

Abstract
Nicotinamidases are metabolic enzymes that hydrolyze nicotinamide to nicotinic acid. These enzymes are widely distributed across biology, with examples found encoded in the genomes of Mycobacteria, Archaea, Eubacteria, Protozoa, yeast and invertebrates but there are none found in mammals. Although recent structural work has improved understanding of these enzymes, their catalytic mechanism is still not well understood. Recent data shows that nicotinamidases are required for growth and virulence of several pathogenic microbes. The enzymes of Saccharomyces cerevisiae, Drosophila melanogaster and Caenorhabditis elegans regulate lifespan in their respective organisms, consistent with proposed roles in the regulation of NAD+ metabolism and organismal aging. In this manuscript, the steady state kinetic parameters of nicotinamidase enzymes from C. elegans, S. cerevisiae, Streptococcus pneumoniae (a pathogen responsible for human pneumonia), Borrelia burgdorferi (the pathogen that causes Lyme Disease) and Plasmodium falciparum (responsible for most human malaria) are reported. Nicotinamidases are generally efficient catalysts with steady state kcat values typically exceeding 1 s−1. The Km values for nicotinamide are low and are in the range from 2 – 110 µM. Nicotinaldehyde was determined to be a potent competitive inhibitor of these enzymes, binding in the low µM to low nM range for all nicotinamidases tested. A variety of nicotinaldehyde derivatives were synthesized and evaluated as inhibitors in kinetic assays. Inhibitions are consistent with reaction of the universally conserved catalytic Cys on each enzyme with the aldehyde carbonyl carbon to form a thiohemiacetal complex which is stabilized by a conserved oxyanion hole. The S. pneumoniae nicotinamidase can catalyse exchange of 18O into the carboxy oxygens of nicotinic acid with 18O-water. The collected data, along with kinetic analysis of several mutants, allowed us to propose a catalytic mechanism that explains nicotinamidase and nicotinic acid 18O exchange chemistry for the S. pneumoniae enzyme involving key catalytic residues, a catalytic transition metal ion and the intermediacy of a thioester intermediate.
The nicotinamidases hydrolyze nicotinamide to nicotinic acid (Scheme 1). They play important roles in nicotinamide salvage in multiple species of bacteria (1), mycobacteria (1–4), yeast (5–7), protozoa (8) and are encoded in genomes of plants (9) and in many metazoan species, such as Drosophila melanogaster (10) and Caenorhabditis elegans (10–12). Nicotinamide salvage is important because NAD(P)+ is chemically unstable to non-enzymatic hydrolysis even at physiologic temperatures (13–15) and nicotinamide is the product of multiple NAD+ consuming enzymes (13, 14), such as sirtuins, that are widely distributed in biology (16). Consistent with their centrality to NAD+ homeostasis, nicotinamidases have been shown to be essential for the viability of several microorganisms that are pathogenic to humans such as Borrelia burgdorferi (which causes Lyme disease) (17–19) and Brucella abortus (20). It is possible that other pathogenic organisms require this enzyme as well, since some of these, including Plasmodium falciparum, do not appear to encode genes for enzymatic components of de novo NAD+ biosynthetic pathways (21). Consequently, it is likely that these organisms are highly reliant on salvage of nicotinamide from the human host. Attractively, human and mammalian genomes do not encode a nicotinamidase, suggesting that small molecule inhibitors of nicotinamidases could serve as antimicrobial agents (21).
Scheme 1.

Nicotinamidase hydrolyzes nicotinamide to give nicotinic acid.
Biologically interesting roles for nicotinamidases have also been identified as increasers of lifespan in D. melanogaster (10) and in C. elegans (11) and in the budding yeast Saccharomyces cerevisiae (22). Specifically, nicotinamidases regulate intracellular nicotinamide concentrations, which are implicated in negative regulation of NAD+ dependent deacetylases (the sirtuins) (16, 23). Overexpression of nicotinamidases increases lifespan and requires sirtuin activity in yeast (24) and in flies (10) consistent with the idea that nicotinamidase overexpression depletes intracellular nicotinamide concentrations, thereby increasing sirtuin activity. Consistently, sirtuin overexpression within D. melanogaster (25, 26) C. elegans (27, 28) and S. cerevisiae (29, 30) also causes increased lifespan. Stresses that increase lifespan in the S. cerevisiae, such as low calorie stress, are inducers of nicotinamidase transcription and translation (22), and oxidative stress increases nicotinamidase expression in D. melanogaster (10). These data suggest that these enzymes are central to phylogenetically conserved adaptive responses to environmental stresses in single-celled eukaryotes and in non-mammalian multicellular eukaryotes. Of additional interest, the nicotinamidase from Mycobacterium tuberculosis is responsible for activation of the pro-drug pyrazinamide to the active antibiotic pyrazinoic acid. Mutations in this enzyme are associated with resistance to the drug (Scheme 2) (4). Recently nicotinamidases have been shown to be important for plant germination as well (31). The available literature supports the view that nicotinamidases are therapeutically and biologically interesting enzymes with diverse functions associated with their enzymatic activity.
Scheme 2.

Nicotinamidase catalyzes the conversion of pyrazinamide to pyrazinoic acid.
The first nicotinamidase activity was reported for the organism Lactobacillus arabinosus (32), but decades later, there is still limited insight into the chemical and enzymatic mechanisms of these enzymes. Two unliganded structures of nicotinamidases have been reported, one of the S. cerevisiae enzyme (6), the other from the archaean thermophile Pyrococcus horikoshii (33). Both structures revealed an active site coordinated metal ion, proposed to be Zn+2, and similar structural folds, although limited insight into catalytic chemistry was provided. A recent structure of the nicotinamidase from Acinetobacter baumanii in complex with nicotinic acid clarified some aspects of the active site design for these enzymes (34). The crystal structure revealed that the pyridine N1 of nicotinic acid is coordinated to the central metal ion, suggesting that coordination of nicotinamide to the metal center organizes the pyridine for attack by a cysteine residue on the amide carbonyl of the substrate (34). This cysteine is universally conserved in all nicotinamidases (Figure 1) and previously had been proposed to perform a nucleophilic function on the enzyme to form a labile thioester of nicotinic acid, which can further hydrolyse to release the free thiol and nicotinic acid (33, 34). This mechanistic proposal was more convincingly demonstrated in a very recent paper, in which several of the current authors contributed, which structurally characterized the thioester intermediate on the nicotinamidase from Streptococcus pneumoniae (35).
Figure 1.

Multiple sequence alignment of the Pyrococcus horikoshii (PDB entry 1IM5), Streptococcus pneumoniae, Escherichia Coli, Saccharomyces cerevisiae, and Borrelia burgdorferi nicotinamidases. The alignment was performed with ClustalW (56) and edited with ESPript (57). Identical residues are highlighted, and the secondary structure elements of the solved P. horikoshii structure (33) are shown above the alignment. Vertical arrows point to the proposed catalytic triad proposed by Du et al. that is composed of an aspartate, cysteine, and lysine residue. The proposed metal-binding residues are shown with an M under the appropriate residue
In this paper, we characterize the steady state kinetic properties for a number of nicotinamidases from multiple sources S. cerevisiae, B. burgdorferi, P. falciparum, S. pneumoniae and two nicotinamidase isoforms from C. elegans. We present a continuous assay to monitor nicotinamidase catalytic function and show that these enzymes have high catalytic efficiency for nicotinamide and can accept a number of additional substituted nicotinamide substrates. The nicotinamidase from S. pneumoniae is shown to exchange 18O into nicotinic acid from 18O water. In addition we found that all of these enzymes are inhibited by nicotinaldehyde with Ki values between 1.4 µM to 11 nM. Kinetic analyses revealed that nicotinaldehyde is a competitive inhibitor with nicotinamide consistent with reversible mechanism-based trapping of the enzyme. Structural variants of nicotinaldehyde were synthesized and biochemically evaluated, establishing the possibility of developing potent yet selective inhibitors for nicotinamidases derived from different sources. Using information provided by nicotinaldehyde inhibition studies, kinetic studies, sequence comparisons, and mutagenesis data, we present mechanisms of nicotinaldehyde inhibition, 18O exchange, and nicotinamide hydrolysis catalyzed by nicotinamidases.
Materials and Methods
Reagents and Instrumentation
Unless otherwise stated in the text, all reagents were purchased from Sigma-Aldrich or VWR and were of the highest purity commercially available. UV analyses were performed on a Hitachi U-3010 spectrophotometer. Fluorescence analyses were performed on a LJL Biosystems Analyst AD 96.384 microplate reader.
Plasmid Construction and Protein Expression
Standard molecular biology techniques were used to clone the genes of interest. The cloning and expression of nicotinamidases from S. pneumoniae, B. burgdorferi and P. falciparum (SpNic, BbNic and PfNic respectively) are reported in the Supporting Information. The nicotinamidase gene from B. burgdorferi was cloned from a construct pBBE22 provided by Steven J. Norris, Ph.D. of University of Texas Health Science Center at Houston (36). C. elegans PNC1 and PNC2 (CePNC1 and CePNC2) were expressed and purified as described (12) and the expression plasmid of S. cerevisiae Pnc1 was received as a gift from Dr. Jeffrey Smith of the University of Virginia and expressed and purified as described previously (24). Mutants of nicotinamidase from S. pneumoniae, R97A, K103A, C136A and C136S were prepared consistent with instructions of QuickChange® Site-Directed Mutagenesis Kit (Agilent Technologies, Genomics), and each mutant was confirmed by sequencing. Primers for the mutagenesis are reported in the Supporting information. Mutants were expressed in the same manner as native SpNic. Proper folding of mutant enzymes was confirmed by native gel electrophoresis. For all of the experiments that follow, the enzyme concentrations were determined by the method of Bradford (37).
GDH Coupled Nicotinamidase Assay
Nicotinamidase activity was monitored by coupling the production of ammonia with the consumption of NAD(P)H by the enzyme bovine glutamate dehydrogenase (GDH, from Sigma). A typical reaction contained 1 mM α-ketoglutarate, 250 µM NAD(P)H, 1.5 units of GDH per 100 µL of reaction volume, and different concentrations of nicotinamide in 100 mM phosphate buffer, pH 7.3 at 25. The components were mixed and placed in cuvettes (for analysis by UV, 340 nm detection wavelength), or into a 96-well plate (for analysis on a plate reader using fluorescence). Reaction was initiated by addition of the appropriate nicotinamidase enzyme. Typical enzyme concentrations were: [BbNic] = 140 nM. [PfNic] = 14 nM. [Pnc1] = 210 nM. [SpNic] = 12 nM. [CePNC1] = 10 nM. [CePNC2] = 133 nM. Absorbance at 340 nm was used to monitor concentration of NAD(P)H with time. In plate reader experiments, fluorescence intensity was used to monitor NADP(H) (excitation 360 nm, emission 490 nm).
HPLC Assay for Nicotinamidase Activity
All HPLC analyses were performed on a Hitachi Elite Lachrom system equipped with diode array detector. After a typical injection on a Macherey-Nagel Nucleosil C-18 250 mm × 4.6 mm column, the mixture was eluted with 20 mM ammonium acetate pH 6.9 at 1 mL/min for 15 min and then eluted with 20 mM ammonium acetate pH 6.9 10% methanol. Authentic nicotinic acid eluted at 4.2 min and nicotinamide eluted at 16 min using 260 nm as a detection wavelength (Typical chromatograms are available in the Supporting Information). In some instances a Waters 300 mm × 7.8 mm Delta-pak C18 column was used for HPLC measurements with slight adjustments to the assay method. A typical reaction was run in 50 µL total volume containing 200 µM nicotinamide in 100 mM phosphate buffer at pH 7.3. Reaction was initiated by addition of nicotinamidase (10–200 nM final concentration) and incubated for 20 min at 25 °C. Reactions were quenched by addition of 6 µL 10 % trifluoroacetic acid and incubated on ice for 30 min. Prior to injection on HPLC, the sample was centrifuged at 13, 000 g for 2 min to remove precipitates.
Steady State Analysis (Initial Rate Method)
For C. elegans PNC1 and PNC2 (CePNC1 and CePNC2), the GDH coupled assay described above was used to determine the Michaelis-Menten parameters. Reactions were run in 150 µL total volume containing varying concentrations of nicotinamide (0, 10, 20, 30, 40, 50, 75, 100, 200, 400 and 600 µM) on a 96-well plate. The reactions were initiated by addition of nicotinamidase (final concentration 500 nM CePNC1, 667 nM CePNC2) and decreasing fluorescence intensity was monitored by a microplate reader for a total of 900 s at 27 s intervals. The initial rates of reactions were derived from the slope of product formed versus time where extent of reaction had not exceeded 15% of substrate in each case. Determined rates were plotted against nicotinamide concentrations and fit with the Michaelis-Menten equation using Kaleidagraph® (Synergy Software).
Steady State Progress Curve Analysis
For SpNic and Pnc1, kinetic parameters were measured by the GDH coupled assay described above and calculated by progress curve analysis. Reactions were run in 600 µL total volume containing 100 µM of nicotinamide in UV cuvettes. The reactions were initiated by addition of nicotinamidase (final concentrations: 200 nM Pnc1, 12 nM SpNic). Absorbance at 340 nm was monitored by UV spectrophotometer over a total of 60 min at 22 s intervals for SpNic, and 15 min at 25 s intervals for Pnc1. Progress curve analysis involved approximating instantaneous rates at a time tx, where product formation at tx−1 and tx+1 was determined and the ΔP/Δt calculated to determine the instantaneous rate. tx represents the mid time point between tx−1 tx+1 and the tx−1 tx+1 represent the start and end points of the time interval. Rates were plotted vs. the substrate concentration at each time calculated for each point and the data fit to the Michaelis-Menten equation to determine Km and kcat values. The method is similar to that published for this approach (38).
Steady State Kinetics-HPLC Analysis
For BbNic and PfNic, HPLC assay described above was used to measure the steady state kinetic parameters. Nicotinamide was incubated at concentrations of 0, 20, 40, 60, 80, 120, 160, 200 and 400 µM. For BbNic 5.4 µM was used as a final concentration. Chromatograms were analyzed at wavelength of 260 nm. Reactions were quantified by integrating areas of peaks corresponding to nicotinamide and nicotinic acid. Rates were plotted versus nicotinamide concentration and best fit of points to the Michaelis-Menten equation was performed by Kaleidagraph®.
For P. falciparum nicotinamidase, reactions containing 0, 2.5, 3.75, 5, 7.5, 10, 20, 25, 50 µM of nicotinamide with 20% [carbonyl-C14]-nicotinamide in 100 mM phosphate buffer pH 7.3 were initiated with the addition of PfNic, reactions were incubated at room temperature for 7 min and quenched by addition of 8 µL of 10% trifluoroacetic acid. After centrifugation at 13,000 g for 2 min, the reactions were injected on HPLC to separate nicotinamide and nicotinic acid. Eluents containing nicotinamide and nicotinic acid were collected and radioactivity was determined by scintillation counting. Rates were determined as cpm/s conversion to nicotinic acid, and then converted to a turnover rate (s−1) by adjustment for specific radioactivity of nicotinamide and enzyme concentration. Rates were plotted versus nicotinamide concentration and best fit of plotted data to the Michaelis-Menten equation was performed using Kaleidagraph®.
Inhibition Analysis
To determine inhibition, reactions were performed using the GDH coupled assay described above. Reactions containing 1 mM α-ketoglutarate, 250 µM NAD(P)H, 1.5 units of GDH per 100 µL of reaction volume, either 200 µM or 2 mM of nicotinamide, and varying concentrations of inhibitors in 100 mM phosphate buffer, pH 7.3, were initiated with the addition of nicotinamidases. Rates were plotted and points fit to equation 1 using Kaleidagraph®:
| (1) |
where νinh. is the inhibited rate for a given concentration of inhibitor, ν0 is the uninhibited rate, [S] is the nicotinamide concentration, Km is the Michaelis constant for nicotinamide for the nicotinamidase, [I] is the inhibitor concentration and Ki is the intrinsic binding constant for competitive inhibition. In most cases the inhibition data was fit to Morrison’s quadratic equation and in all cases where inhibitors were found to have an intrinsic Ki below 5 µM (Equation 2):
| (2) |
where νinh is the inhibited rate for a given concentration of inhibitor, ν0 is the uninhibited rate, [E]T is the total enzyme concentration, [I]T is the total inhibitor concentration, and Kiapp is the apparent inhibition constant as described (39). Ki, the intrinsic binding constant of the inhibitor to the enzyme can be calculated from Kiapp by the relation (Equation 3):
| (3) |
where [S] is the substrate concentration and Km is the Michaelis constant of the substrate to the enzyme as described (39).
Competition Analysis of Inhibitors
Nicotinaldehyde was characterized as an inhibitor versus nicotinamide. Use of the GDH coupled assay described above was used for the competitive analysis. Inhibition reactions were performed in 150 µL volumes containing 1 mM α-ketoglutarate, 250 µM NADPH, 2.25 units of GDH, and different concentrations of nicotinamide (50, 100, 250, 500 and 1000 µM) in 100 mM phosphate buffer, pH 7.3, with inhibitor concentrations from 0 to 2× or 5× estimated Ki as indicated in Figure 5. Reactions were initiated by addition of nicotinamidase enzymes (typically 20 nM-100 nM). Double reciprocal plots of 1/ν versus 1/[Nicotinamide] at different fixed concentrations of the inhibitor were plotted where ν is the observed rate. Intersection of the lines at 1/ν axis indicated competitive inhibition. All data points were fit to the double reciprocal equation for competitive inhibition using Kaleidagraph®.
Figure 5.

The Lineweaver-Burke plots for nicotinamidases from P. falciparum (A), S. pneumoniae (B) and B. burgdorferi (C) in the presence of nicotinaldehyde determined by fluorescence plate reader assay. Curves were obtained in the presence of three different concentrations of nicotinaldehyde, demonstrating the competitive nature of the inhibitor with substrate. The units in the y-axis are expressed as s/A.U. (A.U.: arbitrary units). A control showing that nicotinaldehyde does not interfere with the GDH enzyme activity is provided in the Supporting Information.
Nicotinamidase activity of mutants
To determine the catalytic rate of SPNic and its mutants, reactions containing 200 µM nicotinamide were carried out at pH 7.5 and 37 °C using standard assay conditions for HPLC as described elsewhere in the experimental. 100 nM SPNic wt, 100nM R97A, 5 µM K103A, 5 µM C136A, 5 µM C136S were used in each reaction respectively. Reaction were quenched by 10% TFA. Nicotinic acid was separated from nicotinamide by HPLC and quantified. The catalytic rates of SPNic and the R97A mutant were also determined by plate assay as described elsewhere in the experimental at 37 °C. In HPLC assays, the observed catalytic rates of K103A C136A and C136S were determined from product formation after a 1 h reaction. Similar reaction rate analyses were performed in each case with 1 mM nicotinamide.
18O Isotope Exchange Catalyzed by S. pneumoniae Nicotinamidase
To obtain a fully equilibrated nicotinic acid with 18O content in water, 25µL reactions in 40 mM phosphate buffer pH 7.0 containing 18O-H2O (to final 18O content of 87%), 1 mM nicotinic acid and 6 µM SpNic were incubated for 0 (pre quenched), 2, 10, 30, 60 min at 37 °C and reactions were quenched with 4 µL 10% trifluoroacetic acid. A reaction with no enzyme was run as a control and quenched identically at 60 min. 20 µL of each reaction was injected onto a C-18 reverse phase column (EC250/4.6 NUCLEODUR 100-5 C18, Macherey-Nagel) to separate nicotinic acid using 20 mM ammonia acetate pH 7.0 as eluant. Eluant containing nicotinic acid was dried by lyophilization and redissolved into 10 µL water. 1µL of sample was spotted on a gold MALDI plate and 1µL of matrix composed of 10 mg/mL α-cyano-4-hydroxycinnamic acid (CHCA) and 0.1% TFA (50: 50 v/v) was added and dried. The mass spectrum was acquired by an Applied Biosystems DE-STR MALDI-TOF spectrometer (Rockfeller University, Proteomics Resource Center). For each sample, the mass spectrum was recorded through 200 shots and the measurement repeated three times and averaged. [U-16O]-nicotinic acid was detected at m/z = 124.07(5) which is the mass of the twice-protonated species. [16O, 18O]-nicotinic acid was detected at m/z = 126.07(5) and [U-18O]-nicotinic acid was detected at m/z = 128.07(5). Complete equilibrative exchange of 18O into nicotinic acid was observed after an hour and significant exchange was measured at 2 min. No exchange was observed in the sample that received no enzyme. Formation of 18O nicotinic acids (single and double labeled) was confirmed by HRMS (CUNY Hunter Mass Spectrometry Facility). Predicted mass for single labeled 18O nicotinic acid (C6H5NO[18O]) m/z calculated =125.0363, found 125.0365; for (C6H5N[18O]2) m/z calculated = 127.0405, found 127.0407.
In order to determine the rate of exchange with 18O-H2O, we used the initial rate method. Reactions of volume 25µL in 40 mM phosphate buffer pH 7.0 containing 18O-H2O (to final 18O content of 87%), 1 mM nicotinic acid and 100 nM SpNic were reacted for 0, 20, 40, 60, 120 min and quenched and processed as described above. The 18O isotope-incorporated nicotinic acid at m/z = 126 and at m/z = 128 were detected by MALDI-TOF spectrometer and their combined intensity percentage was fit linearly with time. The exchange rate was corrected to the intrinsic rate of exchange by Equation 4:
| (4) |
where kobs is the experimentally determined rate of exchange, 0.870 is the mole fraction of 18O isotope in water and kobs (corrected) is the observed rate constant corrected for the isotope mole fraction.
The exchange rates of mutant SpNic enzymes were measured using 1 mM NA in reactions of volume 25µL in 40 mM phosphate buffer pH 7.0 containing 18O-H2O (to final 18O content of 87%). 100 or 500 nM SPNic wt, 500 nM R97A, 5 µM K103A, and 5 µM C136A were used in each reaction respectively. The observed exchange rates of SPNic wt and R97A were determined by the initial rate method for a 30 min-reaction at 37 °C; the observed exchange rates of K103A and C136A were determined from a 2 h-reaction at 37 °C. The 18O isotope-incorporated nicotinic acid at m/z of 126 and at m/z of 128 was detected by MALDI-TOF spectrometer and their combined intensity percentage was used to calculate the observed exchange rate with correction for the isotope fraction of 18O in water as described above.
Synthesis of Substrate Analogues
Nicotinamide, 2-chloronicotinamide, 6-chloronicotinamide, 6-methylnicotinamide, 6-aminonicotinamide, isonicotinamide and ethyl nicotinate were purchased from Aldrich. Thionicotinamide was purchased from Acros. We also thank Peter Tyler of IRL New Zealand for several samples of nicotinamides including 5-methylnicotinamide, 6-chloronicotinamide, 6-methylnicotinamide and 6-aminonicotinamide. 5-methylnicotinamide was alternatively synthesized as reported (40).
5-Methoxynicotinamide was synthesized from the corresponding aldehyde (41). 5-Methoxy-3-pyridinecarboxaldehyde (400 mg, 2.92 mmol) was dissolved in 4 mL of methanol and to this solution was added hydroxylamine hydrochloride (264 mg, 3.80 mmol). The reaction mixture was allowed to stir at 25 °C for 4 h. Solvent was removed under reduced pressure, residue was redissolved in 3 mL of dry pyridine and to this solution was added methanesulfonyl chloride (435 mg, 3.80 mmol). The mixture was stirred at 25 °C for 2 h, and then water and ethyl acetate were added. The combined organic layer was dried over Na2SO4. Column chromatography (hexanes: ethyl acetate = 4:1) afforded 320 mg (2.39 mmol, 82% yield) of 3-cyano-5-methoxypyridine as a white solid. 1H NMR (400 MHz, CDCl3), δ ppm: 8.49 (d, J= 2.8 Hz, 1H), 8.46 (d, J= 1.5 Hz, 1H), 7.37 (dd, J= 1.6, 2.8 Hz, 1H), 3.89 (s, 3H). Hydrogen peroxide (4 mL) was added dropwise to a solution of 3-cyano-5-methoxypyridine (320 mg, 2.39 mmol) and K2CO3 (790 mg, 5.72 mmol) in 3 mL of DMSO, the mixture was stirred for one hour before it was diluted with water and lyophilized to dryness. The crude product was purified by column chromatography (ethyl acetate: ethanol = 10:1) followed by recrystallization in ethyl acetate to afford 200 mg (1.32 mmol, 55% yield) of 5-methoxynicotinamide as white solid. 1H NMR (400 MHz, DMSO), δ ppm: 8.62 (d, J= 1.7 Hz, 1H), 8.40 (d, J= 2.9 Hz, 1H), 8.13 (s, br, 1H), 7.74 (dd, J= 1.8, 2.8 Hz, 1H), 7.59 (s, br, 1H), 3.86 (s, 3H); 13C NMR (100 MHz, DMSO), δ ppm: 55.7, 118.8, 130.3, 140.1, 140.7, 155.0, 166.2; HRMS (ESI): calcd for C7H8N2O2 152.0586, found 152.0587.
4-Methoxynicotinamide was synthesized from the corresponding aldehyde (41). 4-Methoxy-3-pyridinecarboxaldehyde (180 mg, 1.31 mmol) was dissolved in 2 mL of methanol and to this solution was added hydroxylamine hydrochloride (118 mg, 1.70 mmol). The reaction mixture was allowed to stir at room temperature for 4 h. Solvent was removed under reduced pressure, residue was redissolved in 2 mL of dry pyridine and to this solution was added methanesulfonyl chloride (195 mg, 1.70 mmol). The mixture was stirred at room temperature for 2 h, and then water and ethyl acetate were added. The combined organic layer was dried over Na2SO4. Column chromatography (hexanes: ethyl acetate = 1:2) afforded 135 mg (1 mmol, 78% yield) of 3-cyano-4-methoxypyridine as white solid. 1H NMR (400 MHz, CDCl3), δ ppm: 8.67 (s, 1H), 8.62 (d, J= 6.0 Hz, 1H), 6.90 (d, J= 6.0 Hz, 1H), 3.99 (s, 3H). Hydrogen peroxide (1.67 mL) was added dropwise to a solution of 3-cyano-4-methoxypyridine (135 mg, 1mmol) and K2CO3 (333 mg, 2.4 mmol) in 3 mL of DMSO, the mixture was stirred for one hour before it was diluted with water and lyophilized to dryness. The crude product was purified by column chromatography (ethyl acetate: ethanol = 10:1) followed by recrystallization in ethyl acetate to afford 100 mg (0.66 mmol, 66% yield) of 4-methoxynicotinamide as white solid. 1H NMR (400 MHz, DMSO), δ ppm: 8.70 (s, 1H), 8.51 (d, J= 5.8 Hz, 1H), 7.64 (s, br, 1H), 7.61 (s, br, 1H), 7.15 (d, J= 5.9 Hz, 1H), 3.93 (s, 3H); 13C NMR (125 MHz, DMSO), δ ppm: 57.0, 108.7, 117.7, 151.5, 154.2, 165.2, 168.9; HRMS (ESI): calcd for C7H8N2O2 152.0586, found 152.0587.
2-Aminoisonicotinamide was synthesized as described before (42) with several modifications. Briefly, acetylation of 2-amino-4-picoline followed by oxidation using KMnO4 gave 2-N-acetylamido-isonicotinic acid. This compound was subjected to amidation in the presence of DCC and ammonia in methanol to yield 2-N-acetylamido-isonicotinamide. Further reaction of 2-N-acetylamido-isonicotinamide with ammonia in methanol provided the desired 2-aminoisonicotinamide in modest yield, 1H NMR matches reported data
Phenyl nicotinate was synthesized from nicotinoyl chloride hydrochloride. To a 100 mL round-bottom flask were added nicotinoyl chloride hydrochloride (770 mg, 4.33 mmol), 5 mL of triethylamine and 20 mL of THF. To this mixture was added 4 mL of pyridine dropwise. After stirring for 30 min at 25 °C, phenol (610 mg, 6.49 mmol) was added. The reaction was kept at 25 °C overnight, precipitate was filtered off and solution was evaporated under reduced pressure. The residue was redissolved in water and extracted with CH2Cl2. The combined organic layer was washed with brine and dried over anhydrous Na2SO4. Solvent was then concentrated in vacuo and crude product was recrystallized from ethyl acetate. 1H NMR (500 MHz, CDCl3), δ ppm: 9.41 (s, 1H), 8.86 (dd, J = 1.7, 4.9 Hz, 1H), 8.46 (dt, J = 2.0, 8.0 Hz, 1H), 7.47 (stack, 3H), 7.32 (t, J = 7.3 Hz, 1H), 7.23 (m, 2H).
1-Methylnicotinamide was synthesized similarly to the method reported by Martin and Hull (43). Briefly, three equivalents of iodomethane was added to a solution of nicotinamide in 2 mL of methanol. The reaction was allowed to stir at room temperature and monitored by TLC. When the reaction was complete (after approximately 30 hours), the solid was filtered off and dried under reduced pressure. The resulting product was purified by recrystallization from methanol. 1H NMR (500 MHz, DMSO), δ ppm: 9.4 (s, 1H), 9.11 (d, J= 6.1 Hz, 1H), 8.90 (d, J= 8.2 Hz, 1H), 8.51 (s, br, 1H), 8.25 (dd, J= 6.1, 8.1 Hz, 1H), 8.14 (s, br, 1H), 4.42 (s, 3H). 1-Methyl-isonicotinamide was prepared similarly.
Synthesis of Inhibitors
3-Pyridinecarboxaldehyde, 5-bromo-3-pyridinecarboxaldehyde, 2-chloro-3-pyridinecarboxaldehyde, 3,5-lutidine, 3,5-dibromopyridine, 4-methoxypyridine, 3-cyanopyridine, 3-acetylpyridine, 3-hydroxypyridine and nicotinic acid were purchased from Aldrich.
5-Methoxy-3-pyridinecarboxaldehyde was synthesized in two steps from 3, 5-dibromopyridine (44). Sodium methoxide in methanol (4 mL, 9.56 mmol) was stirred under reduced pressure at 65 °C for 10 min. The remaining solid was dissolved in 5 mL of DMF. Solid 3, 5-dibromopyridine (1.5 g, 6.33 mmol) was added, and the mixture was stirred at 63~68 °C for one hour. Then it was poured into water, extracted with ethyl acetate. The combined organic layer was washed with brine and dried over anhydrous Na2SO4. After solvent was evaporated under reduced pressure, column chromatography afforded 1 g (5.32 mmol, 85% yield) of 3-bromo-5-methoxypyridine as a white solid. 1H NMR (400 MHz, CDCl3), δ ppm: 8.31 (s, 1H), 8.26 (s, 1H), 7.35 (s, 1H), 3.88 (s, 3H). To a stirred solution of 3-bromo-5-methoxypyridine (1 g, 5.3 mmol) in 20 mL of anhydrous THF at −90 °C was added nBuLi (2.9 mL, 5.8 mmol) over 5 min. The solution was allowed to stir for another 20 min at −90 °C, and then DMF (0.53 mL, 6.9 mmol) was added. The reaction mixture was stirred for 30 min and the cold mixture was poured directly into brine and extracted with ethyl acetate. Combined organic layer was dried over anhydrous Na2SO4. After solvent was evaporated under reduced pressure, column chromatography afforded 0.5 g (3.65 mmol, 68% yield) of 5-methoxy-3-pyridinecarboxaldehyde as a pale yellow solid. 1H NMR (400 MHz, CDCl3), δ ppm: 10.08 (s, 1H), 8.63 (d, J= 1.5 Hz, 1H), 8.53 (d, J= 3 Hz, 1H), 7.58 (dd, J= 1.5, 3 Hz, 1H), 3.89 (s, 3H).
5-Methyl-3-pyridinecarboxaldehyde was synthesized in three steps from 3,5-lutidine. 3,5-lutidine was converted to 5-methylnicotinic acid as described previously (45). 5-methylnicotinic acid (100 mg, 0.73 mmol), triphenylphosphine (382 mg, 1.46 mmol) and methanol (47 mg, 1.46 mmol) were dissolved in 1 mL of anhydrous THF, to this solution was added DIAD (295 mg, 1.46 mmol) at 0 °C dropwise. The reaction mixture was allowed to warm to room temperature and stir overnight. Saturated brine was added to quench the reaction, aqueous layer was extracted with ethyl acetate. Combined organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. Crude product was purified by column chromatography to afford 70 mg (0.46 mmol, 64% yield) of 5-methylnicotinic acid methyl ester as white solid. 1H NMR (500 MHz, CDCl3), δ ppm: 9.01 (s, 1H), 8.59 (s, 1H), 8.09 (s, 1H), 3.93 (s, 3H), 2.38 (s, 3H). To a solution of 5-methylnicotinic acid methyl ester (70 mg, 0.46 mmol) in 2 mL of toluene was added DIBAL-H (3.2 mL, 3.2 mmol) dropwise at −78°C. The reaction was kept at −78°C for one hour before quenched by methanol at −20 °C. To the reaction mixture was then added 0.1 M HCl and ether, aqueous phase was extracted with ether, combined organic phase was washed with saturated NaHCO3, water and brine, and then dried over anhydrous Na2SO4. Solvent was evaporated under reduced pressure and crude product was purified by column chromatography to afford 50 mg (0.41 mmol, 89% yield) of 5-methyl-3-pyridinecarboxaldehyde as colorless oil. 1H NMR (500 MHz, CDCl3), δ ppm: 10.08 (s, 1H), 8.87 (s, 1H), 8.67 (s, 1H), 7.95 (s, 1H), 2.43 (s, 3H).
6-Fluoro-3-pyridinecarboxaldehyde was synthesized from the corresponding carboxylic acid. In general, the acid was reduced with 1.1 equivalents of lithium aluminum hydride in refluxing anhydrous THF overnight. After quenching with ethyl acetate and water, the precipitate was filtered off, solvent was removed under reduced pressure and the alcohol was used for the next step without further purification. The alcohol was added to two equivalents of sodium nitrite followed by one equivalent of acetic anhydride and stirred at 0 °C (46). The reaction was monitored by TLC and upon completion the product was extracted with diethyl ether. The aldehyde was further purified by column chromatography. 1H NMR (500 MHz, CDCl3), δ ppm: 10.08 (s, 1H), 8.75 (d, J= 1.5 Hz, 1H), 8.32 (dt, J= 2.1, 8.2 Hz, 1H), 7.11 (dd, J= 2.5, 8.4 Hz, 1H). 13C NMR (125 MHz, DMSO), δ ppm: 110.6, 110.9, 114.1, 139.3, 140.9, 141.0, 152.0, 152.1, 188.6; HRMS (ESI): calcd for C6H4FNO 125.0277, found 125.0281.
4-Methoxy-3-pyridinecarboxaldehyde was synthesized in one step from 4-methoxypyridine (44). To a solution of tBuLi (1.7 M in pentane, 16.6 mL, 28 mmol) in 30 mL of anhydrous THF at −78°C was added 2.2 g of 2-bromomesitylene (1.66 mL, 11 mmol). After stirring at −78 °C for 1 hour, a pale yellow heterogeneous mixture was formed. To this mixture was then added 1.09 g of 4-methoxypyridine (1 mL, 10 mmol), and stirring was continued at −78 °C for 1 hour, at −23 °C for 1 hour and at room temperature for 1 hour. The mixture was again cooled to −78 °C, and 0.954 g of DMF (1 mL, 13.1 mmol) was added dropwise. After stirring at −78 °C for another hour, the reaction was quenched by saturated NaHCO3 solution. Aqueous layer was extracted with ethyl acetate (3 × 10 mL), and organic layers were combined and dried over anhydrous Na2SO4. After solvent was evaporated under reduced pressure, column chromatography afforded 0.88 g (6.4 mmol, 64% yield) of 4-methoxy-3-pyridinecarboxaldehyde as a pale yellow solid. 1H NMR (400 MHz, CDCl3), δ ppm: 10.42 (s, 1H), 8.86 (s, 1H), 8.61 (d, J= 6 Hz, 1H), 6.91 (d, J= 6 Hz, 1H), 3.98 (s, 3H).
Results
Cloning and Protein Expression
The genes encoding the nicotinamidase enzymes from B. burgdorferi (BbNic), S. pneumoniae (SpNic) and P. falciparum (PfNic) were cloned into appropriate expression vectors and expressed and purified as described in the Materials and Methods and Supporting Information. Expression plasmids for the two C. elegans nicotinamidases, CePNC1 and CePNC2 (12), and S. cerevisiae Pnc1 (24) were previously described. When assayed by HPLC under standard reaction conditions (200 µM nicotinamide, 100 mM phosphate, pH 7.3), all enzymes were found to catalyze the conversion of nicotinamide into nicotinic acid (Supporting Information).
Nicotinamidase Assay Development
In order to monitor the activity of purified enzymes in real time, we developed a robust and straightforward assay that could continuously monitor one of the two products of the nicotinamidase reaction, ammonia. Bovine glutamate dehydrogenase (GDH), which is commercially available, was found to be an ideal enzyme for this purpose. GDH catalyzes the reaction of α-ketoglutarate, ammonia and NADPH (or NADH) to form glutamate and the oxidized dinucleotide (Scheme 3). The ammonia produced by conversion of nicotinamide to nicotinic acid is reacted by the coupling enzyme, which in turn consumes NADPH stoichiometrically. NADPH conversion to NADP+ can be measured by absorbance of NADPH at a wavelength of 340 nm where NADP+ has no absorbance (Δε340nm = 6200 AU. M−1 cm−1). Alternatively, the method was readily adapted to a plate reader which can detect fluorescence emission of NADPH (360 nm excitation, 490 emission). NADPH is measured by amount of emitted light and rates determined by changes in light emission versus time. Both methods were used to monitor nicotinamidase activity in this study.
Scheme 3.

The nicotinamidase assay developed couples the release of ammonia to the consumption of NAD(P)H via glutamate dehydrogenase. The synthesis of glutamate by GDH occurs in the presence of ammonia and α-ketoglutarate and is dependent upon the stoichiometric oxidation of NAD(P)H. The conversion of NAD(P)H to NAD(P)+ can be followed by monitoring the decrease in absorbance at 340 nm or the decrease in fluorescence (excitation 360 nm, emission 490 nm).
The quantity of NADPH consumed correlates well to nicotinamide or ammonia quantities added to a sample as shown in Figure 2 A and 2B. In addition, the rate of nicotinamidase activity scales linearly with amount of enzyme added, demonstrating that the coupling enzyme can be provided so that GDH is not limiting for determination of catalytic velocity (Figure 2C). This assay is effective for monitoring a wide range of nicotinamide concentrations and can accurately detect rates generated by low micromolar concentrations of nicotinamide, particularly because of the low GDH Km for NADPH (28 µM) and an excellent rate of turnover (>9 s−1, calculated from kinetic parameters) (47). Favorably, the equilibrium position very strongly favors the oxidized dinucleotide product at pH 7.0 (48). Interference of GDH from nicotinamide and nicotinic acid were neglible, and the components of the GDH assay did not interfere with nicotinamidase enzymatic activity as determined by HPLC (data not shown). The components of this assay, nicotinamidase and GDH, were recently combined in a coupled assay to detect and measure activity of nicotinamide forming enzymes, such as sirtuins, that degrade NAD+ to nicotinamide (49).
Figure 2.

(A) The GDH assay was performed with varying concentrations of ammonium acetate. (B) The GDH-coupled assay was performed with varying concentrations of nicotinamide. Reactions containing 1 mM α-ketoglutarate, 250 µM of NADPH, 1.5 units GDH per 100 µL of reaction volume in 100 mM phosphate buffer pH 7.3 were initiated with the addition of PfNic to a final concentration of 320 nM. The reactions were measured by monitoring the absorbance at 340 nm over time. (C) SpNic activity scales nicely with varying concentrations of enzyme. Reactions containing 1 mM α-ketoglutarate, 250 µM of NADPH, 1.5 units GDH per 100 µL of reaction volume in 100 mM phosphate buffer pH 7.3 were initiated with the addition of SpNic to the amounts indicated in the figure. The reactions were measured by monitoring the absorbance at 340 nm over time. The slopes for 0, 36, 72 and 108 pmol of SpNic are −2.23 × 10−5, −2.39 × 10−4, −4.79 × 10−4 and −7.30 × 10−4, respectively.
Kinetics Parameters for Nicotinamidase Enzymes
Kinetic properties of nicotinamidase enzymes cloned or isolated from a variety of organisms have been determined previously, including Arabidosis thaliana (9), M. tuberculosis (3) Mycobaceterium smegmatis (2), Lactobaccillus arabinosis (32, 50) Torula cremoris (7) Flavobacterium peregrinum (51) and S. cerevisiae (5, 6, 52, 53). Of the enzymes considered in this study, nicotinamidase activity of the P. falciparum enzyme was previously detected in crude dialyzed lysates derived from P. falciparum infected erythrocytes (8). With a reliable assay available, we sought to determine the kinetic parameters for the S. cerevisiae Pnc1 as the prototype enzyme. In addition, the kinetic parameters for the S. pneumoniae, B. burgdorferi, P. falciparum and both C. elegans nicotinamidases were determined for the first time. Enzymes were expressed in E. Coli and isolated by Ni+2 column chromatography. Although different enzymes from different sources have been reported to have different central metal requirements (3, 9, 34), enzymes were active as purified without reconstituting them from apo-form. However, we did examine the addition of metal ions to the purified enzymes, and in almost every case, no change in activity was observed (See Supporting Information, Table S1). We did not attempt to investigate the identity of the coordinated central metal atom for the respective enzymes, although SpNic was recently reported to prefer Zn+2 (35) and the preferred metal for the yeast enzyme is reportedly Zn+2 as well (6). Interestingly, addition of Zn+2 to reaction mixtures did not appreciably change catalytic activity of any nicotinamidase although the SpNic and CePNC2 enzymes were moderately stimulated by Mn+2 added to reaction mixtures (See Supporting Information).
Employing standard conditions of assay (100 mM potassium phosphate, pH 7.3 with or without GDH components for detection of ammonia formed by nicotinamidase reaction, as described in Materials and Methods), we determined steady state kinetic parameters for each enzyme by varying nicotinamide concentration and plotting initial rates versus concentration of nicotinamide to determine Km and kcat values (Figure 3, and Table 1). kcat was also measured by HPLC assay using at least 10 × Km concentration of nicotinamide for confirmation.
Figure 3.

The steady state saturation curves of nicotinamidases from S. cerevisiae (A), B. burgdorferi (B), P. falciparum (C), and S. pneumoniae (D), C. elegans PNC1 (E) and C. elegans PNC2 (F). A and D were derived from progress curve analysis (see experimental) of the nicotinamidase reaction run in the presence of 100 µM nicotinamide, B and C are from HPLC analysis of reactions run in varying nicotinamide concentrations, E and F are from the GDH coupled fluorescence assay with various concentrations of nicotinamide. In all cases, the data was fit to the Michaelis-Menten equation using KaleidaGraph.
Table 1.
Kinetic parameters of nicotinamidases
| Enzyme | Km (µM)a | kcat (s−1)a |
|---|---|---|
| BbNic | 36.7 ± 7.5 | 0.0030 ± 0.0002b |
| PfNic | 3.1 ± 1.2 | 0.30 ± 0.032b |
| Pnc1 | 10.6 ± 0.5 | 3.8 ± 0.1b |
| SpNic | 2.7 ± 0.3 | 2.1 ± 0.1b |
| CePNC1 | 113 ± 11 | 3.1 ± 0.12 |
| CePNC2 | 76 ± 10 | 0.28 ± 0.013 |
Km and kcat values were determined by fitting the data to the Michaelis-Menten equation using the Kaleidagraph software package (Synergy Software). Error values represent the error in the fit to the data as calculated by the curve-fitting algorithm.
Calculated kcat values were confirmed by HPLC assay for several enzyme concentrations.
For the Pnc1 and SpNic enzymes, progress curve analysis was used (38), which enabled sensitivity to low concentrations of substrate. This was important because of a low Km for nicotinamide for these enzymes. A potential caveat with these analyses is product inhibition, which can increase the apparent Km, but we showed that nicotinamidase rates for all enzymes do not vary when concentrations of nicotinic acid up to 500 µM are present (data not shown) and nicotinic acid is thermodynamically stable relative to substrate, so the reaction invariably goes to completion. Pnc1 and SpNic were determined to have Km values of 10 and 2.7 µM for nicotinamide and corresponding values of kcat of 3.8 s−1 and 2.1 s−1.
For other nicotinamidases, we used the GDH method (CePNC1 or CePNC2) or an HPLC method (PfNic and BbNic) to determine initial reaction rates as a function of nicotinamide concentration. All data are summarized in Table 1. All Km values were found to be below 120 µM, with kcat values in the range 0.3–4 s−1. Only BbNic deviated from the pattern. An examination of the BbNic sequence against the others (Figure 1) shows that the genetically encoded enzyme is approximately 20 residues (51) shorter at the N-terminus. This region contains a highly conserved DXQXD sequence that is highly conserved in nicotinamidases (Figure 1). Thus, we introduced additional sequence at the N-terminus to create active enzyme (as originally reported (19)). It remains possible that BbNic is imperfectly reconstituted in sequence or in other requirements, accounting for its poor catalytic properties (introduction of different metal ions had no effect on activity, see Supporting Information).
Substrate Specificity
We probed the specificity of nicotinamidases to hydrolyze a variety of nicotinamide analogues. Using both HPLC analysis and the GDH coupled assay, we determined the relative rates for a number of compounds showing structural similarity to nicotinamide (Table 2). In the cases of SpNic, Pnc1, and PfNic substitutions at the 5-position were well tolerated, and interestingly, 5-methyl, 1, and 5-O-methyl substitutions, 2, caused up to a 6.5 fold rate enhancement (in kcat, see Table 2) over nicotinamide. Moving the O-methyl group to the 4-position, 7, caused slowing of all enzymes to 5%–12% of the rate observed for nicotinamide (Table 2). Nicotinamidases (BbNic, PfNic, Pnc1 and SpNic) could not hydrolyze isonicotinamides 8, 9, 1-methyl isonicotinamide, 15, or 1-methylnicotinamide derivatives (14, Table 2). These results are consistent with a requirement of coordination of the pyridine to the active site metal center, as observed in recent X-ray structures (34, 35) and a requirement of a 3-substitution pattern of the hydrolyzable acyl group relative to the pyridine N1. 2-chloronicotinamide 3, could not be hydrolyzed, although 6-chloronicotinamide 4 did react as a substrate, albeit slowly, with Pnc1 and SpNic. The steric proximity of the 2 and 6-groups to the coordinating metal as well as weak basicity likely accounts for these substrate behaviors. Consistently, 6-methyl-, 6-amino- and 6-O-methylnicotinamides, 5–7 respectively, were weaker substrates than nicotinamide for all enzymes, probably because of non-optimal substrate binding to the metal center. However these substrates reacted faster than the 6-chloro-derivative probably because they are better metal ligands than the halogenated analogue. Nicotinate esters, such as methylnicotinate, 11, and ethylnicotinate 12, could be hydrolyzed efficiently, although not as well as nicotinamide. A notable exception was methylnicotinate. The rate of turnover for methylnicotinate on PfNic was determined to be 65 times faster than nicotinamide, with a steady state rate constant of 18 s−1. Interestingly, even phenylnicotinate 13 could be hydrolyzed by SpNic, Pnc1 and PfNic, implying some flexibility of the active site to accommodate this bulkier but activated ester (See Supporting Information for NMR study with this substrate and PfNic). Sensitivity to substitutions showed similar trends for the four enzymes for which substrate specificity was broadly evaluated, likely because of structural and mechanistic similarities of these enzymes.
Table 2.
Relative rates of nicotinamidase enzymes for various nicotinamide analogues
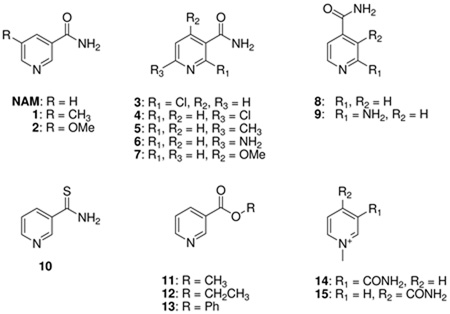 | ||||
|---|---|---|---|---|
| Substrate | BbNic | PfNic | Pnc1 | SpNic |
| NAM | 1.00 | 1.00 | 1.00 | 1.00 |
| 1a | 0.51 | 3.30 | 0.80 | 1.63 |
| 2b | 2.21 | 6.46 | 2.06 | 1.37 |
| 3b | 0 | 0 | 0 | 0 |
| 4b | 0 | 0 | 0.17 | 0.018 |
| 5a | 0.45 | 0.40 | 0 | 0.11 |
| 6a | NDc | 0.45 | 0.20 | 0.36 |
| 7b | 0.20 | 0.12 | 0.050 | 0.065 |
| 8b | NDc | 0 | 0 | 0 |
| 9b | NDc | 0 | 0 | 0 |
| 10 | 0 | 0 | 0.44 | 0 |
| 11b | NDc | 65.0 | 0.15 | 0.035 |
| 12b | NDc | 0.023 | 0.044 | 0.0086 |
| 13b | NDc | 0.13 | 0.031 | 0.031 |
| 14b | NDc | 0 | 0 | 0 |
| 15b | NDc | 0 | 0 | 0 |
Reactions were run in 150 µL total volume containing 1 mM α-ketoglutarate, 250 µM NADPH, 3 units of GDH, and 500 µM of nicotinamide analogue in 100 mM phosphate buffer, pH 7.3, for control experiments, reactions containing 500 µM of nicotinamide were run in parallel. Reactions were initiated by addition of nicotinamidase enzymes, decreasing of fluorescence intensity was monitored by a microplate reader.
Reactions were in 25 µL total volume containing 500 µM of nicotinamide analogue in 100 mM phosphate buffer, pH 7.3, for control experiments, reactions containing 500 µM of nicotinamide were run in parallel. Reactions were initiated by addition of nicotinamidase enzymes, incubated 37 °C and quenched by addition of 10% trifluoroacetic acid. Reactions were quantified by integrating area of peaks corresponding to nicotinic acid analogue.
ND = Not determined. NAM: nicotinamide.
Chemical Exchange of Nicotinic Acid Oxygens with 18O Water
To evaluate for reversibility of product formation on the SpNic enzyme, we co-incubated nicotinic acid with enzyme in buffered 18O water to test for 18O exchange into the carboxy oxygens of nicotinic acid. The cycling of nicotinic acid to a proposed thioester intermediate (33, 34) and back to nicotinic acid would be predicted to cause loss of 16O in the carboxy oxygens and replacement with 18O provided water can equilibrate into the active site. We used mass spectrometry to monitor this exchange. As shown in Figure 4B, nearly immediate loss of unlabeled nicotinic acid was observed when 1 mM nicotinic acid was treated with 6 µM SpNic enzyme. This is evident by loss of the m/z = 124 intensity in the MALDI MS spectrum, and is accompanied by increase in intensities for m/z = 126 and m/z =128. The exchange can be understood to achieve equilibration of the substrate oxygens with the initial 87% 18O content in solvent over the course of an hour, and the relevant equilibria are shown in the scheme accompanying Figure 4. The exchange of isotope can be observed in as little as 2 min (Figure 4B), and the calculated rate for the process is 0.34 s−1 (based on initial rates) indicating it is quite fast at pH 7 (Figure 4C, Table 3). To further confirm that nicotinic acid can be exchanged with 18O water to the extent predicted by the isotopic enrichment, we collected nicotinic acid after incubation with nicotinamidase and 18O water and analyzed it by HRMS, which corroborated these results (See Materials and Methods). The results show that nicotinic acid undergoes facile 18O exchange into the carboxylates catalysed by SpNic, most likely by reversible formation of thioester from nicotinic acid catalyzed by the enzyme.
Figure 4.

Mass spectra demonstrating time dependent 18O exchange into nicotinic acid (NA) catalysed by SpNic. Data is plotted as percent intensity versus time, where the total intensities of peaks in mass spectra at m/z = 124, 126, and 128 are summed to reach 100 percent. 124 is the mass of unlabeled nicotinic acid as a twice protonated cation (M+). 126 is the molecular mass of nicotinic acid incorporating a single 18O label, and 128 is the mass of nicotinic acid incorporating two 18O atoms. Mass intensities of single and double-labeled nicotinic acid are summed. The data in panel B shows that the mass spectrum of nicotinic acid treated with SpNic in the presence of 18O becomes rapidly depleted of intensity for the peak corresponding to m/z = 124, whereas there is a corresponding enrichment in m/z = 126 and m/z = 128 peak intensities, demonstrating 18O incorporation into nicotinic acid. Control does not exchange. A. Schematic exchange reaction catalyzed by SpNic. B. Time course of 18O exchange catalyzed by SPNic. The reaction was carried out in 1 mM NA, 18O-H2O, 6 µM SPNic at 37 °C for the indicated time. C. Determination of the initial rate of 18O exchange. The reaction was carried out in 1 mM NA, 18O-H2O, 100 nM SpNic at 37 °C for the indicated time. The initial rate was determined to be 0.34 s−1 after correction for the mole fraction of 18O present in the reaction (Equation 4, Materials and Methods).
Table 3.
Nicotinamidase and 18O exchange activity of SpNic and mutants
| SPNic | Catalytic ratea (kobs, s−1) | 18O Exchange rated (kobs, s−1) |
|---|---|---|
| wt | 3.4b | 0.34 |
| R97A | 3.2b | 0.27 |
| K103A | 0.0053c | 0.0076 |
| C136A | <10−6c | <10−6 |
| C136S | <10−6c | NDc |
The observed rate of nicotinamidase catalysis of SPNic and mutants measured using 200 µM NAM at 37 °C.
The rates were determined by GDH plate reader assay.
The rates were determined by HPLC assay. The full experimental is available in the Methods section.
The observed 18O exchange into nicotinic acid catalyzed by SpNic and mutants as measured by the initial rate method and mass spectrometry (Figure 4) in the presence of 1 mM nicotinic acid and 18O H2O at 37 °C. Enzyme concentrations were 100 nM SpNic wt and R97A and 5 µM for K103A or C136A mutants. The measured rate was corrected by the mole fraction of 18O in the reaction as described by equation 4 in the Methods. Full experimental is in Methods section. cN/D: not determined.
Mutations of Cys 135, Lys113 and Arg97
To further investigate the catalytic mechanism of 18O exchange and of nicotinamidase catalysis, we prepared several active site mutants of SpNic. These were C136A, C136S, K103A and R97A. The C136S mutant has been solved by X-ray crystallography and nicotinamide co-crystallizes in the active site (35). We found that C136A and C136S were unable to catalyze nicotinamide hydrolysis down to 10−6 s−1 sensitivity, consistent with prior work (35). These two mutants were unable to catalyze 18O exchange into nicotinic acid. The two mutants K103A and R97A were prepared based on the prediction that K103A is an active site Lys, which was recently confirmed by crystallography (35). R97A is a mutant that our sequence predictions indicate is possibly in the active site (Figure 1), but was shown by crystallography to be far from the catalytic site (35). Consistently, we found that R97A mutation was a robust nicotinamidase and was nearly as good as the native enzyme in promoting 18O exchange. The K103A mutant had only 2.2 % of the catalytic activity of the native enzyme in the 18O exchange reaction and 0.15 % of the native rate in the nicotinamidase reaction (Table 3). This mutant is well expressed and the slow rate is attributed to the essentiality of this universally conserved lysine in catalysis.
Inhibition by Nicotinaldehyde
A report that S. cerevisiae nicotinamidase could be weakly inhibited by nicotinaldehyde Ki = 68 µM (52) led us to wonder if nicotinaldehydes might be general inhibitors of nicotinamidases. Yan et al. (52) suggested that nicotinaldehyde inhibited the yeast enzyme by a non-competitive mechanism via Schiff’s base formation. We reinvestigated this inhibition and showed that nicotinaldehyde 16 potently inhibits nicotinamidases from all sources we examined (Table 4). Double reciprocal plots confirmed competitive inhibition with nicotinamide in all cases, in contrast to the Yan report (52). (Three examples are provided in Figure 5, SpNic, PfNic and BbNic, other examples are available in the Supporting Information). Table 4 shows that the determined Ki values were 1.5 µM for the yeast enzyme, whereas Ki values were 34, 11 and 110 nM for the corresponding enzymes from P. falciparum, S. pneumoniae and B. burgdorferi. Nicotinaldehyde competitively inhibited both C. elegans enzymes with 22 nM and 110 nM Ki values determined for PNC2 and PNC1 respectively (Table 4 and Supporting Information).
Table 4.
Ki Values of Nicotinamidase Inhibitors (µM)
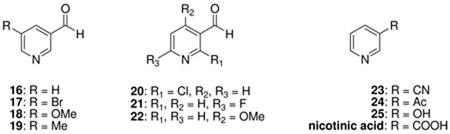 | ||||||
|---|---|---|---|---|---|---|
| Substrate | BbNica | PfNicb | Pnc1c | SpNicd | CePNC1e | CePNC2f |
| 16 g | 0.11± 0.02 | 0.034± 0.007 | 1.4± 0.1 | 0.011± 0.001 | 0.11± 0.01 | 0.022± 0.004 |
| 17 g | 1.3± 0.20 | 0.59±0.14 | 4.0± 0.80 | 0.071± 0.022 | 0.14± 0.02 | 0.088± 0.02 |
| 18 g | 0.85± 0.04 | 0.039± 0.004 | 3.8± 0.54 | 0.14± 0.02 | 0.31± 0.05 | 0.14± 0.02 |
| 19 g | 0.19± 0.03 | 0.023± 0.005 | 0.65± 0.07 | 0.056± 0.002 | NDh | ND |
| 20 | 370i | NIj | 5000i | 110i | ND | ND |
| 21 | 18i | 4.9i | NI | 0.73± 0.29g | ND | ND |
| 22 | 153i | 1.0± 0.11 | 68i | 50i | 5.3± 1.0g | 0.44± 0.022g |
| 23 | 2000i | 1000i | 85i | 500i | ND | ND |
| 24 | 342i | 10i | 46i | 60i | ND | ND |
| 25 | NI | NI | NI | NI | ND | ND |
| nicotinic acid | NI | NI | NI | 2000i | NI | NI |
Typical enzyme concentrations: a[BbNic] = 140 nM.
[PfNic] = 14 nM.
[Pnc1] = 210 nM.
[SpNic] = 12 nM.
[CePNC1] = 10 nM.
[CePNC2] = 133 nM. In most cases the inhibitions were determined by the GDH assay as described in Materials and Methods.
The inhibition data was fit to Morrison’s equation 2 described in the Materials and Methods (See examples of data and fits in Figure 6 and in the Supporting Information). The determined Kiapp values were converted to the Ki values (the intrinsic binding constant of the inhibitor for the enzyme) shown in Table 4 by Equation 3 in the Materials and Methods.
ND= not determined.
The inhibition data were fit to Equation 1 as described in Materials and Methods.
NI = no inhibition detected.
Nicotinaldehydes as Inhibitors of Nicotinamidases
To more completely explore the potency of nicotinaldehydes as inhibitors of nicotinamidases we synthesized a number of substituted analogues of nicotinaldehyde, with variable substituents (as shown in Table 4). The Ki values for inhibition of the nicotinamidases are listed in Table 4. Consistent with observations that 5-substituted nicotinamides are excellent substrates, 5-substituted nicotinaldehyde analogues 17–19 were potent inhibitors of all of nicotinamidases tested (Table 4). Several representative curves demonstrating inhibition of nicotinamidases by 5-O-methyl and 5-methylnicotinaldehydes 18 and 19 respectively are shown in Figure 6. All enzymes tolerated even a bulky 5-bromo substituent (17; Table 4).
Figure 6.

Curves showing the inhibition of the nicotinamidase enzymes by 5-O-methylnicotinaldehyde (A, [PfNic] = 14 nM) and 5-methylnicotinaldehyde (B, [SpNic] = 12 nM C, [PfNic] = 14 nM). The reactions were run in the presence of 1 mM nicotinamide and various concentrations of the inhibitor in 1 mM α-ketoglutarate, 250 µM NADH and 1.5 units of GDH per 100 µL of reaction volume in 100 mM phosphate buffer, pH 7.3. After initiation by addition of the enzyme, the reactions were monitored by fluorescence and initial rates of reaction were calculated. The curves show observed rate with respect to inhibitor concentration. Data points are fit to Morrisons equation (39) and the Ki calculated from the Kiapp value of Morrison’s equation as described in the Materials and Methods. Controls showing that nicotinaldehydes do not interfere with the GDH enzyme activity is provided in the Supporting Information.
Consistent with observations that 4-O-methyl substitution was poorly tolerated in substrates, 4-O-methylnicotinaldehyde 22 was a weak inhibitor with a higher Ki value (Table 4). However, 22 was a fair inhibitor of the CePNC2 enzyme, with a 19 fold selectivity over CePNC1, suggesting that variations in chemical structure of these compounds can lead to modest inhibitor selectivity. (Table 4). We also tested several other compounds for inhibitory activity including acetylpyridine 24, 3-cyanopyridine 23, 3-hydroxypyridine 25 and nicotinic acid (Table 4).
Discussion
Mechanism of Inhibition
We propose that the mechanism of aldehyde inhibition parallels the mechanism of catalysis. Nicotinamide initially coordinates the central metal atom, and is attacked by the universally conserved active site Cys to form a tetrahedral adduct that subsequently decomposes by elimination of ammonia to yield an enzyme stabilized thioester. Subsequent hydrolysis of the thioester completes nicotinic acid formation on the enzyme. This overall general mechanistic proposal was first proposed by Fyfe et al. (34). We propose that nicotinaldehydes bind the enzyme analogously via coordination of the pyridine N1 (Scheme 4). The subsequent attack of the cysteine on the aldehyde occurs analogously to that envisioned for substrate, except that instead of forming an unstable intermediate that can decompose to a thioester, the tetrahedral adduct, a thiohemiacetal, is unable to decompose forward and is stalled by enzymatic features that stabilize its tetrahedral geometry, which is the oxyanion hole identified to be Ala 155 and Cys 159 on the A. baumanii enzyme (34). Sequence comparisons show that the Ala is highly conserved across nicotinamidases, although is found to be a Leu131 in the SpNic enzyme and it has been confirmed by X-ray crystallography that this Leu contributes to the oxyanion hole (Figure 1, Scheme 4 (35)). The Cys residue is universally conserved, and the n, n+4 relationship in sequence is also conserved across nicotinamidases (Figure 1). The ability of aldehydes to trap Cys nucleophiles of amidases is known, and very potent aldehyde inhibitors have been synthesized that target the caspase enzymes, wherein thiohemiacetal adducts have been demonstrated by X-ray crystallography (54). The thiohemiacetal adducts to caspases are stabilized by binding interactions to the oxyanion hole.
Scheme 4.

Mechanism for nicotinaldehyde inhibition of nicotinamidase enzymes. Numbering is from the enzyme from SpNic. Top figure represents reaction coordinate diagram, where plateaus levels are meant to indicate relative energy.
The tetrahedral adduct formed can decompose only by reaction reversal to the initial aldehyde-metal complex (Scheme 4). We propose that thiohemiacetal adducts form reversibly with solution nicotinaldehyde because the inhibitions observed are competitive with nicotinamide present (Figure 5) and no bi-phasic onset is apparent for reaction progress curves, and preincubation does not change inhibitor potency (data not shown). Ki values for nicotinaldehyde are smaller than the Km for nicotinamide by a factor of 91–3450 across all enzymes studied (with the exception of yeast pnc1 where Km/Ki is 7.6). The inhibition data are consistent with oxyanion stabilization of the tetrahedral intermediate formed during catalysis, an energy of binding which can also be captured in the form of enzyme-stabilized thioacetal-adducts. This mechanism of inhibition was recently confirmed on the SpNic enzyme for nicotinaldehyde 16 and 5-O-methylnicotinaldehyde 18 (35). We depict the inhibition as depending upon the initial coordination of the aldehyde to the metal center, characterized by a binding constant Kd, which is further stabilized by a factor 1/Kint, where Kint is the equilibrium constant for the cysteine adduct. The inhibition constant can thus be deduced to be Ki = Kd(Kint)−1. This is also qualitatively visualized in the reaction coordinate presented in Scheme 4.
18O Exchange Mechanism and Catalytic Mechanism of Nicotinamidases
The X-ray crystal structure of nicotinic acid coordinated to the nicotinamidase from A. baumanii (AbPncA) led to a firm mechanistic proposal for catalysis by Fyfe et al (34). However, we have obtained additional insight into the general properties of these enzymes by determining the substrate specificities, the generality of nicotinaldehyde inhibitions as well as the observation that SpNic can catalyse 18O exchange into the product nicotinic acid. Mutational studies have also provided new insights. To explain 18O exchange we propose the mechanism in Scheme 5. Coordination of nicotinic acid to the Zn+2-aqua center places the carboxy of substrate in proximity to three universally conserved residues, Asp8, Cys 136 and Lys103 (using the SpNic sequence information). Crystallographic data suggests that the conserved Asp can interact equally with the Cys thiol as well as the nicotinate oxygen (34, 35), and this structural arrangement is likely preserved for the corresponding SpNic residue Asp 8. We propose that when the universally conserved Asp and the metal coordinated nicotinic acid share a hydrogen bond, with the Cys nucleophile in thiolate form (SpNic sequence), the nicotinic acid can react by Cys thiolate nucleophilic addition to form a tetrahedral adduct. In this process, full proton transfer from Asp occurs. The sp3 center generated on substrate bears a hydroxyl substituent, an oxyanion substituent (which is proposed to sit in the oxyanion hole) and a thioether substituent (from nucleophilic Cys). Subsequent tetrahedral intermediate collapse eliminates hydroxide to form a thioester intermediate.
Scheme 5.

Mechanism for nicotinic acid reversal to the nicotinoyl thioester complex. The mechanism explains the ability of SpNic to catalyse 18O exchange into nicotinic acid. A key feature of the proposed mechanism is the formation of a deprotonation of Lys103 upon thioester formation from nicotinic acid, which then serves as a base catalyst to activate water in the 18O exchange reaction. The numbering system used is for the SpNic enzyme.
Full expulsion of the hydroxide group from the tetrahedral center would be thermodynamically challenging, but could be facilitated by protonation of the leaving group oxygen by Lys103 (SpNic) which is universally conserved in all nicotinamidases and when mutated in M. tuberculosis PncA produces an inactive enzyme (3, 4). Correspondingly, in SpNic the K103A mutant has only 2.2 % of the activity of native enzyme as measured by observed catalytic rate of 18O exchange (Table 3). There is a 4–5 order of magnitude difference in predicted pKa between water and protonated Lys103 (using the free amino acid pKa value), making proton transfer to form water thermodynamically downhill (Scheme 5). The deprotonation of this Lys is supported by the fact that the ε-N is located 4.8 Å to the carboxy oxygen in the AbPncA structure (34) and located only 4.70 Å from the carbonyl carbon in the thioester complex in the SpNic thioester structure determined recently (35), suggesting there is intervening space for a water to accommodated. The first half of the exchange reaction is summarized as follows, a thiolate (pKa =7–8) is reacted to form a Lys (ε-NH2, pKa = 9–10; the free amino acid) with a CysS-C(carbonyl) bond formed and a O-C(carboxy) bond in nicotinic acid cleaved. In addition two strong O-H bonds are formed in the process of generating water at the active site from deprotonation of two acidic groups (AspCOOH and LysNH3+). The overall driving force appears to be formation of water in the active site, making it easier to understand how the barrier to isotope exchange is not significant, as measured by the 0.34 s−1 rate constant for this process. The expulsion of water from the thioester site followed by water rebinding (water exchange) permits 18O leakage into the active site and the reaction run in reverse completes a single 18O exchange into the nicotinic acid substrate. Full exchange occurs after many enzymatic turnovers. The flexibility of the SpNic active site appears to be supported by its ability to hydrolyze even the very bulky phenylnicotinate ester (Table 2).
This mechanistic proposal can be applied to understand the total reaction mechanism and suggests an important role for the conserved Lys in the catalytic mechanism that hydrolyses nicotinamide, supported by our finding that the SpNic K103A mutant loses over 99 % of its catalytic activity in this reaction (Table 3). The proposed overall mechanism is shown in Scheme 6. Initially nicotinamide coordinates the active site Zn+2 complex to form the Michaelis complex. This coordination is proposed to activate substrate, not only by organizing it into a geometry ideal for catalysis, but by electrophilic activation caused by the Lewis acidity of the Zn+2. The universally conserved Asp deprotonates Cys to form a nucleophilic thiolate which can readily proceed to the tetrahedral intermediate. Proton transfer to the amino group of the tetrahedral intermediate facilitates oxyanion electron flow to form the thioester. This mechanism is entirely consistent with the mechanism initially proposed by Fyfe et al (34). However, the ammonia product is quite basic and with no other protons in vicinity (except for an aqua-Zn+2), it can deprotonate the active site Lys to form a free amine (ε-NH2) species prior to its departure from the active site as ammonium. X-ray studies of SpNic suggest that the amide N of substrate is in close proximity to the Lys ε-amino group, with a determined distance of 5.2 Å in the nicotinamide-C136S mutant structure (35). At this point our mechanism differs from the Fyfe et al. mechanism which proposed no active catalytic role for the conserved Lys, except for structural and possibly electrostatic (34). The Fyfe et al. mechanism also proposes that ammonia departs the active site as NH3, leaving AbPncA Lys114 unperturbed (34).
Scheme 6.

Proposed mechanism of nicotinamidases. The ammonia generated in the first half of the reaction coordinate generates deprotonated Lys residue prior to departure from the active site. The intermediacy of this Lys is envisioned to act as a base catalyst that activates a water to complete the second half of the catalytic reaction. The numbering system used is for the SpNic enzyme.
Our proposed mechanism provides a thermodynamic driving force for hydroxide formation at the active site via the availability of the basic Lys(ε-NH2). Deprotonation of water by this base is predicted to be uphill with a ΔpKa of 4–5 (based on pKa value of the isolated amino acid). However, additional driving force for deprotonation of water is obtainable by the formation of a salt bridge between the strictly conserved Lys and the strictly conserved Asp located at 2.66 Å in the AbPncA structure (34) and also present in the recent SpNic structure (35). Hydroxide can then hydrolyze the thioester and complete the catalytic mechanism (Scheme 6). Interestingly, the products formed from nicotinamide to form the thioester exactly reproduce the intermediate and protonation states of active site groups that we invoked to explain the 18O exchange reaction (Scheme 5). As compared with the Fyfe et al. mechanism (34) we do not propose to deprotonate water with an Asp residue, but rather by the Lys amino group which makes the thermodynamic barrier for hydroxide generation more favorable (ΔpKa: ~9 versus ~4–5 respectively). Our proposed reaction mechanism (Scheme 6) is completed in analogy to the proposed mechanism for 18O exchange (Scheme 5). An additional advantage of our mechanism is that it does not require generation of a protonated and Zn+2 coordinated nicotinic acid at the active site as proposed by Fyfe et al (34). Nicotinic acid acidity is characterized by a pKa near 3, and the coordination to an acidifying Zn+2 center would make this protonation thermodynamically unfavorable.
Other mechanistic alternatives could be imagined. For example the role we propose for the Lys could be played by the aqua-Zn+2. Aqua-Zn+2 coordination sites proximal to the substrate carbonyl position have been identified for all four crystallographically determined nicotinamidases, Pnc1 (6), AbPncA (34), P. horikoshii PncA (33) and most recently in the full set of SpNic structures (35). In the SpNic thioester structure, the coordinated water oxygen is located at 5.5 Å to the thioester carboxy oxygen (35). A mechanistic scheme which invokes this role for the proximal aqua-Zn+2 is shown in Scheme 7. Inspection of the X-ray structure of AbPncA indicates that the aqua-Zn+2 center probably is too far from the active site position where protonations and deprotonations are required, but strictly speaking we cannot rule out this possibility. An aqua-Zn+2 species typically has pKa = 7–8 (55) which could provide an effective quench for active site generated ammonia, and the resulting Zn-OH species would have a more favorable pKa tradeoff than Asp for generating hydroxide within the active site (~6–7 versus 9 respectively). There is also the possibility that both aqua-Zn+2 and the conserved Lys provide duplication of function within the active site, since ammonia formed from substrate is small and can be expected to leak out of the enzyme rapidly. We propose that ammonia protonation in the active site drives the reaction forward to thioester formation, and likely prevents reverse commitment, and prepares the active site for hydroxide generation. To guarantee this occurs, two potential routes for ammonia quenches in the active site may have co-evolved. Theoretically, the conjugate bases formed by quench of ammonia (Lys103-NH2, or Zn-OH) are basic enough to activate water to complete catalysis. Further experimental studies are required to sort out these possibilities and to decide which mechanism is most likely for distinct nicotinamidase enzymes.
Scheme 7.

Alternative aqua-Zn+2 mechanism of nicotinamidases. The ammonia generated in the first half of the reaction coordinate generates a Zn-hydroxide by deprotonation prior to departure from the active site. The intermediacy of a Zn-hydroxide is envisioned to act as a base catalyst that activates a solvent derived nucleophilic water in the second half of the catalytic reaction. The numbering system used is for the SpNic enzyme.
The generality of nicotinaldehyde inhibition across broad phylogenetic sources of nicotinamidases, conservation of key catalytic residues (Asp, Cys and Lys), and the requirement of an active site metal ion indicates that, as a class, nicotinamidases are likely to have similar catalytic mechanisms to accomplish nicotinamide hydrolysis chemistry. Inhibitors of nicotinamidases are expected to be useful for investigating the diverse functions of these enzymes in a multitude of biological settings. Investigations of nicotinamidase inhibitors as anti-microbials are also clearly of interest.
Supplementary Material
Acknowledgements
We would like to thank Dr. Haiteng Deng of the Proteomics Resource of Rockefeller University for assistance with obtaining MALDI-MS data. We also would like to thank the Mass Spectrum Facility of Hunter College for HRMS data.
Footnotes
This work was funded by the Tri-Institutional Program in Chemical Biology of Cornell University and by the Milstein Institute for Chemical Biology of Weill Medical College of Cornell University.
Supporting Information Available: Methods of cloning of and expression of enzymes, mutant construction, HPLC demonstration of catalytic activity of nicotinamidases, inhibitions of nicotinamidases, demonstration by NMR of PfNic catalysed hydrolysis of phenylnicotinate and metal ion studies are included. This material is available free of charge via the Internet at http://pubs.acs.org.
Abbreviations: AbPncA, Acinetobacter baumanii nicotinamidase; BbNic, Borrelia burgdorferi nicotinamidase; cADPR, cyclic ADP-ribose; CePNC1, Caenorhabditis elegans nicotinamidase 1; CePNC2, Caenorhabditis elegans nicotinamidase 2; DTT, dithiothreitol; GDH, glutamate dehydrogenase; HR-MS, High resolution mass spectometry; IPTG, isopropyl β-D-1-thiogalactopyranoside; MALDI-TOF, Matrix assisted laser desorption ionization time of flight; NA, nicotinic acid; NAD+, nicotinamide adenine dinucleotide; NADH, nicotinamide adenine dinucleotide reduced form; NAM, nicotinamide; NADP+, nicotinamide adenine dinucleotide phosphate; NADPH, nicotinamide adenine dinucleotide phosphate reduced form; NMN, nicotinamide mononucleotide; NR, nicotinamide riboside; PfNic, Plasmodium falciparum nicotinamidase; Pnc1, Saccharomyces cerevisiae nicotinamidase; PncA, Mycobacterium tuberculosis nicotinamidase; SpNic, Streptococcus pneumoniae nicotinamidase.
References
- 1.Gerdes SY, Scholle MD, D'Souza M, Bernal A, Baev MV, Farrell M, Kurnasov OV, Daugherty MD, Mseeh F, Polanuyer BM, Campbell JW, Anantha S, Shatalin KY, Chowdhury SA, Fonstein MY, Osterman AL. From genetic footprinting to antimicrobial drug targets: examples in cofactor biosynthetic pathways. J Bacteriol. 2002;184:4555–4572. doi: 10.1128/JB.184.16.4555-4572.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boshoff HI, Mizrahi V. Purification, gene cloning, targeted knockout, overexpression, and biochemical characterization of the major pyrazinamidase from Mycobacterium smegmatis. J Bacteriol. 1998;180:5809–5814. doi: 10.1128/jb.180.22.5809-5814.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang H, Deng JY, Bi LJ, Zhou YF, Zhang ZP, Zhang CG, Zhang Y, Zhang XE. Characterization of Mycobacterium tuberculosis nicotinamidase/pyrazinamidase. Febs J. 2008;275:753–762. doi: 10.1111/j.1742-4658.2007.06241.x. [DOI] [PubMed] [Google Scholar]
- 4.Scorpio A, Zhang Y. Mutations in pncA, a gene encoding pyrazinamidase/nicotinamidase, cause resistance to the antituberculous drug pyrazinamide in tubercle bacillus. Nat Med. 1996;2:662–667. doi: 10.1038/nm0696-662. [DOI] [PubMed] [Google Scholar]
- 5.Ghislain M, Talla E, Francois JM. Identification and functional analysis of the Saccharomyces cerevisiae nicotinamidase gene, PNC1. Yeast. 2002;19:215–224. doi: 10.1002/yea.810. [DOI] [PubMed] [Google Scholar]
- 6.Hu G, Taylor AB, McAlister-Henn L, Hart PJ. Crystal structure of the yeast nicotinamidase Pnc1p. Arch Biochem Biophys. 2007;461:66–75. doi: 10.1016/j.abb.2007.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Joshi JG, Handler P. Purification and properties of nicotinamidase from Torula cremoris. J Biol Chem. 1962;237:929–935. [PubMed] [Google Scholar]
- 8.Zerez CR, Roth EF, Jr, Schulman S, Tanaka KR. Increased nicotinamide adenine dinucleotide content and synthesis in Plasmodium falciparum-infected human erythrocytes. Blood. 1990;75:1705–1710. [PubMed] [Google Scholar]
- 9.Wang G, Pichersky E. Nicotinamidase participates in the salvage pathway of NAD biosynthesis in Arabidopsis. Plant J. 2007;49:1020–1029. doi: 10.1111/j.1365-313X.2006.03013.x. [DOI] [PubMed] [Google Scholar]
- 10.Balan V, Miller GS, Kaplun L, Balan K, Chong ZZ, Li F, Kaplun A, VanBerkum MF, Arking R, Freeman DC, Maiese K, Tzivion G. Life span extension and neuronal cell protection by Drosophila nicotinamidase. J Biol Chem. 2008;283:27810–27819. doi: 10.1074/jbc.M804681200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van der Horst A, Schavemaker JM, Pellis-van Berkel W, Burgering BM. The Caenorhabditis elegans nicotinamidase PNC-1 enhances survival. Mech Ageing Dev. 2007;128:346–349. doi: 10.1016/j.mad.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 12.Vrablik TL, Huang L, Lange SE, Hanna-Rose W. Nicotinamidase modulation of NAD+ biosynthesis and nicotinamide levels separately affect reproductive development and cell survival in C. elegans. Development. 2009;136:3637–3646. doi: 10.1242/dev.028431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oppenheimer NJ. Nad Hydrolysis - Chemical and Enzymatic Mechanisms. Molecular and Cellular Biochemistry. 1994;138:245–251. doi: 10.1007/BF00928468. [DOI] [PubMed] [Google Scholar]
- 14.Handlon AL, Xu C, Mullersteffner HM, Schuber F, Oppenheimer NJ. 2'-Ribose Substituent Effects on the Chemical and Enzymatic-Hydrolysis of Nad(+) Journal of the American Chemical Society. 1994;116:12087–12088. [Google Scholar]
- 15.Johnson RW, Marschner TM, Oppenheimer NJ. Pyridine-Nucleotide Chemistry - a New Mechanism for the Hydroxide-Catalyzed Hydrolysis of the Nicotinamide Glycosyl Bond. Journal of the American Chemical Society. 1988;110:2257–2263. [Google Scholar]
- 16.Sauve AA, Wolberger C, Schramm VL, Boeke JD. The biochemistry of sirtuins. Annu Rev Biochem. 2006;75:435–465. doi: 10.1146/annurev.biochem.74.082803.133500. [DOI] [PubMed] [Google Scholar]
- 17.Grimm D, Tilly K, Bueschel DM, Fisher MA, Policastro PF, Gherardini FC, Schwan TG, Rosa PA. Defining plasmids required by Borrelia burgdorferi for colonization of tick vector Ixodes scapularis (Acari: Ixodidae) J Med Entomol. 2005;42:676–684. doi: 10.1093/jmedent/42.4.676. [DOI] [PubMed] [Google Scholar]
- 18.Kawabata H, Norris SJ, Watanabe H. BBE02 disruption mutants of Borrelia burgdorferi B31 have a highly transformable, infectious phenotype. Infect Immun. 2004;72:7147–7154. doi: 10.1128/IAI.72.12.7147-7154.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Purser JE, Lawrenz MB, Caimano MJ, Howell JK, Radolf JD, Norris SJ. A plasmid-encoded nicotinamidase (PncA) is essential for infectivity of Borrelia burgdorferi in a mammalian host. Mol Microbiol. 2003;48:753–764. doi: 10.1046/j.1365-2958.2003.03452.x. [DOI] [PubMed] [Google Scholar]
- 20.Kim S, Kurokawa D, Watanabe K, Makino S, Shirahata T, Watarai M. Brucella abortus nicotinamidase (PncA) contributes to its intracellular replication and infectivity in mice. FEMS Microbiol Lett. 2004;234:289–295. doi: 10.1016/j.femsle.2004.03.038. [DOI] [PubMed] [Google Scholar]
- 21.Sauve AA. NAD+ and vitamin B3: from metabolism to therapies. J Pharmacol Exp Ther. 2008;324:883–893. doi: 10.1124/jpet.107.120758. [DOI] [PubMed] [Google Scholar]
- 22.Anderson RM, Bitterman KJ, Wood JG, Medvedik O, Sinclair DA. Nicotinamide and PNC1 govern lifespan extension by calorie restriction in Saccharomyces cerevisiae. Nature. 2003;423:181–185. doi: 10.1038/nature01578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bitterman KJ, Anderson RM, Cohen HY, Latorre-Esteves M, Sinclair DA. Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J Biol Chem. 2002;277:45099–45107. doi: 10.1074/jbc.M205670200. [DOI] [PubMed] [Google Scholar]
- 24.Gallo CM, Smith DL, Jr, Smith JS. Nicotinamide clearance by Pnc1 directly regulates Sir2-mediated silencing and longevity. Mol Cell Biol. 2004;24:1301–1312. doi: 10.1128/MCB.24.3.1301-1312.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rogina B, Helfand SL. Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc Natl Acad Sci U S A. 2004;101:15998–16003. doi: 10.1073/pnas.0404184101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wood JG, Rogina B, Lavu S, Howitz K, Helfand SL, Tatar M, Sinclair D. Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature. 2004;430:686–689. doi: 10.1038/nature02789. [DOI] [PubMed] [Google Scholar]
- 27.Tissenbaum HA, Guarente L. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature. 2001;410:227–230. doi: 10.1038/35065638. [DOI] [PubMed] [Google Scholar]
- 28.Berdichevsky A, Viswanathan M, Horvitz HR, Guarente L. C. elegans SIR-2.1 interacts with 14-3-3 proteins to activate DAF-16 and extend life span. Cell. 2006;125:1165–1177. doi: 10.1016/j.cell.2006.04.036. [DOI] [PubMed] [Google Scholar]
- 29.Kaeberlein M, McVey M, Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999;13:2570–2580. doi: 10.1101/gad.13.19.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin SJ, Defossez PA, Guarente L. Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science. 2000;289:2126–2128. doi: 10.1126/science.289.5487.2126. [DOI] [PubMed] [Google Scholar]
- 31.Hunt L, Holdsworth MJ, Gray JE. Nicotinamidase activity is important for germination. Plant J. 2007;51:341–351. doi: 10.1111/j.1365-313X.2007.03151.x. [DOI] [PubMed] [Google Scholar]
- 32.Hughes DE, Williamson DH. The synthesis of cozymase from nicotinic acid and its derivatives by Lactobacillus arabinosus 17-5. Biochem J. 1952;51:330–338. doi: 10.1042/bj0510330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Du X, Wang W, Kim R, Yakota H, Nguyen H, Kim SH. Crystal structure and mechanism of catalysis of a pyrazinamidase from Pyrococcus horikoshii. Biochemistry. 2001;40:14166–14172. doi: 10.1021/bi0115479. [DOI] [PubMed] [Google Scholar]
- 34.Fyfe PK, Rao VA, Zemla A, Cameron S, Hunter WN. Specificity and mechanism of Acinetobacter baumanii nicotinamidase: implications for activation of the front-line tuberculosis drug pyrazinamide. Angew Chem Int Ed Engl. 2009;48:9176–9179. doi: 10.1002/anie.200903407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.French JB, Cen Y, Sauve AA, Ealick SE. High-Resolution Crystal Structures of Streptococcus pneumoniae Nicotinamidase with Trapped Intermediates Provide Insights into the Catalytic Mechanism and Inhibition by Aldehydes. Biochemistry. 2010;49:8803–8812. doi: 10.1021/bi1012436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pruser JE, Lawrenz MB, Caimano MJ, Howell JK, Radolf JD, Norris SJ. Mol. Microbiol. 2003;48:753–764. doi: 10.1046/j.1365-2958.2003.03452.x. [DOI] [PubMed] [Google Scholar]
- 37.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 38.Yun SL, Suelter CH. A simple method for calculating Km and V from a single enzyme reaction progress curve. Biochim Biophys Acta. 1977;480:1–13. doi: 10.1016/0005-2744(77)90315-1. [DOI] [PubMed] [Google Scholar]
- 39.Copeland RA. Evaluation of Enzyme Inhibitors in Drug Discovery. Hoboken, New Jersey: Wiley-Interscience; 2005. Tight Binding Inhibition; pp. 185–192. [Google Scholar]
- 40.Sauve AA, Schramm VL. Mechanism-based inhibitors of CD38: a mammalian cyclic ADP-ribose synthetase. Biochemistry. 2002;41:8455–8463. doi: 10.1021/bi0258795. [DOI] [PubMed] [Google Scholar]
- 41.Evans GB, Furneaux RH, Gainsford GJ, Hanson JC, Kicska GA, Sauve AA, Schramm VL, Tyler PC. 8-Aza-immucillins as transition-state analogue inhibitors of purine nucleoside phosphorylase and nucleoside hydrolases. J Med Chem. 2003;46:q155–q160. doi: 10.1021/jm0203332. [DOI] [PubMed] [Google Scholar]
- 42.Boovanahalli SK, Jin X, Jin Y, Kim JH, Dat NT, Hong YS, Lee JH, Jung SH, Lee K, Lee JJ. Synthesis of (aryloxyacetylamino)-isonicotinic/nicotinic acid analogues as potent hypoxia-inducible factor (HIF)-1alpha inhibitors. Bioorg Med Chem Lett. 2007;17:6305–6310. doi: 10.1016/j.bmcl.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 43.Martin RB, Hull JG. Reactions of N1-Methylnicotinamide Cation + Analogues with Dilute Alkali. Journal of Biological Chemistry. 1964;239:1237–1241. [PubMed] [Google Scholar]
- 44.Comins DL, Killpack MO. Lithiation of Methoxypyridines Directed by Alpha-Amino Alkoxides. Journal of Organic Chemistry. 1990;55:69–73. [Google Scholar]
- 45.Carceller E, Merlos M, Giral M, Balsa D, Almansa C, Bartroli J, Garcia-Rafanell J, Forn J. [(3-Pyridylalkyl)piperidylidene]benzocycloheptapyridine derivatives as dual antagonists of PAF and histamine. J Med Chem. 1994;37:2697–2703. doi: 10.1021/jm00043a009. [DOI] [PubMed] [Google Scholar]
- 46.Bandgar BP, Sadavarte VS, Uppalla LS. Selective and rapid oxidation of primary, allylic and benzylic alcohols to the corresponding carbonyl compounds with NaNO2-acetic anhydride under mild and solvent-free conditions. Journal of the Chemical Society-Perkin Transactions. 2000;1:3559–3560. [Google Scholar]
- 47.Rife JE, Cleland WW. Kinetic mechanism of glutamate dehydrogenase. Biochemistry. 1980;19:2321–2328. doi: 10.1021/bi00552a007. [DOI] [PubMed] [Google Scholar]
- 48.Engel PC, Dalziel K. The equilibrium constants of the glutamate dehydrogenase systems. Biochem J. 1967;105:691–695. doi: 10.1042/bj1050691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smith BC, Hallows WC, Denu JM. A continuous microplate assay for sirtuins and nicotinamide-producing enzymes. Anal Biochem. 2009;394:101–109. doi: 10.1016/j.ab.2009.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hughes DE, Williamson DH. The deamidation of nicotinamide by bacteria. Biochem J. 1953;55:851–856. doi: 10.1042/bj0550851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tanigawa Y, Shimoyama M, Ueda I. Nicotinamide deamidase from Flavobacterium peregrinum. Methods Enzymol. 1980;66:132–136. doi: 10.1016/0076-6879(80)66450-7. [DOI] [PubMed] [Google Scholar]
- 52.Yan C, Sloan DL. Purification and characterization of nicotinamide deamidase from yeast. J Biol Chem. 1987;262:9082–9087. [PubMed] [Google Scholar]
- 53.Calbreath DF, Joshi JG. Inhibition of nicotinamidase by nicotinamide adenine dinucleotide. J Biol Chem. 1971;246:4334–4339. [PubMed] [Google Scholar]
- 54.Margolin N, Raybuck SA, Wilson KP, Chen W, Fox T, Gu Y, Livingston DJ. Substrate and inhibitor specificity of interleukin-1 beta-converting enzyme and related caspases. J Biol Chem. 1997;272:7223–7228. doi: 10.1074/jbc.272.11.7223. [DOI] [PubMed] [Google Scholar]
- 55.Groves JT, Olson JR. Models of Zinc-Containing Proteases - Rapid Amide Hydrolysis by an Unusually Acidic Zn2+-Oh2 Complex. Inorganic Chemistry. 1985;24:2715–2717. [Google Scholar]
- 56.Chenna R, Sugawara H, Koike T, Lopez R, Higgins TJ, Desmond G, Thompson JD. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003;31:3497–3500. doi: 10.1093/nar/gkg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gouet P, Robert X, Courcelle E. ESPript/ENDscript; extracting and rendering sequence and 3D information from atomic structures of proteins. Nucleic Acids Res. 2003;31:3320–3323. doi: 10.1093/nar/gkg556. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.