

Abstract
Phosphatidic acid (PA), the primary metabolite of the phospholipase D (PLD)-mediated hydrolysis of phosphatidylcholine, has been shown to act as a tumor promoting second messenger in many cancer cell lines. A key target of PA is the mammalian target of rapamycin (mTOR), a serine-threonine kinase that has been widely implicated in cancer cell survival signals. In agreement with its ability to relay survival signals, it has been reported that both PLD and mTOR are required for the stabilization of the p53 E3 ubiquitin ligase human double minute 2 (HDM2) protein. Thus, by stabilizing HDM2, PLD and mTOR are able to counter the pro-apoptotic signaling mediated by p53 and promote survival. mTOR exists in at least two distinct complexes – mTORC1 and mTORC2 – that are both dependent on PLD-generated PA. Although PLD and its metabolite PA are clearly implicated in the transduction of survival signals to mTOR, it is not yet apparent which of the two mTOR complexes is critical for the stabilization of HDM2. We report here that the PLD/mTOR-dependent stabilization of HDM2 involves mTORC2 and the AGC family kinase serum- and glucocorticoid-inducible kinase 1 (SGK1). This study reveals that mTORC2 is a critical target of PLD-mediated survival signals and identifies SGK1 as a downstream target of mTORC2 for the stabilization of HDM2.
Keywords: Phospholipase D, mTOR, HDM2, serum- and glucocorticoid-induced kinase 1
Introduction
Downstream targets of phosphatidic acid (PA), the product of the transphosphatidylation reaction between phospholipase D (PLD) and phosphatidylcholine (PC), are involved in a multitude of cellular processes. This second messenger has been implicated in regulating diverse cellular processes such as cytoskeletal rearrangement, vesicle trafficking, cell proliferation and survival [1–3]. With regard to cancer cell proliferation and survival, mTOR – the mammalian target of rapamycin – has emerged as the most significant target of PLD-generated PA [2, 4]. mTOR is a serine-threonine kinase that has emerged as a central signaling node for signals that promote cell growth, proliferation, and survival [5]. Many pathways concerned with sensing the nutritional and biochemical status of the cell such as amino acid levels [6], AMP/ATP ratios [7, 8], and growth factor availability [9] provide input to mTOR. These signals are mediated by two mTOR complexes mTORC1 and mTORC2. mTORC1 and mTORC2 are most commonly distinguished from each other by two companion proteins, Raptor [10] and Rictor [11]. These complexes can also be distinguished from one another by their sensitivity to rapamycin, a natural product inhibitor that binds to the immunophilin, FK506 binding protein 12 (FKBP12), and then inserts itself into the FRB (FKBP12 Rapamycin Binding) domain of mTOR to disrupt complex formation [2, 12]. Raptor, Regulatory Associated Protein of mTOR, forms complex 1 and is generally sensitive to rapamycin treatment. Rictor forms complex 2 and this complex has been shown to have varied sensitivity to rapamycin and usually only after long term or upon high dosage treatment [13, 14].
The discovery that PA binds the FRB domain of mTOR [15, 16] – the same site that is bound by rapamycin and FKBP12—has motivated our lab to characterize the nature of this signaling. Our lab has found that much of the oncogenic effects of increased PLD activity, such as survival under stressful conditions, can be attributed its downstream target, mTOR [2]. Recently, we have found that this connection extends to both mTORC1 and mTORC2 [14]. The relationship between mTORC2 and PLD coupled with the finding that mTORC2 is responsible for phosphorylating the AGC family kinase Akt at Ser473 [17] were the stimulus for re-investigating our earlier report linking increased PLD activity with the stabilization of murine double minute (MDM2) [18] – the major regulator of p53 [19–21]. In our previous study, the stabilization of MDM2 was suppressed by rapamycin [18] – suggesting an involvement of mTOR. Since the previous study was done prior to the discovery of mTORC2 and since Akt has been implicated in the regulation of MDM2 [22, 23], we investigated the mTOR dependent regulation of HDM2 – the human orthologue of MDM2 – in a human renal cancer cell line where there are very high levels of PLD activity and HDM2 expression [24]. We report here that the PLD-dependent increase in HDM2 expression in 786-O renal cancer cells is dependent on mTORC2. Surprisingly, HDM2 expression was independent of Akt, but was dependent on another AGC family kinase – the serum- and glucocorticoid-inducible kinase 1 (SGK1).
Materials and Methods
Cells and cell culture conditions
786-O cells (obtained from the laboratory of M. Ohh, University of Toronto) were maintained in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine calf serum (Sigma).
Materials
1-Butanol (1-BtOH) and tert-Butanol (t-BtOH) were purchased from Sigma with purity of 99.8%. Antibodies for Akt1, Akt2, Rictor, Raptor, mTOR and Actin were purchased from Cell Signaling; antibodies for MDM2 which recognizes HDM2 was from Novus Biologicals; and the antibody for SGK1 was purchased from Millipore.
Western blot analysis
Proteins were heated for 3 min at 100 °C prior to separation by SDS–PAGE using an 8 – 12% acrylamide separating gel. After transferring to nitrocellulose membrane, the membrane filters were blocked with 5% non-fat dry milk in phosphate-buffered saline and incubated with the appropriate antibody. Depending on the origin of the primary antibodies, either anti-mouse or anti-rabbit IgG conjugated with horseradish peroxidase was used, and the bands were visualized using the enhanced chemilluminescent detection system (Pierce).
siRNA
Transfections were performed using RNAiMax reagent (Invitrogen) according to the vendor’s instructions. Transfection efficiency was determined by Block-It (Invitrogen) fluorescent control. The percentage of green cells was determined microscopically and was routinely in excess of 70%. Briefly, cells were plated on 6 well plates at 30% confluence in medium containing Optimem (Invitrogen). After one day, cells were transfected with siRNA. After 24 h the media was changed to fresh media containing 10% serum and antibiotics. One to three days later cells were lysed and analyzed for expression by Western Blot. The siRNA primers for the following proteins were used for the knockdowns: Raptor, mTOR, and Rictor (Thermo Scientific Dharmacon), Akt1 (Santa Cruz), Akt2 (Cell Signaling), and SGK1 (Invitrogen).
Results
HDM2 levels are dependent on PLD activity in 786-O kidney cancer cells
We reported previously that elevating the PLD activity in rat fibroblasts increased the level of MDM2 [18]. The human renal cancer cell line 786-O has constitutively elevated PLD activity that is required for the survival of these cells when serum is withdrawn [24]. We therefore examined whether there was elevated HDM2 expression in these cells as a consequence of the elevated PLD activity. As shown in Fig. 1, the 786-O cells express high levels of HDM2 and that suppressing the PLD mediated production of PA with 1-BtOH, which results in the generation of phosphatidyl-BtOH at the expense of PA [14], suppresses the expression of HDM2. t-BtOH, which was used as a negative control, does not participate in this reaction and had no effect. These data indicate that the elevated HDM2 expression in the 786-O cells was dependent on the elevated PLD activity.
Fig. 1.
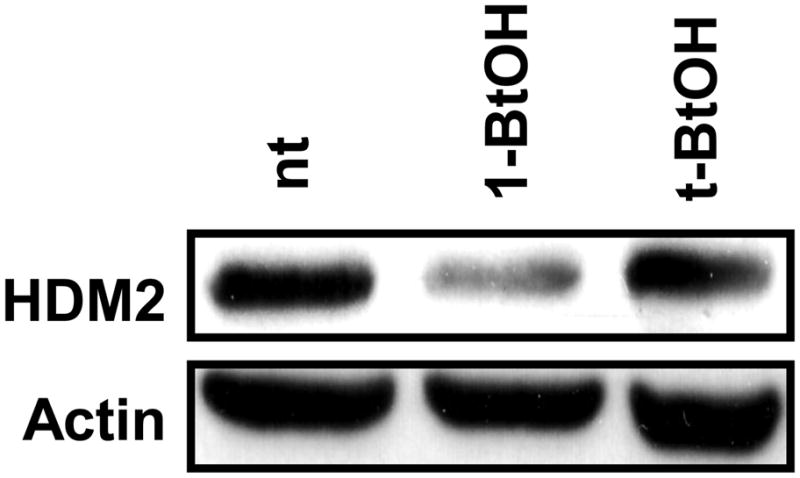
HDM2 levels are suppressed by preventing PA production. 786-O cells were plated at 70% confluence in 60mm plates in 10% serum and allowed to grow overnight. 24 hrs later, the cells were either left untreated (nt) or treated with 0.8% 1-BtOH or 0.8% t-BtOH as a negative control for 3 hrs. The cells were then lysed and subjected to Western blot analysis with antibodies that recognize HDM2 or actin as a loading control.
HDM2 expression is dependent on mTORC2 and not mTORC1
A critical downstream target of PLD-generated PA is mTOR, which like PLD, has been widely implicated in cancer cell survival signals [2, 12]. We recently reported that PA was required for the assembly and activity of both mTORC1 and mTORC2 [14]. We first examined the dependence of HDM2 expression on mTOR. As shown in Fig. 2, suppression of mTOR expression with siRNA resulted in the loss of HDM2 expression. To distinguish whether the mTOR dependence was due to mTORC1 or mTORC2, we introduced siRNAs for Raptor and Rictor, which are essential companion proteins for mTORC1 and mTORC2, respectively. As shown in Fig. 2, the suppression of Raptor had no effect on HDM2 expression. In contrast, suppression of Rictor reduced the expression of HDM2 almost as effectively as did the suppression of mTOR. These data indicate that the expression of HDM2 is dependent on mTORC2.
Fig. 2.

Knockdown of mTORC2 and not mTORC1 results in loss of HDM2. 786-O cells were plated at 30% confluence in 6-well plates. The cells were transfected with siRNA for Raptor, mTOR, or Rictor for 48hrs. As controls cells were left not treated (nt), treated with reagent only (re), or transfected with scrambled siRNA (scr). Actin was used as a loading control. The data are representative of three independent experiments.
HDM2 levels are independent of Akt1 and Akt2 and dependent on SGK1
Though other kinases have been shown or proposed to regulate HDM2 [25], the major kinase downstream of mTORC2 responsible for phosphorylating HDM2 at ser166 and ser188 is considered to be Akt [26,27]. These two sites lead to the protection of HDM2 from self-ubiquitination and allows this E3 ligase to enter the nucleus and prevent normal activity of wildtype p53. We investigated the role of Akt in regulating HDM2 in this cell line by knocking it down as shown in Fig. 3A. Knockdown of the two major isoforms of Akt expressed in 786-O cells had little or no effect on the levels of HDM2. The third Akt isoform, Akt3, was not knocked down as it is undetectably expressed, if expressed at all, in this cell line [28]. To address the concern that Akt may not be active in this cell line, we blotted for the active form of Akt that is phosphorylated at Ser473. We found that Akt is highly phosphorylated at this site [14], however, as shown in Fig. 3A, siRNA knockdown of either Akt1 or Akt2 does not result in a concomitant loss of HDM2.
Fig. 3.

Knockdown of AGC member SGK1 but not Akt1 or Akt2 results in loss of HDM2. (A) 786-O cells were plated at 30% confluence in 6-well plates. The cells were transfected with siRNA for Akt1 and Akt2 for 48hrs. (B) 786-O cells were plated as in A and then transfected with siRNA for SGK1 for 48hrs. As controls wells were left untreated (nt), treated with reagent (re), or transfected with scrambled siRNA (scr). Actin was used as a loading control. The blots are representative of at least three independent experiments.
HDM2 is phosphorylated by Akt on two sites that have a well-known and conserved consensus sequence [26,27]. A natural candidate that may mimic the kinase activity of Akt is a protein that can bind to similar amino acid sequence motifs. SGK1 was the best candidate for the following reasons: it is downstream of mTORC2 [29] and it is in the same family of AGC kinases as Akt that have the defining feature of being able to recognize similar motifs. As shown in Fig. 3B, knockdown of SGK1 results in loss of HDM2 levels in a siRNA concentration dependent manner. These data surprisingly reveal that HDM2 expression is independent of Akt, but instead is dependent on another AGC kinase family member SGK1.
Discussion
We previously reported that the stabilization of MDM2 was dependent on PLD activity and its downstream target mTOR. However, at the time of that study, it was not known that there were two mTOR complexes and that it was mTORC2 that phosphorylated Akt at the critical regulatory site at Ser473. In this report that we have provided evidence that mTORC2 is required for HDM2 stabilization. Surprisingly we found that Akt was not required for HDM2 stabilization and that another AGC kinase and mTORC2 substrate – SGK1 – was required. The unexpected finding that SGK1 is involved in this pathway provides additional support for the complexity and, perhaps, the flexibility of signal transduction circuits when they are co-opted by cancer cells. A schematic for signals mediated by PLD, mTOR and AGC kinases is presented in Fig. 4.
Fig. 4.

Schematic of the signaling cascade from PLD to HDM2.
While the finding that SGK1, rather than Akt linked mTORC2 to HDM2, it was recently reported that SGK1 is responsible for the MDM2-dependent ubiquitination of p53 in RKO colon carcinoma cells [30]. And it was also recently reported that mTORC2 activates epithelial sodium Na+ channel-dependent Na+ transport by phosphorylating SGK1 [31]. Thus, there is precedent for an indirect connection between mTORC2 and HDM2 via SGK1, and in fact, SGK1 is a rational substitute for Akt as the two proteins have structural and functional similarities [32,33]. Not only do these proteins share a common upstream regulator [29,34] but they both possess the ability to recognize and phosphorylate a similar amino acid sequence motif [35]. Although the most studied function of SGK1 is as an upregulator of epithelial sodium channels in response to aldosterone and insulin [32], there are reports to its functioning as a pro-survival signal [30,36–38]. The related AGC kinase SGK3 was also reported to function as a downstream signal in cancer cells with mutations to phosphatidylinositol-3-kinase [39]. The evidence provided here that the mTORC2-dependent stabilization of HDM2 is dependent on SGK1 reinforces the concept that AGC-family kinases, like Akt, can function as a survival kinase in cancer cells [40].
Conclusions
In this report, we have provided evidence that the mTOR-dependent stabilization of HDM2 involves mTORC2 and SGK1. There are two surprising outcomes – the first being that mTORC2, not mTORC1 is required; and second, that SGK1, rather than Akt is involved. Both Akt and SGK1 are members of the AGC family of protein kinases and are substrates of mTORC2. This study reveals an alternative target of mTORC2 that can contribute to the survival signals mediated by PLD and mTOR.
Acknowledgments
This work was supported by National Cancer Institute grant CA046677 from the National Institutes of Health (to D. A. F.). Research Centers in Minority Institutions (RCMI) award RR-03037 from the National Center for Research Resources of the National Institutes of Health, which supports infrastructure and instrumentation in the Biological Sciences Department at Hunter College, is also acknowledged. Donggon Lyo was supported by a Gene Center Fellowship provided by the RCMI.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Donaldson JG. Phospholipase D in endocytosis and endosomal recycling pathways. Biochim Biophys Acta. 2009;1791:845–849. doi: 10.1016/j.bbalip.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Foster DA. Phosphatidic acid signaling to mTOR: Signals for the survival of human cancer cells. Biochem Biophys Acta. 2009;1791:949–955. doi: 10.1016/j.bbalip.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee CS, Kim KL, Jang JH, Choi YS, Suh PG, Ryu SH. The roles of phospholipase D in EGFR signaling. Biochim Biophys Acta. 2009;1791:862–868. doi: 10.1016/j.bbalip.2009.04.007. [DOI] [PubMed] [Google Scholar]
- 4.Sun Y, Chen J. mTOR signaling: PLD takes center stage. Cell Cycle. 2008;7:3118–3123. doi: 10.4161/cc.7.20.6881. [DOI] [PubMed] [Google Scholar]
- 5.Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 6.Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, Sabatini DM. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008;320:1496–1501. doi: 10.1126/science.1157535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Inoki K, Ouyang H, Zhu T, Lindvall C, Wang Y, Zhang X, Yang Q, Bennett C, Harada Y, Stankunas K, Wang CY, He X, MacDougald OA, You M, Williams BO, Guan KL. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell. 2006;126:955–968. doi: 10.1016/j.cell.2006.06.055. [DOI] [PubMed] [Google Scholar]
- 9.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163–175. doi: 10.1016/s0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- 11.Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14:1296–1302. doi: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- 12.Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. 2006;6:729–734. doi: 10.1038/nrc1974. [DOI] [PubMed] [Google Scholar]
- 13.Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–168. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 14.Toschi A, Lee E, Xu L, Garcia A, Gadir N, Foster DA. Regulation of mTORC1 and mTORC2 complex assembly by phosphatidic acid: competition with rapamycin. Mol Cell Biol. 2009;29:1411–1420. doi: 10.1128/MCB.00782-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fang Y, Vilella-Bach M, Bachmann R, Flanigan A, Chen J. Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science. 2001;294:1942–1945. doi: 10.1126/science.1066015. [DOI] [PubMed] [Google Scholar]
- 16.Veverka V, Crabbe T, Bird I, Lennie G, Muskett FW, Taylor RJ, Carr MD. Structural characterization of the interaction of mTOR with phosphatidic acid and a novel class of inhibitor: compelling evidence for a central role of the FRB domain in small molecule-mediated regulation of mTOR. Oncogene. 2008;27:585–595. doi: 10.1038/sj.onc.1210693. [DOI] [PubMed] [Google Scholar]
- 17.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 18.Hui L, Abbas T, Pielak RM, Joseph T, Bargonetti J, Foster DA. Phospholipase D elevates the level of MDM2 and suppresses DNA damage-induced increases in p53. Mol Cell Biol. 2004;24:5677–5686. doi: 10.1128/MCB.24.13.5677-5686.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ashcroft M, Vousden KH. Regulation of p53 stability. Oncogene. 1999;18:7637–7643. doi: 10.1038/sj.onc.1203012. [DOI] [PubMed] [Google Scholar]
- 20.Freedman DA, Wu L, Levine AJ. Functions of the MDM2 oncoprotein. Cell Mol Life Sci. 1999;55:96–107. doi: 10.1007/s000180050273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Momand J, Wu HH, Dasgupta G. MDM2--master regulator of the p53 tumor suppressor protein. Gene. 2000;242:15–29. doi: 10.1016/s0378-1119(99)00487-4. [DOI] [PubMed] [Google Scholar]
- 22.Ashcroft M, Ludwig RL, Woods DB, Copeland TD, Weber HO, MacRae EJ, Vousden KH. Phosphorylation of HDM2 by Akt. Oncogene. 2002;21:1955–1962. doi: 10.1038/sj.onc.1205276. [DOI] [PubMed] [Google Scholar]
- 23.Brazil DP, Park J, Hemmings BA. PKB binding proteins. Getting in on the Akt. Cell. 2002;111:293–303. doi: 10.1016/s0092-8674(02)01083-8. [DOI] [PubMed] [Google Scholar]
- 24.Toschi A, Edelstein J, Rockwell P, Ohh M, Foster DA. HIFα expression in VHL-deficient renal cancer cells is dependent on phospholipase D. Oncogene. 2008;27:2746–2753. doi: 10.1038/sj.onc.1210927. [DOI] [PubMed] [Google Scholar]
- 25.Kulikov R, Boehme KA, Blattner C. Glycogen synthase kinase 3-dependent phosphorylation of Mdm2 regulates p53 abundance. Mol Cell Biol. 2005;25:7170–7180. doi: 10.1128/MCB.25.16.7170-7180.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mayo LD, Donner DB. A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc Natl Acad Sci U S A. 2001;98:11598–11603. doi: 10.1073/pnas.181181198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou BP, Liao Y, Xia W, Zou Y, Spohn B, Hung MC. HER-2/neu induces p53 ubiquitination via Akt-mediated MDM2 phosphorylation. Nat Cell Biol. 2001;3:973–982. doi: 10.1038/ncb1101-973. [DOI] [PubMed] [Google Scholar]
- 28.Toschi A, Lee E, Gadir N, Ohh M, Foster DA. Differential dependence of hypoxia-inducible factors 1 alpha and 2 alpha on mTORC1 and mTORC2. J Biol Chem. 2008;283:34495–34499. doi: 10.1074/jbc.C800170200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garcia-Martinez JM, Alessi DR. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1) Biochem J. 2008;416:375–385. doi: 10.1042/BJ20081668. [DOI] [PubMed] [Google Scholar]
- 30.Amato R, D’Antona L, Porciatti G, Agosti V, Menniti M, Rinaldo C, Costa N, Bellacchio E, Mattarocci S, Fuiano G, Soddu S, Paggi MG, Lang F, Perrotti N. Sgk1 activates MDM2-dependent p53 degradation and affects cell proliferation, survival, and differentiation. J Mol Med. 2009;87:1221–1239. doi: 10.1007/s00109-009-0525-5. [DOI] [PubMed] [Google Scholar]
- 31.Lu M, Wang J, Jones KT, Ives HE, Feldman ME, Yao LJ, Shokat KM, Ashrafi K, Pearce D. mTOR Complex-2 Activates ENaC by Phosphorylating SGK1. J Am Soc Nephrol. 2010 doi: 10.1681/ASN.2009111168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lang F, Bohmer C, Palmada M, Seebohm G, Strutz-Seebohm N, Vallon V. (Patho)physiological significance of the serum- and glucocorticoid-inducible kinase isoforms. Physiol Rev. 2006;86:1151–1178. doi: 10.1152/physrev.00050.2005. [DOI] [PubMed] [Google Scholar]
- 33.Loffing J, Flores SY, Staub O. Sgk kinases and their role in epithelial transport. Annu Rev Physiol. 2006;68:461–490. doi: 10.1146/annurev.physiol.68.040104.131654. [DOI] [PubMed] [Google Scholar]
- 34.Yan L, Mieulet V, Lamb RF. mTORC2 is the hydrophobic motif kinase for SGK1. Biochem J. 2008;416:e19–21. doi: 10.1042/BJ20082202. [DOI] [PubMed] [Google Scholar]
- 35.Pearce LR, Komander D, Alessi DR. The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol. 2010;11:9–22. doi: 10.1038/nrm2822. [DOI] [PubMed] [Google Scholar]
- 36.Amato R, Menniti M, Agosti V, Boito R, Costa N, Bond HM, Barbieri V, Tagliaferri P, Venuta S, Perrotti N. IL-2 signals through Sgk1 and inhibits proliferation and apoptosis in kidney cancer cells. J Mol Med. 2007;85:707–721. doi: 10.1007/s00109-007-0205-2. [DOI] [PubMed] [Google Scholar]
- 37.Hong F, Larrea MD, Doughty C, Kwiatkowski DJ, Squillace R, Slingerland JM. mTOR-raptor binds and activates SGK1 to regulate p27 phosphorylation. Mol Cell. 2008;30:701–711. doi: 10.1016/j.molcel.2008.04.027. [DOI] [PubMed] [Google Scholar]
- 38.Nasir O, Wang K, Foller M, Gu S, Bhandaru M, Ackermann TF, Boini KM, Mack A, Klingel K, Amato R, Perrotti N, Kuhl D, Behrens J, Stournaras C, Lang F. Relative resistance of SGK1 knockout mice against chemical carcinogenesis. IUBMB Life. 2009;61:768–776. doi: 10.1002/iub.209. [DOI] [PubMed] [Google Scholar]
- 39.Vasudevan KM, Barbie DA, Davies MA, Rabinovsky R, McNear CJ, Kim JJ, Hennessy BT, Tseng H, Pochanard P, Kim SY, Dunn IF, Schinzel AC, Sandy P, Hoersch S, Sheng Q, Gupta PB, Boehm JS, Reiling JH, Silver S, Lu Y, Stemke-Hale K, Dutta B, Joy C, Sahin AA, Gonzalez-Angulo AM, Lluch A, Rameh LE, Jacks T, Root DE, Lander ES, Mills GB, Hahn WC, Sellers WR, Garraway LA. AKT-independent signaling downstream of oncogenic PIK3CA mutations in human cancer. Cancer Cell. 2009;16:21–32. doi: 10.1016/j.ccr.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Toker A. mTOR and signaling in cancer: SGK cycles in. Mol Cell. 2008;31:6–8. doi: 10.1016/j.molcel.2008.06.007. [DOI] [PubMed] [Google Scholar]