gases
Samples were immediately measured with a blood gas analyser (ABL 5, Radiometer, Copenhagen, Denmark) for the measurement of pH, haemoglobin (Hb), partial pressures of oxygen/carbon dioxide (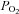 /
/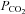 ) and oxyhaemoglobin saturation (
) and oxyhaemoglobin saturation (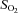 ). Haematocrit (Hct) was determined manually following microcentrifugation. Calculation of the alveolar to arterial
). Haematocrit (Hct) was determined manually following microcentrifugation. Calculation of the alveolar to arterial 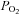 difference [[A-a]O2diff] (Fenn et al. 1946) assumed that
difference [[A-a]O2diff] (Fenn et al. 1946) assumed that 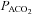 was equal to
was equal to 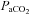 and the respiratory quotient was 0.85.
and the respiratory quotient was 0.85.
Metabolic analyses
Oxidative stress
Antioxidants. For ascorbic acid, 900 μl of 5% metaphosphoric acid was added to 100 μl plasma and assayed via fluorimetry based on the condensation of dehydroascorbic acid with 1,2-phenylenediamine (Vuilleumier & Keck, 1993). Plasma α-tocopherol, was determined by HPLC (Catignani et al. 1983; Thurnham et al. 1988). The intra- and inter-assay c.v. for both assays were <10%.
Ascorbate free radical (A·−). Plasma (1 ml) was injected into a high-sensitivity multiple-bore sample cell (AquaX, Bruker Instruments Inc., Billerica, MA, USA) housed within a TM110 cavity of an electron paramagnetic resonance (EPR) spectrometer operating at X-band (9.87 GHz) (Bailey et al. 2009a). Samples were analysed using a modulation frequency of 100 kHz, modulation amplitude of 0.65 gauss (G), microwave power of 10 mW, receiver gain of 2 × 105, time constant of 41 ms, magnetic field centre of 3477 G and scan width of ±50 G for three incremental scans (Buettner & Kiminyo, 1992). After identical baseline correction and filtering, each of the spectral peak-to-trough line heights were normalised relative to 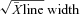 and the mean considered a measure of the relative concentration of A·−. The intra- and inter-assay c.v. were both <5%.
and the mean considered a measure of the relative concentration of A·−. The intra- and inter-assay c.v. were both <5%.
Lipid hydroperoxides. Serum lipid hydroperoxides (LOOH) were determined using the ferrous iron/xylenol orange (FOX) assay (Wolff, 1994) with modification. The intra- and inter-assay c.v. were both <4%.
Inflammatory stress
Cytokines. Plasma tumour necrosis factor (TNF)-α and interleukin (IL)-6 were measured by ELISA (R&D Systems, Minneapolis, MN, USA). The intra/inter-assay c.v. were both <10%.
Nitrosative stress
NO metabolites. Plasma (200 μl) was injected into tri-iodide reagent for the measurement of nitrite (NO2−) +S-nitrosothiols (RSNOs) by ozone-based chemiluminescence (Model 280i, NOA, Sievers, Boulder, CO, USA) (Rassaf et al. 2004). Acidified sulphanilamide (5%) was added to a separate sample and left to incubate in the dark at 21°C for 15 min to permit differentiation of NO2− from RSNOs. RBC (100 μl) were injected into modified tri-iodide (Rogers et al. 2005) for the measurement of total RBC-bound NO (NO2−+ nitrosyl haemoglobin (HbNO) +S-nitrosohemoglobin (HbSNO)). All calculations were performed using Origin Peak Analysis software (OriginLab Corp., Northampton, MA, USA). The intra- and inter-assay c.v. for all NO metabolites were <10%.
3-Nitrotyrosine. Plasma 3-nitrotyrosine (3-NT) was measured by ELISA (Hycult Biotechnology, b.v., Uden, The Netherlands) and the intra- and inter-assay c.v. were both <10%.
Transpulmonary exchange kinetics
Transpulmonary exchange kinetics was determined according to the Fick Principle:
 |
By convention, a positive value reflects net output or gain and a negative value indicates net uptake or loss across the pulmonary vascular bed.
Altitude illness
HAPE was suspected clinically and confirmed by chest radiography (Vock et al. 1989) if at least one lung quadrant demonstrated patchy opacities compatible with interstitial or alveolar oedema. AMS was evaluated using the Lake Louise (LL-cumulative Self Report + Clinical Scores) (Roach et al. 1993) and Environmental Symptoms Questionnaire Cerebral (ESQ-C) (Sampson et al. 1983) scoring systems. Clinical AMS (moderate to severe) was confirmed by a combined total LL score of ≥5 points and ESQ-C score ≥0.7 points (Maggiorini et al. 1998) upon waking after the first night at HA (HA-20h).
Statistical analysis
Distribution normality was assessed using Shapiro–Wilk W tests (SPSS Version 17.0; SPSS Inc., Chicago, IL, USA). Exchange and a-cvD data were analysed using a combination of one-factor (Location: SL vs. HA-3h vs. HA-20h) and two-factor (Location: SL vs. HA-3h vs. HA-20h × Sample Site: Radial Artery vs. Central Venous) repeated measures ANOVA with post hoc Bonferroni-corrected paired samples t test. Relationships were examined using a Pearson product moment correlation. Non-parametric equivalents were applied to TNF-α, IL-6 and ESQ-C data only. Significance was established at P < 0.05 and data presented as mean ±s.d. Where indicated, a main effect refers to a pooled (Arterial + Venous) difference between SL vs. HA-3h vs. HA-20h (P < 0.05) or pooled (SL + HA-3h + HA-20h) difference between arterial vs. venous (P < 0.05).
Results
Altitude illness
LL and ESQ-C scores increased from 1 ± 1 and 0.1 ± 0.2 points respectively at SL to 4 ± 3 and 0.8 ± 0.7 points at HA-3h (P < 0.05 vs. SL) to 6 ± 2 and 1.0 ± 0.8 points at HA-20h (P < 0.05 vs. SL). Fourteen subjects were diagnosed with AMS (AMS+) by HA-20h, three of whom subsequently developed HAPE after 27 h, 37 h and 44 h at HA with radiographic scores of 7, 12 and 5 points, respectively, at the time of diagnosis. Three of the four subjects originally identified as HAPE-S at SL developed HAPE. The remaining 12 subjects reported only mild symptoms of AMS.
Pulmonary gas exchange and haemodynamics
As anticipated, HA was associated with marked hypoxaemia and respiratory alkalosis (Table 1) whereas PBF or PPF did not change relative to SL. PASP was elevated throughout the duration of HA and the increase (value at HA-20h minus the SL control value) was greater (P < 0.05) in HAPE subjects (n= 3, +27 ± 12 mmHg) whose individual PASP values were 66, 52 and 41 mmHg compared to the remaining (i.e. non-HAPE) controls (n= 23, +13 ± 6 mmHg).
Table 1.
Pulmonary gas exchange and haemodynamics
| Location: | SL | HA-3h | HA-20h | |||
|---|---|---|---|---|---|---|
| Sample site | Radial arterial | Central venous | Radial arterial | Central venous | Radial arterial | Central venous |
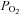 (mmHg) (mmHg) |
95 ± 11 | 38 ± 5* | 38 ± 3† | 28 ± 2*† | 41 ± 4†‡ | 29 ± 3*† |
| Main effects (P < 0.05): HA < SL + Arterial > Venous; interaction effect (P < 0.05) | ||||||
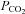 (mmHg) (mmHg) |
40 ± 3 | 48 ± 4* | 29 ± 3† | 33 ± 4*† | 28 ± 2† | 32 ± 2*† |
| Main effects (P < 0.05): HA < SL + Arterial < Venous; interaction effect (P < 0.05) | ||||||
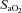 (%) (%) |
97 ± 1 | 70 ± 6* | 72 ± 5† | 52 ± 5*† | 74 ± 5† | 50 ± 6*† |
| Main effects (P < 0.05): HA < SL + Arterial > Venous; interaction effect (P < 0.05) | ||||||
| pH (units) | 7.41 ± 0.03 | 7.36 ± 0.01* | 7.48 ± 0.02† | 7.46 ± 0.02*† | 7.47 ± 0.02†‡ | 7.44 ± 0.02*†‡ |
| Main effects (P < 0.05): HA > SL + Arterial > Venous; interaction effect (P < 0.05) | ||||||
| [A-a]O2diff (mmHg) | 13 ± 7 | 10 ± 4 | 9 ± 5 | |||
| PBF (l min−1) | 6.4 ± 1.4 | 6.9 ± 1.2 | 6.2 ± 1.0‡ | |||
| PPF (l min−1) | 3.7 ± 0.8 | 3.9 ± 0.7 | 3.5 ± 0.5‡ | |||
| PASP (mmHg) | 23 ± 4 | 39 ± 10† | 38 ± 10† | |||
Values are means ±s.d.; SL, sea level; HA, high-altitude; 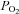 /
/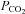 partial pressure of oxygen/carbon dioxide;
partial pressure of oxygen/carbon dioxide; 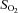 , oxyhaemoglobin saturation; [A-a]O2diff, alveolar to arterial
, oxyhaemoglobin saturation; [A-a]O2diff, alveolar to arterial 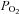 difference; PBF/PPF, pulmonary blood/plasma flow; PASP, pulmonary artery systolic pressure.
difference; PBF/PPF, pulmonary blood/plasma flow; PASP, pulmonary artery systolic pressure.
P < 0.05 vs. radial arterial for given location
P < 0.05 vs. SL for given sample site
P < 0.05 vs. HA-3h for given sample site.
Oxidative stress
HA was associated with a general reduction in ascorbate and α-tocopherol (Table 2) whereas no relationships were observed between transpulmonary loss (value at HA-20h minus the SL control value) and both the absolute values or increase in PASP (P > 0.05). HA was associated with a marked elevation in A·− and LOOH, taken to reflect an increase in both the systemic and transpulmonary rates of free radical-mediated lipid peroxidation. The underlying reaction and typical EPR spectra of the A·− (g= 2.00518; aH4≈ 1.76 G) are illustrated in Fig. 1A and B. The latter provides a visual example of the differences observed in two separate subjects exposed to HA; one who remained healthy and the other diagnosed with HAPE. The increase (value at HA-20h minus the SL control value) in the transpulmonary gain of A·− and LOOH was directly proportional to both the absolute PASP values (r= 0.60 and r= 0.57, P < 0.05) and the increase observed by HA-20h (Fig. 2A–B, P < 0.05). Furthermore, pulmonary gain was selectively elevated (P < 0.05) in HAPE subjects (+2132 ± 354 AU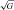 /min+1.98±0.63 μmol min−1) compared to controls (+351 ± 527 AU
/min+1.98±0.63 μmol min−1) compared to controls (+351 ± 527 AU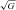 /min+0.99 ± 0.67 μmol min−1).
/min+0.99 ± 0.67 μmol min−1).
Table 2.
Oxidative stress
| Location | SL | HA-3h | HA-20h | |||
|---|---|---|---|---|---|---|
| Sample site | Radial arterial | Central venous | Radial arterial | Central venous | Radial arterial | Central venous |
| Ascorbate (μmol l−1) | 63.3 ± 9.7 | 62.2 ± 8.8 | 57.5 ± 7.7 | 60.2 ± 7.7 | 52.7 ± 5.5 | 55.2 ± 10.9 |
| Main effect (P < 0.05): HA < SL | ||||||
| a-cvD (μmol l−1) | +1.1 ± 4.9 | −2.7 ± 8.0† | −2.5 ± 8.3 | |||
| Exchange (μmol min−1) | +4.3 ± 16.9 | −11.7 ± 25.8† | −10.1 ± 31.1 | |||
| α-Tocopherol (μmol l−1) | 27.8 ± 8.0 | 28.4 ± 7.5 | 26.0 ± 6.6 | 27.4 ± 6.7 | 25.7 ± 7.6 | 26.6 ± 7.9 |
| Main effect (P < 0.05): HA < SL | ||||||
| a-cvD (μmol l−1) | −0.6 ± 2.9 | −1.3 ± 6.0 | −0.9 ± 3.6 | |||
| Exchange (μmol min−1) | −2.4 ± 10.9 | −5.3 ± 24.2 | −3.9 ± 12.5 | |||
A·− (AU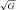 ) ) |
1572 ± 237 | 1536 ± 214 | 1982 ± 352† | 1740 ± 257*† | 1934 ± 387† | 1736 ± 272*† |
| Main effects (P < 0.05): HA > SL + Arterial > Venous; interaction effect (P < 0.05) | ||||||
a-cvD (AU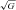 ) ) |
+36 ± 111 | +242 ± 270† | +198 ± 222† | |||
Exchange (AU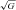 min−1) min−1) |
+137 ± 398 | +988 ± 960† | +715 ± 852† | |||
| LOOH (μmol l−1) | 0.49 ± 0.08 | 0.54 ± 0.09* | 0.84 ± 0.29† | 0.64 ± 0.15*† | 0.95 ± 0.29†‡ | 0.69 ± 0.14*† |
| Main effects (P < 0.05): HA > SL + Arterial > Venous; interaction effect (P < 0.05) | ||||||
| a-cvD (μmol l−1) | −0.06 ± 0.07 | +0.19 ± 0.20† | +0.26 ± 0.22†‡ | |||
| Exchange (μmol min−1) | −0.20 ± 0.21 | +0.77 ± 0.77† | +0.91 ± 0.81† | |||
A·−, ascorbate radical expressed in arbitrary units (AU) 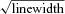 in gauss (G); LOOH, lipid hydroperoxides; a-cvD, radial arterial minus central venous concentration difference.
in gauss (G); LOOH, lipid hydroperoxides; a-cvD, radial arterial minus central venous concentration difference.
P < 0.05 vs. radial arterial for given location
P < 0.05 vs. SL for given sample site
P < 0.05 vs. HA-3h for given sample site.
Figure 1.
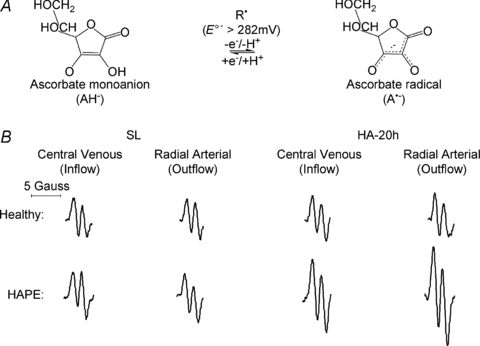
A, oxidation of the ascorbate monoanion (AH−) by an initiating species (R·) with a one electron reduction potential (E°′) greater than +282 mV to yield the domesticated ascorbate radical (A·−). The unpaired electron is delocalised over a highly conjugated tricarbonyl system rendering it resonance-stabilised, thus facilitating direct detection by electron paramagnetic resonance (EPR) spectroscopy. B, typical changes in the EPR spectral intensity of A·− in a healthy control and subject with high-altitude pulmonary oedema (HAPE) who presented with the highest PASP value by HA-20h (66 mmHg) and most severe radiographic score (12 points) with oedema present in all 4 quadrants of the lungs.
Figure 2. Relationship between changes (Δ, calculated as the value at HA-20h minus the SL control value) in pulmonary artery systolic pressure (PASP) and transpulmonary exchange of the ascorbate radical (A·−) (A) and lipid hydroperoxides (LOOH) (B).
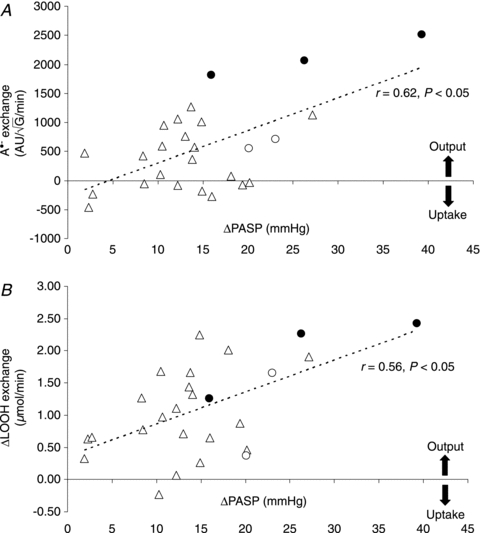
Filled circles represent subjects who developed HAPE (n= 3, one of whom was not identified as HAPE susceptible (HAPE-S) at SL) and open circles represent additional subjects identified as HAPE-S but who did not develop HAPE (n= 2).
Inflammatory stress
An increased transpulmonary gain of TNF-α was observed by HA-20h and the transpulmonary gradient of IL-6 shifted from net loss to gain by HA-20h (Table 3). No relationships were observed between changes (value at HA-20h minus the SL control value) in the output of any of these biomarkers and both the absolute values or increase in PASP (P > 0.05). Exchange was not different in HAPE subjects compared to controls (P > 0.05).
Table 3.
Inflammatory stress
| Location | SL | HA-3h | HA-20h | |||
|---|---|---|---|---|---|---|
| Sample site | Radial arterial | Central venous | Radial arterial | Central venous | Radial arterial | Central venous |
| TNF-α (pg ml−1) | 1.47 ± 1.54 | 1.35 ± 1.31 | 1.46 ± 1.23 | 1.38 ± 1.26 | 1.58 ± 1.24 | 1.40 ± 1.13* |
| a-cvD (pg ml−1) | +0.12 ± 0.36 | +0.08 ± 0.17 | +0.18 ± 0.42 | |||
| Exchange (pg min−1) | +0.48 ± 1.48 | +0.30 ± 0.69 | +0.64 ± 1.37 | |||
| IL-6 (pg ml−1) | 0.50 ± 0.21 | 0.64 ± 0.38* | 1.82 ± 1.29† | 1.92 ± 1.23*† | 3.28 ± 2.91†‡ | 3.06 ± 1.61*†‡ |
| a-cvD (pg ml−1) | −0.14 ± 0.23 | −0.10 ± 0.24 | +0.22 ± 2.61‡ | |||
| Exchange (pg min−1) | −0.58 ± 1.00 | −0.36 ± 0.98 | +0.77 ± 8.62‡ | |||
TNF, tumour necrosis factor; IL, interleukin.
P < 0.05 vs. radial arterial for given location
P < 0.05 vs. SL for given sample site
P < 0.05 vs. HA-3h for given sample site.
Nitrosative stress
A progressive decrease in arterial NO2− was observed at HA, which had the effect of suppressing transpulmonary gain (Table 4). The plasma RSNO and RBC-NO responses were generally antagonistic to that of NO2−. The arterial concentration of all bioactive NO metabolites decreased with increasing exposure to HA, which had the effect of shifting the transpulmonary gradient from net gain to loss. No relationships were observed between changes (value at HA-20h minus the SL control value) in the exchange of any of these biomarkers and both the absolute values or the increase in PASP (P > 0.05). Likewise, exchange was not different in HAPE subjects compared to controls (P > 0.05). Arterial 3-NT (insufficient venous blood precluded exchange measurements) increased from 110.2 ± 136.0 nmol l−1 at SL to 145.5 ± 168.2 nmol l−1 at HA-3h (P > 0.05 vs. SL) and 225.0 ± 282.8 nmol l−1 at HA-20h (P < 0.05 vs. SL). The increase in 3-NT at HA-20h correlated with the increase in pulmonary A·− (r= 0.63, P < 0.05) and LOOH (r= 0.48, P < 0.05) output and both the absolute values (r= 0.55, P < 0.05) and the increase in PASP (r= 0.57, P < 0.05). Furthermore, the increase in 3-NT was selectively elevated (P < 0.05) in HAPE subjects (+560.3 ± 545.7 nmol l−1) compared to controls (+59.7 ± 75.9 nmol l−1).
Table 4.
Nitrosative stress
| Location: | SL | HA-3h | HA-20h | |||
|---|---|---|---|---|---|---|
| Sample site: | Radial arterial | Central venous | Radial arterial | Central venous | Radial arterial | Central venous |
| Plasma NO2− (nmol l−1) | 432.8 ± 107.1 | 265.0 ± 52.9* | 331.2 ± 56.3† | 285.9 ± 68.2* | 263.6 ± 61.2†‡ | 249.7 ± 84.5 |
| Main effects (P < 0.05): HA < SL + Arterial > Venous; interaction effect (P < 0.05) | ||||||
| a-cvD (nmol l−1) | +167.8 ± 103.8 | +45.3 ± 68.1† | +14.0 ± 94.7† | |||
| Exchange (nmol min−1) | +609.5 ± 414.6 | +163.1 ± 242.1† | +65.2 ± 331.0† | |||
| Plasma RSNO (nmol l−1) | 2.3 ± 0.6 | 19.2 ± 3.8* | 3.8 ± 0.6† | 18.5 ± 4.6* | 3.3 ± 1.0†‡ | 16.5 ± 5.6*† |
| Main effects (P < 0.05): HA < SL + Arterial < Venous; interaction effect (P < 0.05) | ||||||
| a-cvD (nmol l−1) | −16.9 ± 3.7 | −14.6 ± 4.4† | −13.2 ± 5.4† | |||
| Exchange (nmol min−1) | −62.7 ± 22.1 | −57.8 ± 20.3 | −45.0 ± 18.1†‡ | |||
| RBC-NO (nmol l−1) | 51.4 ± 30.7 | 106.5 ± 28.6* | 90.8 ± 42.1† | 183.8 ± 59.1*† | 78.1 ± 41.0† | 191.4 ± 62.7*† |
| Main effects (P < 0.05): HA > SL + Arterial < Venous; interaction effect (P < 0.05) | ||||||
| a-cvD (nmol l−1) | −55.1 ± 44.9 | −93.0 ± 37.5† | −113.3 ± 44.8† | |||
| Exchange (nmol min−1) | −143.4 ± 131.3 | −277.7 ± 114.0† | −323.6 ± 153.6† | |||
| Total NO (nmol l−1) | 486.5 ± 114.2 | 390.7 ± 66.6* | 425.9 ± 84.3† | 488.2 ± 113.5*† | 345.0 ± 66.1‡ | 457.6 ± 117.7*† |
| Main effects (P < 0.05): HA < SL + Arterial < Venous; interaction effect (P < 0.05) | ||||||
| a-cvD (nmol l−1) | +95.8 ± 112.5 | −62.4 ± 83.9† | −112.6 ± 109.5† | |||
| Exchange (nmol min−1) | +620.6 ± 778.0 | −451.9 ± 573.7† | −684.2 ± 661.2† | |||
NO2−, nitrite; RSNO, S-nitrosothiols; RBC-NO (red blood cell-bound nitric oxide); Total NO refers to the cumulative concentration of (plasma) NO2−+ RSNO + RBC-NO (excludes nitrate).
P < 0.05 vs. radial arterial for given location
P < 0.05 vs. SL for given sample site
P < 0.05 vs. HA-3h for given sample site.
Discussion
The current study has highlighted several important findings and identifies the pulmonary circulation as a contributory source of oxidative–nitrosative–inflammatory stress at HA due in part to inadequate antioxidant defence. Consistent with our original hypothesis, HA increased the pulmonary output of free radicals that was accompanied by an equivalent output of lipid peroxidants, pro-inflammatory cytokines and loss of bioactive NO metabolites that correlated directly against both the absolute values and the increases observed in 3-NT and PASP. Together, these findings suggest that HA-induced pulmonary hypertension may be related to a free radical-mediated reduction in pulmonary NO bioavailability.
Given the low reduction potential of the A·−/ascorbate monanion (AH−) couple (E°′= 282 mV) (Williams & Yandell, 1982), virtually every oxidising free radical that could arise within the pulmonary circulation will react with AH− to form A·− (R·+ AH−→ A·−+ R-H) (Buettner, 1993). Thus, the EPR detection of A·− and concomitant loss of ascorbate in the current study provides direct evidence for an increased transpulmonary flux of free radicals during hypoxia. The loss of α-tocopherol would have also been expected to constrain peroxidative stress since the phenolic-OH group located in its chromanol ring scavenges lipid-derived peroxyl radicals (LOO·) faster than the equivalent reaction with lipid side-chains (α−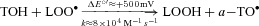 vs.
vs.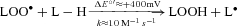 ) (Wang & Quinn, 1999). However, consumption of these chain-breaking donor antioxidants ultimately failed to terminate chain propagation as indicated by the increased pulmonary output of LOOH, one of the major reactants of lipid peroxidation.
) (Wang & Quinn, 1999). However, consumption of these chain-breaking donor antioxidants ultimately failed to terminate chain propagation as indicated by the increased pulmonary output of LOOH, one of the major reactants of lipid peroxidation.
Our data provide unique insight into the corresponding rates of pulmonary free radical-mediated lipid peroxidation, albeit within the constraints of a single arterio-venous transit. The arterial concentration of A·− and LOOH increased by 363 ± 356 AU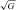 and 0.46 ± 0.28 μmol l−1 respectively at HA-20h. Thus, assuming steady-state kinetics and dilution within the extracellular space (∼12 litres) (Bailey et al. 2006a), the total amount of A·− and LOOH formed during the sojourn at HA would have been substantial, equating to 4356 AU
and 0.46 ± 0.28 μmol l−1 respectively at HA-20h. Thus, assuming steady-state kinetics and dilution within the extracellular space (∼12 litres) (Bailey et al. 2006a), the total amount of A·− and LOOH formed during the sojourn at HA would have been substantial, equating to 4356 AU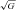 and 9.20 μmol l−1 respectively implying that net pulmonary ‘output’ was ∼197- and 119-fold greater than the respective rates of ‘accumulation’.
and 9.20 μmol l−1 respectively implying that net pulmonary ‘output’ was ∼197- and 119-fold greater than the respective rates of ‘accumulation’.
These calculations emphasise the extraordinarily high turnover of free radicals across the hypoxic human pulmonary circulation, which is not surprising given the multiple potential sites of ROS formation and inherent susceptibility to peroxidation (Connolly & Aaronson, 2010). Likewise, similar kinetics have been observed across the hypoxic human brain (Bailey et al. 2009b) suggesting that transvascular free radical output is likely to be common to ‘all’ organ systems when challenged by hypoxaemia. While not disassociating cause from effect, the linear, albeit modest, relationships observed between oxidant output and rise in PASP are consistent with recent observations in vitro. Current evidence (Ward & McMurtry, 2009) suggests that cellular hypoxia triggers mitochondrial O2·− formation at complex III of the electron transport chain subsequent to increased electron transfer from UQ·− to O2 (Guzy & Schumacker, 2006). The resultant increase in ROS signalling is thought to initiate pulmonary arterial smooth muscle contraction through Ca2+ release from ryanodine-sensitive sarcoplasmic reticulum stores (Waypa & Schumacker, 2008).
Our findings suggest that a free radical-mediated reduction in pulmonary NO bioavailability may prove of equal significance by potentiating HPV. The metal-catalysed reductive decomposition of LOOH can yield secondary LO· (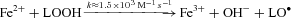 ) that have previously been detected in hypoxic human blood (Bailey et al. 2004, 2006b, 2009b). In combination with O2·−, these radicals have the potential to deplete pulmonary NO stores through a diffusion-controlled reaction that yields peroxynitrite (
) that have previously been detected in hypoxic human blood (Bailey et al. 2004, 2006b, 2009b). In combination with O2·−, these radicals have the potential to deplete pulmonary NO stores through a diffusion-controlled reaction that yields peroxynitrite (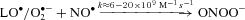 ) (Nauser & Koppenol, 2002) which can promote nitration of tyrosine residues to form the stable reactant 3-NT, our molecular ‘footprint’ of ROS-mediated NO inactivation (Pacher et al. 2007).
) (Nauser & Koppenol, 2002) which can promote nitration of tyrosine residues to form the stable reactant 3-NT, our molecular ‘footprint’ of ROS-mediated NO inactivation (Pacher et al. 2007).
Thus, the blunted systemic and pulmonary output of NO2− combined with the systemic rise in 3-NT in the current study suggests that NO bioavailability is likely to have been decreased at HA subsequent to oxidative inactivation. This finding agrees with previous reports documenting a reduction of NO in expired air (Duplain et al. 2000), loss of NO2− and nitrate in broncho-alveolar lavage fluid (Swenson et al. 2002) and impaired endothelium-dependent vasodilatation in the systemic circulation during hypoxia, especially in HAPE susceptible individuals (Berger et al. 2005). Additional NO2− loss may be attributable to reduced NO synthesis subsequent to limited O2 substrate bioavailability (Le Cras & McMurtry, 2001) though this has recently been questioned (Steiner et al. 2002). Alternatively, it may reflect increased consumption catalysed by deoxyHb/deoxyMb and/or xanthine oxidoreductase/aldehyde oxidase-mediated reduction to NO and subsequent re-apportionment towards longer-lived species notably Hb-NO and Hb-SNO driven by the quite substantial differences in transpulmonary O2 gradients (van Faassen et al. 2009). Whether the dynamic interchange of NO from the plasma to the RBC compartment serves to improve pulmonary oxygenation during hypoxia remains to be determined.
It remains to be established whether pulmonary oxidative–nitrosative stress was the cause or simply the consequence of HPV and HAPE. The early and progressive accumulation of oxidants–nitrosants lends tentative support for a causal role, which is not unreasonable given that these molecules have the thermodynamic potential to promote autocatalytic destruction of the pulmonary lipid membrane structure through cross-linking polymerisation and breakage of fatty acid side chains (Roubal & Tappel, 1966). These species may be responsible for the degradation of proteogylcans and subsequent disruption of the endothelial basement membrane and pulmonary extracellular matrix observed in hypoxia (Miserocchi et al. 2001). When combined with elevated intravascular hydrostatic pressure, this can result in capillary stress failure, destruction of tissue matrix structures, increased alveolar–capillary membrane permeability and alveolar haemorrhage (Bartsch & Gibbs, 2007). Localised inflammation is likely to be a secondary event as suggested by the delayed pulmonary output of cytokines, which agrees with previous observations (Swenson et al. 2002).
Alternatively, these biomolecules have the capacity to serve as integral components of the signal transduction cascade that are physiologically essential in controlled though as of yet undefined amounts, capable of initiating protective adaptation in the face of hypoxic stress for the maintenance of homeostasis (Bailey et al. 2001; Guzy & Schumacker, 2006; Pryor et al. 2006). Though speculative, the oxidative-nitrosative stress response that may ultimately predispose to pulmonary hypertension at HA may represent a physiological attempt to regain homeostasis, a concept that warrants investigation in future studies.
In conclusion, these findings are the first to demonstrate a net pulmonary output and systemic accumulation of oxidative–nitrosative–inflammatory stress biomarkers at HA due in part to inadequate antioxidant defence. A free radical-mediated reduction in pulmonary NO bioavailability may prove the unifying mechanism underlying HA-induced pulmonary hypertension and subsequent susceptibility to HAPE which has broader implications for other human pulmonary diseases characterised by arterial hypoxaemia such as chronic obstructive pulmonary disorder (COPD). The improved clinical outcome of COPD patients given supplemental O2 may be due to the suppression of hypoxia-mediated pulmonary ROS formation (Zouaoui Boudjeltia et al. 2009). Follow-up interventional studies need to consider combined ascorbate and α-tocopherol prophylaxis in order to optimise the chain-breaking antioxidant capacity of the pulmonary circulation at HA.
Acknowledgments
We acknowledge all subjects for their kind participation; the hut keepers and the Sezione Varallo of the Club Alpino Italiano for supporting research at the Capanna Regina Margherita; Sonja Engelhardt, Christiane Herth, Verena Faulkner, Anette Frank and Kathy Pogue for skilled technical assistance and Peter Vock, Guido Robotti, Semanta Greppi, Chantal Imesch for their expertise taking chest radiographs and VIASYS Healthcare (now CareFusion) for financial support.
Glossary
Abbreviations
- AMS
acute mountain sickness
- HA
high altitude
- HAPE
high-altitude pulmonary oedema
- HAPE-S
susceptible to high-altitude pulmonary oedema
- HPV
hypoxic pulmonary vasoconstriction
- LA
low altitude
- PASP
pulmonary artery systolic pressure
- PBF
pulmonary blood flow
- PPF
pulmonary plasma flow
- SL
sea level
Author contributions
Study conception and design: D.M.B., C.D., A.M.L., E.M., E.R.S., H.B., P.B., M.M.B.; data analysis, collection and interpretation: D.M.B., C.D., A.M.L., E.M., M.G., K.A.E., S.T., P.E.J., J.M., I.S.Y., E.R.S., H.B., P.B., M.M.B.; drafting of the article: D.M.B., E.R.S., H.B., P.B., M.M.B.; critical revision of the article: D.M.B., C.D., A.M.L., E.R.S., H.B., P.B., M.M.B. All authors approved the final version for publication.
References
- Allemann Y, Sartori C, Lepori M, Pierre S, Melot C, Naeije R, Scherrer U, Maggiorini M. Echocardiographic and invasive measurements of pulmonary artery pressure correlate closely at high altitude. Am J Physiol Heart Circ Physiol. 2000;279:H2013–2016. doi: 10.1152/ajpheart.2000.279.4.H2013. [DOI] [PubMed] [Google Scholar]
- Bailey D, Ainslie P, Jackson S, Richardson R, Ghatei M. Evidence against redox regulation of energy homoeostasis in humans at high altitude. Clin Sci (Lond) 2004;107:589–600. doi: 10.1042/CS20040085. [DOI] [PubMed] [Google Scholar]
- Bailey D, Raman S, McEneny J, Young I, Parham K, Hullin D, Davies B, McKeeman G, McCord J, Lewis M. Vitamin C prophylaxis promotes oxidative lipid damage during surgical ischemia-reperfusion. Free Radic Biol Med. 2006a;40:591–600. doi: 10.1016/j.freeradbiomed.2005.09.024. [DOI] [PubMed] [Google Scholar]
- Bailey DM, Davies B, Young IS. Intermittent hypoxic training: implications for lipid peroxidation induced by acute normoxic exercise in active men. Clin Sci (Lond) 2001;101:465–475. [PubMed] [Google Scholar]
- Bailey DM, Evans KA, James PE, McEneny J, Young IS, Fall L, Gutowski M, Kewley E, McCord JM, Moller K, Ainslie PN. Altered free radical metabolism in acute mountain sickness: implications for dynamic cerebral autoregulation and blood–brain barrier function. J Physiol. 2009a;587:73–85. doi: 10.1113/jphysiol.2008.159855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey DM, Roukens R, Knauth M, Kallenberg K, Christ S, Mohr A, Genius J, Storch-Hagenlocher B, Meisel F, McEneny J, Young IS, Steiner T, Hess K, Bartsch P. Free radical-mediated damage to barrier function is not associated with altered brain morphology in high-altitude headache. J Cereb Blood Flow Metab. 2006b;26:99–111. doi: 10.1038/sj.jcbfm.9600169. [DOI] [PubMed] [Google Scholar]
- Bailey DM, Taudorf S, Berg RMG, Lundby C, McEneny J, Young IS, Evans KA, James PE, Shore A, Hullin DA, McCord JM, Pedersen BK, Moller K. Increased cerebral output of free radicals during hypoxia: implications for acute mountain sickness? Am J Physiol Regul Integr Comp Physiol. 2009b;297:R1283–1292. doi: 10.1152/ajpregu.00366.2009. [DOI] [PubMed] [Google Scholar]
- Bärtsch P, Gibbs JS. Effect of altitude on the heart and the lungs. Circulation. 2007;116:2191–2202. doi: 10.1161/CIRCULATIONAHA.106.650796. [DOI] [PubMed] [Google Scholar]
- Bärtsch P, Maggiorini M, Ritter M, Noti C, Vock P, Oelz O. Prevention of high-altitude pulmonary edema by nifedipine. N Engl J Med. 1991;325:1284–1289. doi: 10.1056/NEJM199110313251805. [DOI] [PubMed] [Google Scholar]
- Bärtsch P, Shaw S, Franciolli M, Gnadinger MP, Weidmann P. Atrial natriuretic peptide in acute mountain sickness. J Appl Physiol. 1988;65:1929–1937. doi: 10.1152/jappl.1988.65.5.1929. [DOI] [PubMed] [Google Scholar]
- Berger MM, Dehnert C, Bailey DM, Luks AM, Menold E, Castell C, Schendler G, Faoro V, Mairbaurl H, Bartsch P, Swenson ER. Transpulmonary plasma ET-1 and nitrite differences in high altitude pulmonary hypertension. High Alt Med Biol. 2009;10:17–24. doi: 10.1089/ham.2008.1053. [DOI] [PubMed] [Google Scholar]
- Berger MM, Hesse C, Dehnert C, Siedler H, Kleinbongard P, Bardenheuer HJ, Kelm M, Bartsch P, Haefeli WE. Hypoxia impairs systemic endothelial function in individuals prone to high-altitude pulmonary edema. Am J Respir Crit Care Med. 2005;172:763–767. doi: 10.1164/rccm.200504-654OC. [DOI] [PubMed] [Google Scholar]
- Buettner GR. The pecking order of free radicals and antioxidants: lipid peroxidation, α-tocopherol, and ascorbate. Arch Biochem Biophys. 1993;300:535–543. doi: 10.1006/abbi.1993.1074. [DOI] [PubMed] [Google Scholar]
- Buettner GR, Kiminyo KP. Optimal EPR detection of weak nitroxide spin adduct and ascorbyl free radical signals. J Biochem Biophys Methods. 1992;24:147–151. doi: 10.1016/0165-022x(92)90054-e. [DOI] [PubMed] [Google Scholar]
- Busch T, Bartsch P, Pappert D, Grunig E, Hildebrandt W, Elser H, Falke KJ, Swenson ER. Hypoxia decreases exhaled nitric oxide in mountaineers susceptible to high-altitude pulmonary edema. Am J Respir Crit Care Med. 2001;163:368–373. doi: 10.1164/ajrccm.163.2.2001134. [DOI] [PubMed] [Google Scholar]
- Catignani GL, Bieri JG. Simultaneous determination of retinol and Alpha-tocopherol in serum or plasma by liquid chromatography. Clin Chem. 1983;29:708–712. [PubMed] [Google Scholar]
- Connolly MJ, Aaronson PI. Cell redox state and hypoxic pulmonary vasoconstriction: Recent evidence and possible mechanisms. Respir Physiol Neurobiol. 2010 doi: 10.1016/j.resp.2010.08.016. (in press) [DOI] [PubMed] [Google Scholar]
- Corsten SA, van Dijk B, Bakker NC, de Lange JJ, Scheffer GJ. Central venous catheter placement using the ECG-guided Cavafix-Certodyn SD catheter. J Clin Anesth. 1994;6:469–472. doi: 10.1016/0952-8180(94)90086-8. [DOI] [PubMed] [Google Scholar]
- Dehnert C, Luks AM, Schendler G, Menold E, Berger MM, Mairbaurl H, Faoro V, Bailey DM, Castell C, Hahn G, Vock P, Swenson ER, Bartsch P. No evidence for interstitial lung oedema by extensive pulmonary function testing at 4,559 m. Eur Respir J. 2010;35:812–820. doi: 10.1183/09031936.00185808. [DOI] [PubMed] [Google Scholar]
- Duplain H, Sartori C, Lepori M, Egli M, Allemann Y, Nicod P, Scherrer U. Exhaled nitric oxide in high-altitude pulmonary edema: role in the regulation of pulmonary vascular tone and evidence for a role against inflammation. Am J Respir Crit Care Med. 2000;162:221–224. doi: 10.1164/ajrccm.162.1.9908039. [DOI] [PubMed] [Google Scholar]
- Duplain H, Vollenweider L, Delabays A, Nicod P, Bartsch P, Scherrer U. Augmented sympathetic activation during short-term hypoxia and high-altitude exposure in subjects susceptible to high-altitude pulmonary edema. Circulation. 1999;99:1713–1718. doi: 10.1161/01.cir.99.13.1713. [DOI] [PubMed] [Google Scholar]
- Fenn WO, Rahn H, Otis AB. A theoretical study of the composition of the alveolar air at altitude. Am J Physiol. 1946;146:637–653. doi: 10.1152/ajplegacy.1946.146.5.637. [DOI] [PubMed] [Google Scholar]
- Guzy RD, Schumacker PT. Oxygen sensing by mitochondria at complex III: the paradox of increased reactive oxygen species during hypoxia. Exp Physiol. 2006;91:807–819. doi: 10.1113/expphysiol.2006.033506. [DOI] [PubMed] [Google Scholar]
- Ingram TE, Pinder AG, Bailey DM, Fraser AG, James PE. Low-dose sodium nitrite vasodilates hypoxic human pulmonary vasculature by a means which is not dependent upon a simultaneous elevation in plasma nitrite. Am J Physiol Heart Circ Physiol. 2010;298:H331–H339. doi: 10.1152/ajpheart.00583.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang CC, Karlin P, Haythe J, Tsao L, Mancini DM. Ease of noninvasive measurement of cardiac output coupled with peak VO2 determination at rest and during exercise in patients with heart failure. Am J Cardiol. 2007;99:404–405. doi: 10.1016/j.amjcard.2006.08.047. [DOI] [PubMed] [Google Scholar]
- Le Cras TD, McMurtry IF. Nitric oxide production in the hypoxic lung. Am J Physiol Lung Cell Mol Physiol. 2001;280:L575–582. doi: 10.1152/ajplung.2001.280.4.L575. [DOI] [PubMed] [Google Scholar]
- Maggiorini M, Muller A, Hofstetter D, Bartsch P, Oelz O. Assessment of acute mountain sickness by different score protocols in the Swiss Alps. Aviat Space Environ Med. 1998;69:1186–1192. [PubMed] [Google Scholar]
- Miserocchi G, Negrini D, Passi A, De Luca G. Development of lung edema: interstitial fluid dynamics and molecular structure. News Physiol Sci. 2001;16:66–71. doi: 10.1152/physiologyonline.2001.16.2.66. [DOI] [PubMed] [Google Scholar]
- Nauser T, Koppenol WH. The rate constant of the reaction of superoxide with nitrogen monoxide: approaching the diffusion limit. J Phys Chem A. 2002;106:4084–4086. [Google Scholar]
- Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryor WA, Houk KN, Foote CS, Fukuto JM, Ignarro LJ, Squadrito GL, Davies KJA. Free radical biology and medicine: it's a gas, man. Am J Physiol Regul Integr Comp Physiol. 2006;291:R491–511. doi: 10.1152/ajpregu.00614.2005. [DOI] [PubMed] [Google Scholar]
- Rassaf T, Feelisch M, Kelm M. Circulating NO pool: assessment of nitrite and nitroso species in blood and tissues. Free Radic Biol Med. 2004;36:413–422. doi: 10.1016/j.freeradbiomed.2003.11.011. [DOI] [PubMed] [Google Scholar]
- Roach RC, Bartsch P, Hackett PH, Oelz O. The Lake Louise acute mountain sickness scoring system. In: Sutton JR, Coates J, Houston CS, editors. Hypoxia and Molecular Medicine. Burlington: Queen City Printers; 1993. pp. 272–274. [Google Scholar]
- Rogers SC, Khalatbari A, Gapper PW, Frenneaux MP, James PE. Detection of human red blood cell-bound nitric oxide. J Biol Chem. 2005;280:26720–26728. doi: 10.1074/jbc.M501179200. [DOI] [PubMed] [Google Scholar]
- Roubal WT, Tappel AL. Polymerization of proteins induced by free-radical lipid peroxidation. Arch Biochem Biophys. 1966;113:150–155. doi: 10.1016/0003-9861(66)90168-8. [DOI] [PubMed] [Google Scholar]
- Sampson JB, Cymerman A, Burse RL, Maher JT, Rock PB. Procedures for the measurement of acute mountain sickness. Aviat Space Environ Med. 1983;54:1063–1073. [PubMed] [Google Scholar]
- Scherrer U, Vollenweider L, Delabays A, Savcic M, Eichenberger U, Kleger GR, Fikrle A, Ballmer PE, Nicod P, Bartsch P. Inhaled nitric oxide for high-altitude pulmonary edema. N Engl J Med. 1996;334:624–629. doi: 10.1056/NEJM199603073341003. [DOI] [PubMed] [Google Scholar]
- Steiner DRS, Gonzalez NC, Wood JG. Interaction between reactive oxygen species and nitric oxide in the microvascular response to systemic hypoxia. J Appl Physiol. 2002;93:1411–1418. doi: 10.1152/japplphysiol.00251.2002. [DOI] [PubMed] [Google Scholar]
- Swenson ER, Maggiorini M, Mongovin S, Gibbs JSR, Greve I, Mairbaurl H, Bartsch P. Pathogenesis of high-altitude pulmonary edema: inflammation is not an etiologic factor. JAMA. 2002;287:2228–2235. doi: 10.1001/jama.287.17.2228. [DOI] [PubMed] [Google Scholar]
- Thurnham DI, Smith E, Flora PS. Concurrent liquid-chromatographic assay of retinol, alpha-tocopherol, beta-carotene, alpha- carotene, lycopene, and beta-crytoxanthin in plasma, with tocopherol acetate as internal standard. Clin Chem. 1988;34:377–381. [PubMed] [Google Scholar]
- van Faassen EE, Bahrami S, Feelisch M, Hogg N, Kelm M, Kim-Shapiro DB, Kozlov AV, Li H, Lundberg JO, Mason R, Nohl H, Rassaf T, Samouilov A, Slama-Schwok A, Shiva S, Vanin AF, Weitzberg E, Zweier J, Gladwin MT. Nitrite as regulator of hypoxic signaling in mammalian physiology. Med Res Rev. 2009;29:683–741. doi: 10.1002/med.20151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vock P, Fretz C, Franciolli M, Bartsch P. High-altitude pulmonary edema: findings at high-altitude chest radiography and physical examination. Radiology. 1989;170:661–666. doi: 10.1148/radiology.170.3.2916019. [DOI] [PubMed] [Google Scholar]
- Von Euler U, Liljestrand G. Observations on the pulmonary arterial blood pressure in the cat. Acta Physiol Scand. 1946;12:301–320. [Google Scholar]
- Vuilleumier JP, Keck E. Fluorimetric assay of vitamin C in biological materials using a centrifugal analyser with fluorescence attachment. J Micronutr Anal. 1993;5:25–34. [Google Scholar]
- Wang J, Brown MA, Tam SH, Chan MC, Whitworth JA. Effects of diet on measurement of nitric oxide metabolites. Clin Exp Pharmacol Physiol. 1997;24:418–420. doi: 10.1111/j.1440-1681.1997.tb01212.x. [DOI] [PubMed] [Google Scholar]
- Wang X, Quinn PJ. Vitamin E and its function in membranes. Prog Lipid Res. 1999;38:309–336. doi: 10.1016/s0163-7827(99)00008-9. [DOI] [PubMed] [Google Scholar]
- Ward JP, McMurtry IF. Mechanisms of hypoxic pulmonary vasoconstriction and their roles in pulmonary hypertension: new findings for an old problem. Curr Opin Pharmacol. 2009;9:287–296. doi: 10.1016/j.coph.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waypa GB, Schumacker PT. Oxygen sensing in hypoxic pulmonary vasoconstriction: using new tools to answer an age-old question. Exp Physiol. 2008;93:133–138. doi: 10.1113/expphysiol.2007.041236. [DOI] [PubMed] [Google Scholar]
- Williams NH, Yandell JK. Outer-sphere electron-transfer reactions of ascorbic anions. Aust J Chem. 1982;35:1133–1144. [Google Scholar]
- Wolff SP. Ferrous ion oxidation in presence of ferric ion indicator xylenol orange for measurement of hydroperoxides. Methods Enzymol. 1994;233:183–189. [Google Scholar]
- Zouaoui Boudjeltia K, Tragas G, Babar S, Moscariello A, Nuyens V, Van Antwerpen P, Gilbert O, Ducobu J, Brohee D, Vanhaeverbeek M, Van Meerhaeghe A. Effects of oxygen therapy on systemic inflammation and myeloperoxidase modified LDL in hypoxemic COPD patients. Atherosclerosis. 2009;205:360–362. doi: 10.1016/j.atherosclerosis.2009.01.028. [DOI] [PubMed] [Google Scholar]