

Abstract
RAS GTPases control a major signaling network implicated in several cellular functions, including cell fate determination, proliferation, survival, differentiation, migration, and senescence. Within this network, signal flow through the RAF-MEK-ERK pathway, the first identified mitogen-associated protein kinase (MAPK) cascade, mediates early and late developmental processes controlling morphology determination, organogenesis, synaptic plasticity and growth. Signaling through the RAS-MAPK cascade is tightly controlled, and its enhanced activation represents a well-known event in oncogenesis. Unexpectedly, in the past few years, inherited dysregulation of this pathway has been recognized as the cause underlying a group of clinically related disorders sharing facial dysmorphism, cardiac defects, reduced postnatal growth, ectodermal anomalies, variable cognitive deficits and susceptibility to certain malignancies as major features. These disorders are caused by heterozygosity for mutations in genes encoding RAS proteins, regulators of RAS function, modulators of RAS interaction with effectors or downstream signal transducers. Here, we provide an overview of the phenotypic spectrum associated with germline mutations perturbing RAS-MAPK signaling, the unpredicted molecular mechanisms converging towards the dysregulation of this signaling cascade, and major genotype-phenotype correlations.
Keywords: RAS signaling, Noonan syndrome, LEOPARD syndrome, cardio-facio-cutaneous syndrome, Costello syndrome, neurofibromatosis type 1, Legius syndrome, Noonan-like syndrome with loose anagen hair
Introduction
RAS proteins are small guanosine triphosphate (GTP)/guanosine diphosphate (GDP)-binding GTPases that function as molecular switches controlling a major intracellular signaling network that, depending on the cellular context, guides diverse biological functions such as proliferation, migration, survival, cell fate determination, differentiation and senescence.1,2 RAS signaling is switched on upon stimulation by numerous cytokines, hormones and growth factors, and mediates the appropriate cell response to external stimuli through the regulation of transcription, cytoskeletal rearrangement and metabolism. Within this network, signal flow through the RAF-MEK-ERK protein kinase pathway (Figure 1), the first described mitogen-associated protein kinase (MAPK) cascade, controls early and late developmental processes, including determination of morphology, organogenesis, synaptic plasticity and growth.3,4 Signal transmission via this cascade is initiated by the activation of cell surface receptors by extracellular ligands, which creates intracellular docking sites for adaptor molecules and signal-relay proteins recruiting and activating guanine nucleotide-exchange factors (GEFs). In turn, GEF proteins promote the release of GDP from RAS, favoring binding to the more prevalent GTP and activation of the GTPase. Activated GTP-bound RAS interacts with the RAF serine/threonine kinases (RAF1, BRAF and ARAF) favoring their catalytic activation, which results in the phosphorylation and activation of their substrates, the MAPK/ERK kinases (MEK1 and MEK2). Upon activation, MEKs act as dual specificity kinases to phosphorylate regulatory residues of the extracellular signal-regulated kinases (ERK1 and ERK2), which function as serine/threonine proteine kinases to modulate the activity of a large number of cytoplasmic and nuclear substrates, including proteins implicated in the control of gene expression. Several feedback mechanisms negatively control signal flow through the RAS-MAPK pathway, including the functional inactivation of RAS by GTPase activating proteins (GAPs), which stimulate their intrinsically low GTPase activity.
Figure 1. Schematic diagram showing the RAS-MAPK signal transduction pathway and affected disease genes in disorders of the neuro-cardio-facial-cutaneous syndrome family.

The double ovals in dark grey and the light grey ovals represent generic dimerized cell-surface receptors binding to their ligand. Abbreviations: CFCS, cardio-facio-cutaneous syndrome; CS: Costello syndrome; LS, LEOPARD syndrome; NF1, neurofibromatosis type 1; NFLS, Neurofibromatosis type 1-like syndrome (also termed Legius syndrome); NFNS, Neurofibromatosis-Noonan syndrome; NS, Noonan syndrome; NS/LAH, Noonan-like syndrome with loose anagen hair; WS, Watson syndrome.
Due to RAS’s nodal role in signal transduction and cell function, signal traffic through it is tightly controlled. Loss of that control leading to enhanced flow contributes significantly to oncogenesis.5–7 Activating somatic RAS gene mutations occur in approximately 30% of human cancers, but the upregulation of this signaling pathway can also result from enhanced function of upstream signal transducers or RAS effectors, as well as from inefficient function of feedback mechanisms. The recent, unpredicted discovery of germline mutations in a number of genes coding for proteins participating in RAS signaling, however, has established a novel scenario in which aberrant signal flow through RAS is causally linked to a group of clinically related developmental disorders characterized by facial dysmorphism, a wide spectrum of cardiac disease, reduced growth, variable cognitive deficits, ectodermal and musculoskeletal anomalies, and increased risk for certain malignancies (Figure 1).8,9 This group of diseases can now be viewed as a family of disorders of upregulated RAS-MAPK signaling. Based on the shared pathogenic mechanism and clinical overlap, these genetic diseases have been tentatively grouped as neuro-cardio-facial-cutaneous syndromes (NCFCS) or RAS-opathies.10,11
In this review, we outline the phenotypic spectrum associated with perturbed RAS-MAPK signaling, the molecular mechanisms converging towards the dysregulation of this pathway, and major genotype-phenotype correlations.
The RAS-opathies
Noonan syndrome
Noonan syndrome (NS, OMIM 163950) is a disorder characterized by particular facial dysmorphic features, congenital heart defects (CHD), hypertrophic cardiomyopathy (HCM), short stature, skeletal anomalies, and webbing of the neck. Other relatively common features are bleeding diathesis, ectodermal anomalies, lymphatic dysplasias, cryptorchidism, and cognitive deficits.12–14 NS is characterized by marked variability, which is, in part, explained by the underlying molecular lesion (see below). Facial features and signs include high forehead, hypertelorism, downslanting palpebral fissures, epicantal folds, ptosis, low-set and/or posteriorly rotated ears (Figure 2). Besides the short and/or webbed neck, a low posterior hairline commonly occurs. Affected individuals exhibit a wide spectrum of cardiac disease.15,16 Pulmonic stenosis (PS) and HCM occur most commonly, but other lesions are also observed. Feeding problems are noted in the majority of affected infants and can cause failure to thrive.17 While feeding difficulties resolve in most patients by around age 18 months, the inadequacy in oral intake may be severe enough to necessitate placement of gastrostomy tubes in some.
Figure 2. Dysmorphic facial features in disorders of the neuro-cardio-facial-cutaneous syndrome family caused by RAS-MAPK signaling dysregulation.

Series of affected individuals heterozygous for mutations in different disease genes are shown [courtesy of G. Zampino (Università Cattolica del Sacro Cuore, Rome, Italy), B. Dallapiccola and M.C. Digilio (Ospedale “Bambino Gesù”, Rome, Italy), G. Mancini (Erasmus Medical Center, Rotterdam, The Netherlands), G.B. Ferrero (Università di Torino, Turin, Italy), and M. Zenker (University Hospital, Magdeburg, Germany)].
Developmental delay and learning problems are quite common. Some motor delay can be attributed to the hypotonia often observed in affected infants. An increased prevalence of attention deficit/hyperactivity disorder and frank mental retardation are also observed. Available data indicate that the heterogeneity in cognitive abilities observed in NS is at least partially ascribed to the specifics of the causative gene mutation.18,19 Skeletal anomalies most frequently consist of pectus deformities, cubitus valgus, and vertebral defects. Of note, two childhood leukemias, juvenile myelomonocytic leukemia (JMML, OMIM 607785) and acute lymphoblastic leukemia (ALL), arise at increased prevalence in NS although they affect only a small percentage of patients.20–22 The myeloproliferative disorder in children with NS (NS/JMML) may regress without treatment, follow an aggressive clinical course or evolve to acute myeloid leukemia. This condition is strictly associated with a narrow spectrum of mutations affecting the PTPN11 gene (see below).23,24
NS is thought to be relatively common, although its prevalence has not been determined accurately to date. Most authors cite the figure of 1 in 1,000–2,500 live births, but that estimate is not based on a population study.25 NS is a Mendelian trait transmitted in an autosomal dominant manner, and, typical for that genetic mechanism, 1/3 to ½ of cases arise from spontaneous mutations. Thus far, seven genes (PTPN11, SOS1, KRAS, NRAS, RAF1, BRAF and MEK1) have been causally related to this disorder. Mutations in these genes account for a large percentage of cases (approximately 70–75%, in our experience), but is highly dependent on the diagnostic criteria used to make the diagnosis. A recent review provides an up-to-date and thorough survey of the clinical issues and pathogenetic mechanisms for this disorder.26
LEOPARD syndrome
LEOPARD syndrome (LS, OMIM 151100) is an autosomal dominant trait that overlaps phenotypically with NS. It is also allelic with NS, with a restricted spectrum of mutations in PTPN11 accounting for the vast majority of affected individuals (see below).27,28 In a small proportion of cases, LS has been causally linked to mutations in RAF1 or BRAF.29–31 The acronymic name refers to the major features: Lentigines, ECG conduction abnormalities, Ocular hypertelorism, Pulmonic stenosis, Abnormal genitalia, Retardation of growth, and sensorineural Deafness.32 Similar to NS, there are age-dependent aspects of the phenotype. Facial dysmorphism is similar to that of NS but is usually milder,33 and the neck is short but not webbed. Multiple lentigines present as dispersed flat, black-brown macules, mostlyon the face, neck, and upper part of the trunk, but sparing the mucosae. In general, lentigines appear at the age of 4–5 years and increase until puberty into the thousands. Café-au-lait spots are also observed, alone or in association withlentigines, in up to 70–80% of the patients,33 and usually precede the appearance of lentigines, being present from the first months of life. Growth retardation (height below the 3rd centile) is observed in 25% of affected individuals, and final height in 85% of subjects is below the 25th centile.34,35 Approximately half of affected individuals have heart defects, which are similar to those recurring in NS but recur with different frequencies. ECG anomalies, progressive conduction defects and HCM are the most frequent features. Of note, HCM is detected in up to 80% of LS patients with heart defects, most commonly appearing during infancy. It is progressive and paralleled by the appearance of lentigines.36 Diagnostic criteria for LS have been outlined by Voron and colleagues.34 A clinical diagnosis is not always feasible, particularly in young patients with no lentigines, due to the overlap with NS and neurofibromatosis-Noonan syndrome (see below). In these patients, the clinical differentiation from NS may be difficult during infancy and early childhood. The occurrence of distinct associated signs, including HCM, sensorineural deafness, and café-au-lait spots represents an important diagnostic handle in these young patients.33 Sarkozy et al.37 provide an updated review of the clinical and molecular genetics aspects of this disorder.
Noonan syndrome-like with loose anagen hair
Subjects with Noonan syndrome-like with loose anagen hair (NS/LAH; OMIM 607721) show features that are at first view reminiscent of NS (Figure 2). The phenotype of these subjects, however, is notable for reduced growth associated with proven GH deficiency, cognitive deficits, distinctive hyperactive behaviour, and easily pluckable, sparse, thin, slow growing hair in the anagen phase but lacking an inner and outer root sheaths.38,39 Such hair anomalies fit a well-known condition termed loose anagen hair (LAH) syndrome, which is clinically heterogeneous and has been reported to occur typically in young children and, in most cases, spontaneously improves clinically as early as adolescence.40 In these subjects, LAH can be confirmed by microscopic examination of plucked hairs.41 Most affected individuals exhibit hairless and darkly pigmented skin with eczema or ichthyosis, and a tendency to pruritus. Ectodermal anomalies also include sparse eyebrows and dystrophic or thin nails. The voice is characteristically hoarse or hypernasal. Cardiac anomalies are observed in the majority of cases, with dysplasia of the mitral valve and septal defects significantly overrepresented compared with the general NS population. So far, NS/LAH appears to be genetically homogeneous, as all affected individuals share the same 4A>G missense change (Ser2Gly) in SHOC2, which encodes a scaffold protein required for the efficient transmission of information from RAS to the MAPK cascade.39
Noonan-like/multiple giant cell lesion syndrome
NS is rarely accompanied by multiple giant cell lesions (MGCLs) of the jaw and/or other bone/soft tissues. This finding was originally introduced as a distinct nosologic entity named Noonan-like/multiple giant cell lesion syndrome (NL/MGCLS) or Noonan syndrome with cherubism (OMIM 163955).42 Now, it is apparent that MGCLs are an associated feature of NS, LS and cardiofaciocutaneous syndrome and that NL/MGCLS is not a separate disorder. This view is supported by the observation that this feature has been documented to occur in different conditions and be caused by mutations affecting PTPN11, SOS1 and MEK1 genes (see below) previously reported in subjects with those syndromes without any evidence of bony involvement.43–50 Based on these considerations, the use of NL/MGCLS should be avoided.
Cardiofaciocutaneous syndrome
Cardiofaciocutaneous syndrome (CFCS, OMIM 115150) is a rare, sporadic multiple congenital anomalies/mental retardation syndrome characterized by failure to thrive, severe feeding problems, developmental delay, reduced growth, distinctive dysmorphic facial features, abnormalities of the skin, gastrointestinal tract and central nervous system, and cardiac defects.51,52 CFCS has considerable clinical overlap with NS, and “borderline” cases are commonly observed, which justified the long debated question of whether it was a separate nosologic entity or an extreme phenotype/variant of NS.53–55
Recurrent facial/head features include relative/absolute macrocephaly, which is usually associated with high forehead, bitemporal narrowing and facial dysmorphia that is coarser compared to NS (Figure 2). The cutaneous involvement includes dry and hyperkeratotic skin (face, arms and legs), ichthyosis, eczema, sparse, friable and curly hair, absent/sparse eyebrows and eyelashes, pigmentary changes (such as café-au-lait spots, naevi or lentigines) and hemangiomata. Heart defects occur in the majority of affected individuals and consist of pulmonic stenosis most commonly, HCM and septal defects.52 Cognitive deficits are usually moderate to severe. Hypotonia results in marked motor delay. Structural brain abnormalities include cerebral atrophy, frontal lobe, corpus callosum or cerebellar vermis hypoplasia, and hydrocephalus.52 Seizures are also frequently observed. As with Costello syndrome (see below), life expectancy is shortened on average due to the early death of affected individuals with severe cardiac involvement.
CFCS is genetically heterogeneous, with mutations in the KRAS, BRAF, MEK1 and MEK2 genes occurring in approximately 60–90% of affected individuals.56–60
Costello syndrome
Costello syndrome (CS; OMIM 218040) is the eponymous name for the disorder originally described in 1971, and further delineated by the same author in 1977, as a condition characterized by prenatal overgrowth followed by postnatal feeding difficulties and severe failure to thrive, distinctive coarse facial features (Figure 2), mental retardation, short stature, cardiac defects (most commonly hypertrophic cardiomyopathy, septal defects, valve thickening and/or dysplasia, and arrhythmias), and musculoskeletal and skin abnormalities.61–64 Children and young adults with CS are predisposed to malignancies.65 Solid tumors, including rhabdomyosarcoma and less frequently neuroblastoma and bladder carcinoma, have been estimated to occur in approximately 15% of affected individuals throughout childhood, while benign cutaneous papillomata in the perinasal or/and perianal region(s) represent a distinctive and common feature associated with the disorder.
CS is caused by germline missense mutations in the HRAS proto-oncogene (OMIM 190020).66 Data from published surveys indicate that HRAS mutations account for 83% to 100% of subjects with a confirmed diagnosis of CS.66–70 This finding has been explained as either locus heterogeneity or by the difficulties of diagnosis in cases exhibiting overlapping features with clinically related disorders or a phenotype that evolves with time. In our opinion, CS should be used only for those subjects who are heterozygous for a HRAS mutation. The absence of a mutation in the gene in a subject with phenotype apparently fitting CS would be predictive of a clinically related but nosologically distinct condition. Clinical re-evaluation of HRAS mutation-negative cases is often helpful diagnostically.
Neurofibromatosis type I, including Neurofibromatosis-Noonan syndrome and Watson syndrome
Neurofibromatosis type 1 (NF1; OMIM 162200) is one of the most common autosomal dominant diseases (1 in 3000–4000 live births). Its main clinical characteristics include cutaneous, subcutaneous and/or plexiform neurofibromas, café-au-lait spots, axillary and/or inguinal freckling, Lisch nodules in the iris, skeletal deformities, vascular defects, learning disabilities and behavioral problems, short stature, macrocephaly and a predisposition for developing benign and malignant neoplasias, most commonly myeloid malignancies, including JMML, optic gliomas, pheochromocytomas, rhabdomyosarcomas and neuroblastomas.71,72 The diagnosis of NF1 is based on clinical criteria established at the NIH Consensus Development Conference in 1988.73 It should be noted, however, that subjects diagnosed as having NF1 using those criteria may carry heterozygous mutations in SPRED1, which underlie Legius syndrome (see below). NF1 is caused by heterozygous loss-of-function mutations or deletions of the NF1 gene, encoding the RAS-specific GTPase, neurofibromin.74 Loss-of-function NF1 mutations occur in the vast majority of affected individuas (> 90%). NF1 is essentially fully penetrant but there is considerable variation in the clinical features of the disease, even within families transmitting the trait. Fernel75 and Lee and Stephenson76 provide updated reviews of the clinical and molecular genetic aspects of this disorder.
Neurofibromatosis-Noonan syndrome (NFNS, OMIM 601321) is a clinical trait first noted in 1985 by Allanson and co-workers who described subjects with features of both NF1 and NS (Figure 2).77 It has been long questioned whether NFNS was a variant of either NF1 or NS, a chance association, or a distinct disorder.78,79 Recent reports have provided some insights but have not completely resolved that issue. NF1 mutation analysis performed in three independent cohorts of patients with features fitting NFNS documented occurrence of heterozygous NF1 mutations in 23 of 28 subjects.80–82 Lesions included nonsense mutations, out-of-frame deletions, missense changes (mostly affecting the GAP domain), and small in-frame deletions. Those groups also searched for PTPN11 mutations in their subjects but failed to find any. In contrast, two cases of NFNS with NF1 and PTPN11 mutations have been reported.83,84 Finally, NF1 mutations have co-segregated with the NFNS trait in a few kindreds large enough to make second mutations highly unlikely. Taken together, the current evidence indicates that most individuals with NFNS harbor an NF1 mutation and that a single mutation appears to be sufficient to engender the trait. Double heterozygosity for NF1 and PTPN11 mutations rarely causes NFNS, and it is not yet clear how frequently mutations in other NS disease genes co-occur with NF1 defects in the disorder. These findings support the view that NFNS is genetically distinct from NS and emphasize the extreme phenotypic variability associated with lesions in the NF1 gene. The identification of specific NF1 alleles recurring in NFNS, the fact that these alleles cosegregate in families with the condition, and the observation of a peculiar mutational spectrum strongly suggest that the term “NFNS” primarily characterizes a phenotypic variant of NF1, which manifests with a lower incidence of plexiform neurofibromas, skeletal anomalies, and internal tumors, in association with hypertelorism, ptosis, low-set ears, and CHDs.79–82,85 It should be noted, however, that some of the mutations identified in patients with NFNS have also been reported in NF1 without any feature suggestive of NS.
A second trait closely related to NF1 is Watson syndrome (WS, OMIM 193520), an autosomal dominant condition reported by Watson in 1967,86 characterized by pulmonic stenosis, café-au-lait spots, reduced growth and decreased intellectual ability.86–89 This phenotype was originally considered distinct from NF1 and LS based on the absence of neurofibromata, Lisch nodules, lentigines, and deafness.86,87 Allanson and co-workers, however, made observations expanding the clinical phenotype to include relative macrocephaly, Lisch nodules, and neurofibromata as recurrent features of the disorder.88,89 Consistent with the clinical overlap with NF1, linkage to the NF1 locus was documented,89,90 and allelism between these two traits was proven by the identification of a 80-kb NF1 deletion in a patient,91 and an in-frame tandem duplication of 42 bases in exon 28 of the gene in an additional family.92 The available records indicate that subjects with WS show only a mild expression of NF1 and lack the typical facial appearance of NS. As in the case of NFNS, WS should be considered as part of the broad clinical spectrum associated with NF1 mutations.
Legius syndrome
Legius syndrome (OMIM 611431), previously termed neurofibromatosis type 1-like syndrome, is an autosomal dominant condition characterized by multiple axillary freckling, café-au-lait spots, macrocephaly, and a NS-like facial dysmorphism in some individuals.93–96 Some patients have learning difficulties and/or hyperactive behavior. Despite the clinical overlap with NF1, affected individuals do not have Lisch nodules, cutaneous or plexiform neurofibromata, typical NF1 osseous lesions, or symptomatic optic gliomas, while lipomas represent a relatively common feature.93,94,96 Additional studies are required, however, to establish susceptibility of affected individuals to malignancies.
Legius syndrome is caused by loss-of-function mutations of the SPRED1 gene (see below).93 Messiaen and co-workers observed that, among 42 SPRED1 mutation-positive individuals, approximately half of the cases fulfilled NIH’s diagnostic criteria for NF1.96 In the same study, mutational analysis of a large cohort of patients fulfilling NIH criteria for diagnosis of NF1 revealed a SPRED1 mutation in 2% of cases (i.e., approximately 8% of NF1 mutation-negative cases).96
RAS-MAPK signaling dysregulation and phenotypic consequences
PTPN11 gene
The PTPN11 gene (OMIM 176876) encodes SHP2, a widely expressed cytoplasmic protein tyrosine phosphatase that positively modulate RAS signaling.97–100 It is characterized by two tandemly arranged N-terminal src-homology 2 domains (N-SH2 and C-SH2), a single catalytic domain (PTP) and a C-terminal tail of uncharacterized function. SHP2 binds to phosphotyrosyl-containing signaling partners through the SH2 domains, which control both SHP2’s subcellular localization and function.101
We discovered PTPN11 as a major NS disease gene using a positional candidacy approach in 2001.102 PTPN11 mutations account for approximately 50% of NS, and are more prevalent among subjects with pulmonary valve stenosis and short stature, and less common in individuals with HCM and/or severe cognitive deficits.43,103 These mutations are almost always missense changes and perturb SHP2’s function through distinct mechanisms.104–106 Most mutations affect residues involved in the N-SH2/PTP interdomain binding network that stabilizes SHP2 in its catalytically inactive conformation (Figure 3). These mutations are predicted to up-regulate SHP2’s function by impairing the switch between the active and inactive conformations, favoring a shift in the equilibrium toward the former. There are, however, changes affecting residues located in the phosphopeptide binding pocket of the N-SH2 and C-SH2 domains or in the linker stretch connecting these domains. There is evidence supporting the idea that these substitutions promote SHP2 gain of function by increasing binding affinity, altering the binding specificity for the signaling partners, or altering the flexibility of the N-SH2 domain in a manner that inhibits the N-SH2/PTP interaction.
Figure 3. PTPN11 gene organization, SHP2 domain structure and location of affected residues in human disease.

(A) The PTPN11 gene and its encoded protein product. The numbered, filled boxes at the top indicate the coding exons; the positions of the ATG and TGA codons are shown. Exon location of disease-associated mutations is indicated (the number of asterisks is an index of the relative prevalence of mutations within each exon). SHP2’s functional domains, consisting of two tandemly arranged SH2 domains at the N-terminus (N-SH2 and C-SH2) followed by a protein tyrosine phosphatase (PTP) domain, are shown below. The numbers below that cartoon indicate the amino acid boundaries of those domains. (B) Location of mutated residues in the three dimensional structure of SHP2 in its catalytically inactive conformation.94 Residues affected by germline (left) or somatically acquired (right) mutations are shown with their lateral chains (yellow, mutations affecting the N-SH2/PTP interaction; green, mutations affecting the N-SH2/PTP interaction and possibly substrate specificity and/or catalysis; red, mutations promoting increased SH2 phosphopeptide binding affinity or affecting specificity; blue, mutations affecting SH2 orientation or mobility; black, unclassified). The identity of affected residues and amino acid substitutions, and the prevalence of mutations are listed in Tartaglia et al.98
A distinct class of PTPN11 mutations has been identified to underlie LS.27,28 Tyr279Cys and Thr468Met represent the most common defects, although additional mutations have been documented.37 An impaired catalytic activity has been established as the biochemical behavior shared by these SHP2 mutants.105–108 Their role in SHP2 functional dysregulation still requires further study, since they do not appear to perturb intracellular signaling by a merely dominant negative effect,106,109,110 as supposed in the past.108
Children with NS are predisposed to a spectrum of hematologic abnormalities and malignancies, including JMML, a clonal myeloproliferative disorder of childhood,111 and children with NS/JMML are heterozygous for PTPN11 germline mutations in the majority of cases, with one mutation rarely observed in NS (218C>T, Thr73Ile) occuring in a large percentage of cases.23,24,112 Remarkably, a distinct class of mutations in this gene, acquired as a somatic event, occurs in approximately one-third of children with JMML without NS as well as variable proportions of other myeloid and lymphoid malignancies of childhood.23,24, 112–115 As observed in NS, the vast majority of PTPN11 lesions identified in this heterogeneous group of hematologic malignancies are missense changes that alter residues located at the interface between the N-SH2 and PTP domains (Figure 3), but appear to be more activating.104,105,112,116,117
De novo duplications of the chromosomal region encompassing PTPN11 (12q24) have been documented in two subjects with clinical features suggestive of NS without mutations in any known NS genes.118,119 These findings suggest that increased dosage of SHP2 may have dysregulating effects on intracellular signaling and consequences on developmental processes. The screening of a relatively large of cohort of NS cases negative for mutations in previously identified genes, however, indicate that PTPN11 gene duplication represents an uncommon cause of NS.
SOS1 gene
Cell-surface tyrosine kinase receptors activate RAS proteins by recruiting GEFs to the cytoplasmic side of the plasma membrane. These proteins catalyze the release of GDP from RAS, facilitating the conversion of the inactive GDP-bound form of RAS to its active GTP-bound state.120 Among the GEFs, two members of the son of sevenless (SOS) family, SOS1 and SOS2, promote guanine nucleotide exchange on RAS proteins, but not on RAS-related family members, such as the RAP and RHO proteins.121,122
Based on their modulatory role in RAS signaling, we and others discovered that SOS1 (OMIM 182530) is mutated in a relatively large percentage of subjects with NS.123,124 This gene encodes a large multidomain protein (Figure 4).125 The N-terminal portion of the protein contains a histone domain (HD) that is characterized by two tandemly arranged histone folds, and is followed by Dbl homology (DH) and pleckstrin homology (PH) domains, which are implicated in the activation of RAC, a small GTPase of the RHO/CDC42 family.126 The C-terminal half of the protein contains the RAS exchanger motif (REM) domain and the Cdc25 domain, which are required for the RAS-specific nucleotide exchange activity of SOS1. Finally, the region at the C-terminus contains recognition sites for SH3 domains and mediates interaction of SOS1 with SH3 domain-containing adaptor proteins that deliver SOS1 to the membrane upon receptor activation. Additional anchorage sites on the membrane are provided by the phosphatidylinositol phosphate-binding site within the PH domain,127 and an extended positively charged surface of the HD domain.128
Figure 4. SOS1 domain structure and location of affected residues in NS.

(A) SOS1 missense mutations are positioned below the cartoon of the SOS1 protein with its functional domains indicated above. Abbreviations: DH, Dbl homology domain; PH, plekstrin homology domain; Rem, RAS exchanger motif; PxxP, proline-rich region. (B) Location of the mutated residues on the three-dimensional structure of SOS1. The functional domains are color coded as follows: Histone folds, cyan; DH, magenta; PH, orange; Rem, green; Cdc25, yellow. Residues affected by mutations are indicated with their lateral chains (histone folds, violet; DH, blue; PH, green; helical linker, red; Rem, orange; Cdc25, cyan). Based on Sondermann et al.121, which utilized structural data and computational modeling.
The available data indicate that SOS1 is the second most frequently mutated NS disease gene, accounting for approximately 10% of subjects.123,124,129 The vast majority of the mutations identified are missense changes and affect multiple domains (Figure 4). Approximately half of the SOS1 defects affect residues located in the short helical linker connecting the PH and REM domains, with substitutions of residue Arg552 accounting for approximately 30% of total mutations. A second mutation cluster is located within the PH domain (residues 432 to 434), while a third functional cluster resides at the interacting regions of the DH (Thr266 and Met269) and REM (Trp729 and Ile733) domains. A single amino acid change (Glu846Lys) within the Cdc25 domain accounts for more than 10% of defects.
Subjects heterozygous for a mutated SOS1 allele tend to exhibit a distinctive phenotype characterized by ectodermal abnormalities generally associated with an absence of cognitive deficits and normal growth.124,129 A few SOS1 mutation-positive individuals with ectodermal manifestations and distinctive facial dysmorphia that might be suggestive of CFCS have been reported (Figure 2).130,131 In these subjects, cognitive deficits were generally absent or minor.
The GEF activity of SOS1 is controlled by an allosteric site, involving the Cdc25 and REM domains, that stimulates SOS1 exchange activity through the binding of nucleotide-bound RAS.132 Basally, the interaction between the DH and REM domains stabilizes SOS1 in its catalytically inactive conformation by masking the allosteric binding site for RAS. Following SOS1’s translocation to the membrane, the inhibitory effect of the DH domain is relieved by still undefined events allowing RAS binding to the allosteric site, which in turn promotes a conformational change of the REM and Cdc25 domains, and RAS binding to the catalytic site.133 Most NS-causing SOS1 mutations reside in regions within the molecule that are predicted to contribute structurally to the maintenance of the catalytically autoinhibited conformation (Figure 4). Biochemical data confirmed such predictions, demonstrating that NS-causing SOS1 mutations promote gain of function and enhance RAS and ERK activation.123,124
A possible contribution of activating SOS1 mutations in cancer has been evaluated (our unpublished data).134,135 Mutational analysis performed on genomic DNA from more than 1,000 solid tumors (pancreatic, lung, breast, colon, thyroid cancers) and hematological malignancied (myeloproliferative disorders and acute myeloid and lymphoblastic leukemias) indicate that SOS1 is not mutated in most cancers, suggesting that NS patients heterozygous for a SOS1 mutation are not at increased risk of developing cancer.
HRAS, KRAS and NRAS genes
The three members of the RAS subfamily, HRAS (OMIM 190020), KRAS (OMIM 190070) and NRAS (OMIM 164790), are monomeric GTPases that use GDP/GTP-regulated molecular switching to control intracellular signal flow.1,136 They exhibit high affinity for GDP and GTP, low GTPase activity, and cycle from a GDP-bound inactive state to a GTP-bound active state, the latter favoring their interaction with multiple downstream transducers. As anticipated, GDP/GTP cycling is controlled by GEFs, such as SOS1, which promote release of GDP, and GAPs, which accelerate the intrinsic GTPase activity. RAS proteins share a structure that includes a conserved domain (residues 1 to 165), known as the G domain, which is required for its signaling function, and a less conserved C-terminal tail, called the hypervariable region, that guides post-translational processing and plasma membrane anchoring (Figure 5). Within the G domain, conserved sequence elements direct GTP/GDP binding and GTP hydrolysis. Furthermore, two tracts, denoted as Switch I and Switch II, undergo major conformational changes upon GTP/GDP exchange and mediate binding to RAS effectors, GAPs and GEFs.1,137 Heterozygous germline mutations in each of the three members of the RAS subfamily have been identified to be causally related with NS, CFCS or CS.
Figure 5. Domain structure of RAS proteins and location of affected residues in NS, CFCS and CS.

(A) Schematic diagram and tridimensional representation of the structural and functional domains defined within RAS proteins. The motifs required for signaling function (PM1 to PM3 indicate residues involved in binding to the phosphate groups, while G1 to G3 are those involved in binding to the guanine base) are indicated. The hypervariable region is shown in grey, together with the C-terminal motifs that direct post-translational processing and plasma membrane anchoring (dark grey). The GTP/GDP binding pocket is shown in cyan (guanine ring binding surface) and yellow (triphosphate group binding surface) together with the Switch I (forest green) and Switch II (magenta) domains, according to the GTP-bound RAS conformation. (B) Location of the KRAS residues mutated in NS and CFCS. Residues affected by mutations are indicated with their lateral chains (triphosphate group binding surface, orange; guanine ring binding surface, blue; Switch I, green; other regions; black). (C) Location of the HRAS residues mutated in CS. Residues affected by mutations are indicated with their lateral chains as reported above. (C) Location of the NRAS residues mutated in NS. Residues affected by mutations are indicated with their lateral chains as reported above.
Based on the link between the functions of SHP2 and RAS, Aoki and co-workers used a candidate gene approach to discover that a small number of missense mutations affecting residues Gly12 and Gly13 in HRAS underlie CS.66 These residues represent hot spots for oncogenic somatic mutations.138 Defects affecting those residues render the protein insensitive to GAP-promoted GTP hydrolysis, resulting in a constitutively GTP-bound, active state.5,138 Other less common amino acid substitutions involving residues in the purine ring binding pocket of the protein (His117 and Ala146) are predicted to affect the stability of HRAS binding to GDP/GTP,69,70 favoring a shift in the equilibrium towards the GTP-bound, active form of RAS, which bypasses the requirement for a GEF.139–142 Recently, two different three-nucleotide insertions resulting in the duplication of Glu37 within the switch I region have been reported.143 This small in-frame insertion promotes a weak hyperactivation of signaling resulting from a balancing effect between a profound GAP insensitivity and inefficient binding to effector proteins.
In CS, the Gly12Ser substitution in HRAS accounts for approximately 85% of affected individuals. This change recurs with considerably lower prevalence in human cancers (approximately 5% of total HRAS amino acid substitutions) (COSMIC; http://www.sanger.ac.uk/genetics/CGP/cosmic/). Experimental data indicate that Gly12Ser has a lower transformation potential than the Gly12Val mutant,144 which is the most common somatic HRAS defect in human tumors (about 45% of total somatic HRAS gene mutations). The latter has been documented in only two individuals with CS who exhibited a relatively severe phenotype (approximately 2% of cases with an HRAS gene mutation).66,145 Similarly, no HRAS mutation affecting residue Gln61, which constitute approximately 33% of total HRAS lesions in human cancer, has been documented in CS to date. Such a striking difference in prevalence and spectrum of HRAS amino acid substitutions suggests that CS-causative mutations have less potency for deregulating protein function than cancer-contributing ones and that the latter might exert a stronger perturbing effect on development when inherited.
Based on the identification of HRAS gene mutations in CS, we and others discovered that germline KRAS mutations can underlie NS or CFCS (Figure 2).56,146,147 Mutations account for a relatively small percentage of affected subjects in both disorders (< 2–5%), and, in NS, are generally associated with a highly variable but generally severe phenotype. The diversity of mutations associated with these developmental disorders as well as their phenotypic spectrum have been investigated further, refining the picture of a clustered distribution of germline disease-associated KRAS defects, and confirming the high clinical variability.59,148,149 The KRAS gene (OMIM 190070) produces two transcripts through alternative splicing, resulting in two proteins called KRASA and KRASB,150 which differ at the C-terminus. Most KRAS mutations cluster in the first and second coding exons of the gene, which are shared by the two KRAS isoforms. Mutations affecting exon 6, however, have been documented in one-third of NS or CFCS subjects with a KRAS germline mutation. This exon codes for the C-terminal tail of KRASB but not KRASA. These findings document that isolated KRASB gain of function is sufficient for disease pathogenesis, which is also consistent with the available evidence about the relative roles of the KRAS isoforms on development from knock-out animal models.151–153
As observed for the somatically acquired oncogenic RAS gene mutations and germline transmitted HRAS lesions, the NS/CFCS-causing KRAS defects were found to up-regulate protein function by either affecting the intrinsic and/or GAP-stimulated GTPase activity and, consequently, impairing the switch between the active and inactive conformation, or determining a dramatically increased rate of guanine nucleotide dissociation.147,154 Although these studies documented a complex pattern of intrinsic biochemical properties, expression of these mutants in COS-7 cells promoted higher levels of phosphorylated MEK and ERK proteins, indicating hyperactivation of the MAPK cascade. Of note, available records indicate that germline KRAS mutations are rarely observed in human cancers. This finding is consistent with functional data indicating a reduced perturbing effect of these mutations on protein function compared with the cancer-associated ones.147,154 This is also supported by the observation that malignancies have been rarely observed in subjects with NS or CFCS due to a KRAS mutation.56,59,147,148
More recently, two germline missense mutations (Thr50Ile and Gly60Glu amino acid changes) have been reported to account for a few NS cases.155 A germline KRAS mutation affecting Gly60 (Gly60Arg) has been reported in CFCS,56 and the somatic NRAS mutation Gly60Glu rarely has been observed to occur in malignancies (COSMIC database). Of note, mutations of the neighbouring Gln61 are frequent oncogenic RAS alterations and confer impaired GTPase function. In contrast, no somatic or germline RAS gene mutation affecting Thr50 has been described so far. Thr50 is an exposed residue located in the β2-β3 loop connecting the two switch regions but is not predicted to be directly involved in GTP/GDP binding, GTP hydrolysis, or effector interaction. Functional characterization of these disease-associated mutations documented that, similarly to what was observed for KRAS and HRAS mutation, they confer enhanced stimulus-dependent MAPK activation but apparently through different molecular mechanisms.155
NF1 gene
The NF1 gene (OMIM 16200) encodes neurofibromin, a cytoplasmic multifunction protein ubiquitously expressed with highest expression in neurons, Schwann cells, oligodendrocytes, astrocytes and leukocytes.156 The NF1 coding sequence spans over 61 exons distributed over more than 300 Kb. A number of variant transcripts have been identified, which originate from alternative splicing, exon skipping and short sequence insertions. While neurofibromin is a large protein, it has no clearly defined functional domains, except the SEC14 and RasGAP domains, the latter mediating the RAS-specific GAP function of the protein. Besides its role as a negative modulator of RAS signaling, neurofibromin is involved in the modulation of other signaling pathways, including the protein kinase A (PKA)-cAMP pathway, and in microtubule-associated processes.157–160 Moreover, a possible function within the nucleus has been hypothesized.161 For a general overview see Trovó-Marqui and Tajara.156
In the majority of affected individuals, disease-causing NF1 mutations consist of intragenic frameshift, nonsense, splicing or missense mutations arrayed along the entire length of the gene. A small proportion of affected individuals (< 5%) carry a heterozygous deletion of approximately 1.5 Mb, which is the result of unequal homologous recombination and encompasses the entire NF1 and a few adjacent genes.162–164 There is poor correlation between classes of mutations and disease features, with the exception of patients with large deletions who tend to have more severe phenotype and higher numbers of neurofibromata. Phenotypic heterogeneity is high among affected individuals, even within families co-inheriting the same NF1 mutation.
NF1 is a tumor suppressor gene, and loss of the wild-type allele has been demonstrated to occur somatically as a second hit in malignancies associated with the trait.165–167
SPRED1 gene
The SPROUTY-related ENA/VASP homology-1 (EVH1) domain-containing protein 1 (SPRED1; OMIM 609291) is a member of the SPROUTY/SPRED family of membrane-associated proteins that function as modulators of growth factor–induced activation of the RAS-MAPK pathway. SPRED1 is characterized by an N-terminal EVH-1 domain, which binds to proline-rich sequences, a central KIT–binding domain (KBD) with unknown function, and a C-terminal SPRY domain implicated in protein translocation to the plasma membrane, RAF binding and heterodimerization with other SPRED proteins.168 SPRED1 negatively controls MAPK signaling and operates downstream of RAS by suppressing RAF phosphorylation and activation,169 although a target upstream of RAS has also been suggested.170 Heterozygous loss-of-function mutations in SPRED1 cause Legius syndrome.93 The mutational spectrum is wide, including nonsense, frameshift and missense mutations, as well as small in-frame deletions.93–96 Most mutated alleles produce unstable proteins that are likely to be quickly degraded in the cell, but others encode non-functional proteins. It has been demonstrated that disease-causing mutations resulted in increased RAF1 kinase activity, enhanced phosphorylation levels of MEK and ERK proteins, and increased activity of ELK1, a transcription factor functioning downstream the MAPK cascade.93 Of note, as documented in NF1, second hits in the gene have been documented in melanocytes from café-au-lait spots of affected subjects, indicating that this feature is caused by biallelic inactivation of SPRED1, as previously demonstrated for NF1.171 Mutational screening performed on kidney and cell nervous system cancers do not suggest a major contribution of somatic SPRED1 mutations to these cancers (COSMIC database).
RAF1 and BRAF genes
The RAF1 (OMIM 164760), BRAF (OMIM 164757)and ARAF (OMIM 311010) proteins are members of a small family of serine-threonine kinases that function as RAS effectors.7,172,173 These kinases phosphorylate and activate the dual specificity kinases MEK1 and MEK2, which in turn promote the activation of the MAPKs, ERK1 and ERK2. The three members of the RAF family play different roles in the activation of the RAS-MAPK signaling cascade. BRAF has considerably higher MEK kinase activity compared to RAF1 and ARAF. These proteins also differ in their expression profiles as well as in the regulatory mechanisms controlling their function, and appear to have unique roles during development.174–176 Consistent with these observations, somatic BRAF mutations frequently occur in malignant melanomas and in thyroid, colorectal and ovarian cancers (COSMIC database), while ARAF and RAF1 missense changes are observed rarely in malignancies. These kinases are characterized by three functional domains, known as conserved regions 1 to 3 (CR1-3) (Figure 6). The N-terminal CR1 contains the region involved in GTP-RAS binding and a cysteine-rich sequence that mediates protein interaction with the cytosolic surface of the cell membrane. CR2 contains a negative regulatory domain controlling protein translocation to the membrane and its catalytic activation, while the C-terminal CR3 comprises the kinase domain of the protein.172
Figure 6. RAF1 and BRAF domain structure and location of affected residues in human disease.
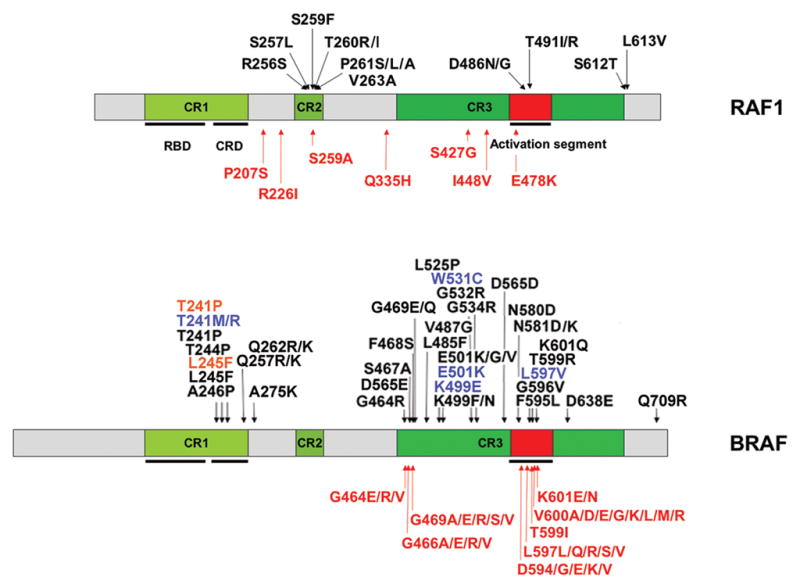
The domains of the RAF1 (above) and BRAF (below) proteins are indicated (CR, conserved region; RBD, RAS binding domain, CRD, cysteine-rich domain). Germline (RAF1: NS and LS; BRAF: NS, LS and CFCS) and somatic (associated with cancer) mutations are reported above and below each cartoon, respectively. Only somatic BRAF mutations with prevalence ≥ 1.5%, according the COSMIC database (http://www.sanger.ac.uk/genetics/CGP/cosmic/) are reported. The BRAF Thr599Ile substitution, which rarely occurs in cancer and is homologous to the NS-causing RAF1 Thr491Ile change, is also reported. BRAF missense changes associated with a phenotype fitting NS or LS are colored blue and orange, respectively.
We and another group identified missense mutations in RAF1 (OMIM 164760) in subjects with NS with a prevalence of mutations ranging between 5–15% (Figure 2).29,177 Mutations in the same gene were also identified in two of six subjects with LS without a mutation in PTPN11.29 RAF1 mutations affect residues clustering in three regions of the protein (Figure 6). The first cluster (70% of total RAF1 defects) involves the N-terminal consensus 14-3-3 recognition sequence or adjacent residues within the CR2 region. Of note, Arg256, Ser257, Ser259 and Pro261, which are the invariant residues within this motif, were all found to be mutated. The second group (15% of RAF1 lesions) includes mutations affecting residues within the activation segment region of the kinase domain (Asp486 and Thr491). Of note, several BRAF missense mutations detected in solid tumors alter the activation segment, including some (Asp594Gly and Thr599Ile) homologous to those of RAF1 identified in subjects with NS. Finally, the third cluster (15% of RAF1 mutations) affect two adjacent residues (Ser612 and Leu613) located at the C-terminus. Phenotype analysis of the NS and LS subjects with RAF1 mutations is notable for the observation that a large percentage of cases (75%) exhibit HCM, compared to HCM prevalence of 18% in the general NS population.29,177 Moreover, this genotype-phenotype correlation seems to be allele-specific as HCM appears to be associated with mutations affecting the N-terminal 14-3-3 consensus site or the C-terminus. In addition to the two cases with LS, multiple nevi, lentigines and/or café-au-lait spots were detectable in one-third of NS patients with RAF1 mutations, suggesting a predisposition to hyperpigmented cutaneous lesions.
In RAF1’s inactive state, the N-terminal portion of the protein interacts with and inactivates the kinase domain at the C-terminus. This autoinhibited conformation is stabilized by 14-3-3 protein dimers that bind to phosphorylated Ser259 and Ser621.7,172,173 Dephosphorylation of Ser259, which is possibly mediated by protein phosphatase 1C (PP1C) or protein phosphatase 2A (PP2A), is required for stable interaction with GTP-RAS, allowing protein translocation to the plasma membrane and phosphorylation of regulatory residues required for RAF1 catalytic activation. Functional characterization of a selected panel of RAF1 mutants supports the idea that mutations can differentially perturb protein function and intracellular signaling. In particular, mutations affecting the N-terminal 14-3-3 binding motif or the C-terminus of the protein, promote enhanced kinase activity and increased activation of the MAPK cascade compared to wild-type protein.29,177 In contrast, Asp486Asn and Thr491Ile, representing the mutation cluster in the activation segment, were observed to be kinase impaired and differentially perturb signaling through the MAPK cascade. Pandit and co-workers provided evidence supporting the model that the increased activation promoted by the amino acid substitutions affecting the N-terminal 14-3-3 binding site results from a loss of 14-3-3-mediated inactivation,29 while the mechanism through which mutations at the C-terminus activate RAF1’s kinase activity remains to be explained.
Two groups independently discovered that BRAF is a major disease gene for CFCS (50–75% of cases) (Figure 2).56,57 More recently, germline BRAF mutations have been documented also in a small percentage of subjects with phenotypes fitting or suggestive of NS or LS (< 2% of cases) (Figure 2).30,31,177,178 Of note, NS-associated mutations largely do not overlap with those occurring in CFCS, suggesting a genotype-phenotype correlation. The recurrence of the Trp531Cys amino acid substitution as well as the clustering of mutations at the Thr241 further supports the idea that the phenotype resulting from germline BRAF defects might be allele specific. Among subjects with NS, the available clinical data indicate an association with neonatal growth failure and feeding difficulties, mild-to-moderate cognitive deficits, and hypotonia, and a higher prevalence of multiple nevi and dark colored lentigines.30 As adults, they display the full-blown phenotype of NS, which is more severe compared to those associated with PTPN11 and SOS1 mutations. In these individuals, however, polyhydramnios, HCM, and CFCS-related skin features are uncommon or absent,52,179 and cardiac defects, neurological impairment and feeding problems appear to be less severe compared to what is generally observed in CFCS.30 While the occurrence of distinct BRAF gene mutations in subjects with a phenotype fitting NS would indicate that some features might be allele-specific, the observation that a subgroup of BRAF mutation-positive subjects exhibits an “intermediate” phenotype suggests that a clinical continuum characterized by a differential combination and severity of features is associated with defects in the BRAF gene.30 These findings emphasize the difficulty in identifying efficient clinical criteria to define CFCS, LS or NS nosologically, and make evident the usefulness of a molecular-based definition of these clinically overlapping conditions that might direct clinicians towards the appropriate management of patients. Of note, the observation of a patient with LS and normal intelligence who was found to carry a novel sequence change in BRAF,31 further illustrates that the phenotypic spectrum caused by BRAF mutations is broader than previously assumed and that mental retardation is not an invariably associated feature.
More than 40 germline BRAF mutations have been identified (Figure 6). All are missense defects that are not randomly distributed, clustering in the cysteine-rich domain and in the amino-terminal portion and activation segment of the kinase domain. Most mutations are recurrent, with substitutions of residues Gln257 and Glu501 accounting for approximately 40% of all defects. Germline BRAF mutations are rarely observed as somatic mutations contributing to oncogenesis and NIH-3T3 cell colony focus formation assay data indicate that CFCS-associated BRAF mutants have a reduced transforming capability compared with the recurrent oncogenic Val600Glu BRAF protein, and that the NS- and LS-associated BRAF mutants do not confer enhanced transformation to cells.30 While these results support the view that the mutations transmitted through the germline have less potency in deregulating BRAF’s function, they also suggest that NS- and LS-associated mutations are less activating compared to those associated with CFCS, which parallels observations indicating that phenotypes arising from germline BRAF defects might be allele specific. Of note, in vitro functional studies indicate that germline and somatic amino acid changes can confer either increased or decreased activity upon the mutant protein,30,57,180 suggesting that multiple alternative mechanisms are likely to be involved in the functional dysregulation of the kinase.
SHOC2 gene
We recently discovered that the invariant 4A>G missense (Ser2Gly) change in the SHOC2 gene (OMIM 602775), which encodes a cytoplasmic leucine-rich repeat-containing protein that positively modulates RAS-MAPK signal flow,181–185 underlies NS/LAH.39 This amino acid change was demonstrated to promote N-myristoylation of the protein, and causes aberrant targeting of SHOC2 to the plasma membrane and impaired translocation to the nucleus upon growth factor stimulation (Figure 7).39 N-myristoylation is an irreversible form of protein fatty acylation occurring during protein synthesis, in which myristate, a 14-carbon saturated fatty acid, is covalently added to an N-terminal glycine residue after excision of the initiator methionine residue by methionylaminopeptidase.186–188 It is a relatively common lipid modification of many signal transducers, and contributes to direct protein anchoring to cellular membranes, which is required for their proper function.
Figure 7. The disease-causing 4A > G (Ser2Gly) change in SHOC2 promotes protein myristoylation and cell membrane targeting.

(A) SHOC2 genomic organization and protein structure. The coding exons are shown at the top as numbered filled boxes. Intronic regions are reported as dotted lines. SHOC2 motifs comprise an N-terminal lysine-rich region (grey coloured) followed by 19 leucine-rich repeats. Numbers above the domain structure indicate the amino acid boundaries of those domains. (B) Confocal laser scanning microscopy analysis documents that SHOC2wt (red) is uniformly distributed in the cytoplasm and nucleus (left) of transiently transfected Cos-1 cells (DMEM supplemented with 10% heat-inactivated FBS), while SHOC2S2G (red) co-localizes with ganglioside M1 (green) to the cell membrane.39 Nuclei are visualized by DAPI staining (blue). Bars indicate 20μm.
SHOC2 is a widely expressed protein composed almost entirely by leucine-rich repeats (LRR) and has a lysine-rich sequence at the N-terminus (Figure 7). Since LRRs can provide a structural framework for protein-protein interactions, SHOC2 is believed to function as a scaffold linking RAS to downstream signal transducers. It has recently been reported that SHOC2 functions as a regulatory subunit of the catalytic subunit of protein phosphatase 1 (PP1C).184 By binding GTP-MRAS, SHOC2 promotes PP1C translocation to the membrane, allowing PP1C-mediated RAF1 dephosphorylation at residue Ser259, which is required for stable RAF1 translocation to the plasma membrane and catalytic activation. According to this model, constitutive membrane translocation of the disease-causing SHOC2S2G protein is expected to promote prolonged PP1C-mediated RAF1 dephosphorylation at Ser259 and, consequently, sustained RAF1-stimulated MAPK activation. Initial functional characterization of the mutant protein indicates that the expression of SHOC2S2G mutant enhances ERK activation in a cell type-specific fashion.39 The observations that subcellular localization of SHOC2 is restricted to the nucleus following EGF stimulation and that SHOC2S2G ectopic expression in C. elegans engenders protruding vulva, a neomorphic phenotype previously associated with aberrant signaling, suggest that the disease-causing mutant might exert a wider perturbing effect on intracellular signaling.
MEK1 and MEK2 genes
MEK1 (OMIM 176872) and the functionally related MEK2 (OMIM 601263), belong to a family of dual specificity kinases that phosphorylate ERK proteins at tyrosine and serine/threonine residues.189 The MEK proteins share a conserved structure including a negative regulatory domain at the N-terminus and a single protein kinase domain. MEK1 and MEK2 are both effectors of RAF proteins, but appear to play non-redundant roles. In particular, genetic evidence from mouse models indicates that MEK1 function is required during embryonic development,190 while MEK2 is dispensable.191 Of note, even though MEK kinase activity is necessary for cell transformation via the MAPK cascade,192 and constitutively active MEK mutants promote transformation,193 mutations in these genes have not been reported in human cancers so far (COSMIC database). Missense mutations and in-frame deletions in MEK1 and MEK2 account for approximately 20% of CFCS.57–60,178,194 Recently, one germline mutation in MEK1, predicting the Asp67Asn amino acid substitution, has been documented in two unrelated subjects exhibiting a phenotype fitting NS.59 According to this study, MEK1 gene mutations would account for approximately 3% of PTPN11- and SOS1-mutation negative NS cases. In NS and CFCS, MEK gene mutations are mostly missense changes affecting residues located in the regulatory region and the N-terminal portion of the catalytic domain. Functional characterization of a panel of CFCS-causing mutants documented that they are more active than the wild-type protein in stimulating ERK phosphorylation.57,195 No datum on the effect of the Asp67Asn change on MEK1 function and MAPK signaling is currently available.
Concluding remarks
While dysregulation of the RAS-MAPK signaling pathway has been known for decades as a major event in oncogenesis, discoveries derived from a massive disease gene hunting effort have only recently established a novel scenario in which the upregulation of this signaling cascade underlies a group of clinically related developmental disorders. Based on the relatively high prevalence of some of these disorders, the dysregulation of this signaling pathway also represents one of the most common events affecting developmental processes. These discoveries have also provided us with novel molecular mechanisms causing enhanced signaling.
During the last years, screening efforts and genotype-phenotype correlation analyses have documented that mutations in identified disease genes are associated with distinct phenotypes, explaining, in part, the wide clinical diversity characterizing these disorders. It should be considered that the overlapping features observed among disorders of the NCFCS family, the wide breadth of phenotypes within each trait, and absence of clinical features with pathognomic value and consensus on specific and routinely used diagnostic criteria make diagnosis of these diseases challenging, particularly during early infancy and in adulthood. Such a failure has direct impact on patient management because of the prognostic relevance of diagnosis (prevalence and extent of cardiac disease, degree of cognitive and growth impairment, cancer predisposition). Molecular diagnosis now offers the opportunity to overcome the weakness of subjective clinical criteria and represents a highly informative prognostic tool, with direct impact on counseling and appropriate patient management.
Acknowledgments
The authors apologize to colleagues whose work was not cited due to limited space. Research in the authors’ laboratories is supported in part by grants from Telethon-Italy (GGP10020), ERARE 2009 (NSEuroNet) and “Associazione Italiana Sindromi di Costello e Cardiofaciocutanea” to M.T., and from the National Institutes of Health (HL71207) and March of Dimes (06-1047) to B.D.G.
References
- 1.Mitin N, Rossman KL, Der CJ. Signaling interplay in Ras superfamily function. Curr Biol. 2005;15:R563–574. doi: 10.1016/j.cub.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 2.Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nat Rev Mol Cell Biol. 2008;9:517–531. doi: 10.1038/nrm2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sundaram MV. RTK/Ras/MAPK signaling. WormBook. 2006;11:1–19. [Google Scholar]
- 4.McKay MM, Morrison DK. Integrating signals from RTKs to ERK/MAPK. Oncogene. 2007;26:3113–3121. doi: 10.1038/sj.onc.1210394. [DOI] [PubMed] [Google Scholar]
- 5.Bos JL. Ras oncogenes in human cancer: a review. Cancer Res. 1989;49:4682–4689. [PubMed] [Google Scholar]
- 6.Rajalingam K, Schreck R, Rapp UR, Albert S. Ras oncogenes and their downstream targets. Bioch Biophys Acta. 2007;1773:1177–1195. doi: 10.1016/j.bbamcr.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 7.Leicht DT, Balan V, Kaplun A, Singh-Gupta V, Kaplun L, Dobson M, Tzivion G. Raf kinases: function, regulation and role in human cancer. Biochim Biophys Acta. 2007;1773:1196–1212. doi: 10.1016/j.bbamcr.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer. 2007;7:295–308. doi: 10.1038/nrc2109. [DOI] [PubMed] [Google Scholar]
- 9.Rauen KA, Schoyer L, McCormick F, Lin AE, Allanson JE, Stevenson DA, Gripp KW, Neri G, Carey JC, Legius E, et al. Proceedings from the 2009 genetic syndromes of the Ras/MAPK pathway: From bedside to bench and back. Am J Med Genet A. 2010;152A:4–24. doi: 10.1002/ajmg.a.33183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bentires-Alj M, Kontaridis MI, Neel BG. Stops along the RAS pathway in human genetic disease. Nat Med. 2006;12:283–285. doi: 10.1038/nm0306-283. [DOI] [PubMed] [Google Scholar]
- 11.Tidyman WE, Rauen KA. The RASopathies: developmental syndromes of Ras/MAPK pathway dysregulation. Curr Opin Genet Dev. 2009;19:230–236. doi: 10.1016/j.gde.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Noonan JA. Noonan syndrome. An update and review for the primary pediatrician. Clin Pediatr (Phila) 33:548–555. doi: 10.1177/000992289403300907. [DOI] [PubMed] [Google Scholar]
- 13.Allanson JE. Noonan syndrome. Am J Med Genet C Semin Med Genet. 2007;145C:274–279. doi: 10.1002/ajmg.c.30138. [DOI] [PubMed] [Google Scholar]
- 14.van der Burgt I. Noonan syndrome. Orphanet J Rare Dis. 2007;2:4. doi: 10.1186/1750-1172-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burch M, Sharland M, Shinebourne E, Smith G, Patton M, McKenna W. Cardiologic abnormalities in Noonan syndrome: Phenotypic diagnosis and echocardiographic assessment of 118 patients. J Am Coll Cardiol. 1993;22:1189–1192. doi: 10.1016/0735-1097(93)90436-5. [DOI] [PubMed] [Google Scholar]
- 16.Marino B, Digilio MC, Toscano A, Giannotti A, Dallapiccola B. Congenital heart diseases in children with Noonan syndrome: An expanded cardiac spectrum with high prevalence of atrioventricular canal. J Pediatr. 1999;135:703–706. doi: 10.1016/s0022-3476(99)70088-0. [DOI] [PubMed] [Google Scholar]
- 17.Shah N, Rodriguez M, Louis DS, Lindley K, Milla PJ. Feeding difficulties and foregut dysmotility in Noonan’s syndrome. Arch Dis Child. 1999;81:28–31. doi: 10.1136/adc.81.1.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cesarini L, Alfieri P, Pantaleoni F, Vasta I, Cerutti M, Petrangeli V, Mariotti P, Leoni C, Ricci D, Vicari S, et al. Cognitive profile of disorders associated with dysregulation of the RAS/MAPK signaling cascade. Am J Med Genet A. 2009;149A:140–146. doi: 10.1002/ajmg.a.32488. [DOI] [PubMed] [Google Scholar]
- 19.Pierpont EI, Pierpont ME, Mendelsohn NJ, Roberts AE, Tworog-Dube E, Seidenberg MS. Genotype differences in cognitive functioning in Noonan syndrome. Genes Brain Behav. 2009;8:275–282. doi: 10.1111/j.1601-183X.2008.00469.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bader-Meunier B, Tchernia G, Mielot F, Fontaine JL, Thomas C, Lyonnet S, Lavergne JM, Dommergues JP. Occurrence of myeloproliferative disorder in patients with Noonan syndrome. J Pediatr. 1997;130:885–889. doi: 10.1016/s0022-3476(97)70273-7. [DOI] [PubMed] [Google Scholar]
- 21.Fukuda M, Horibe K, Miyajima Y, Matsumoto K, Nagashima M. Spontaneous remission of juvenile chronic myelomonocytic leukemia in an infant with Noonan syndrome. J Pediatr Hematol Oncol. 1997;19:177–179. doi: 10.1097/00043426-199703000-00019. [DOI] [PubMed] [Google Scholar]
- 22.Choong K, Freedman MH, Chitayat D, Kelly EN, Taylor G, Zipursky A. Juvenile myelomonocytic leukemia and Noonan syndrome. J Pediatr Hematol Oncol. 1999;21:523–527. [PubMed] [Google Scholar]
- 23.Tartaglia M, Niemeyer CM, Fragale A, Song X, Buechner J, Jung A, Hählen K, Hasle H, Licht JD, Gelb BD. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat Genet. 2003;34:148–150. doi: 10.1038/ng1156. [DOI] [PubMed] [Google Scholar]
- 24.Kratz CP, Niemeyer CM, Castleberry RP, Cetin M, Bergsträsser E, Emanuel PD, Hasle H, Kardos G, Klein C, Kojima S, Stary J, Trebo M, Zecca M, Gelb BD, Tartaglia M, Loh ML, et al. The mutational spectrum of PTPN11 in juvenile myelomonocytic leukemia and Noonan syndrome/myeloproliferative disease. Blood. 2005;106:2183–2185. doi: 10.1182/blood-2005-02-0531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nora JJ, Nora AH, Sinha AK, Spangler RD, Lubs HA. The Ullrich-Noonan syndrome (Turner phenotype) Am J Dis Child. 127:48–55. doi: 10.1001/archpedi.1974.02110200050007. [DOI] [PubMed] [Google Scholar]
- 26.Tartaglia M, Zampino G, Gelb BD. Noonan syndrome: clinical aspects and molecular pathogenesis. Mol Syndromol. 2010;1:2–26. doi: 10.1159/000276766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Digilio MC, Conti E, Sarkozy A, Mingarelli R, Dottorini T, Marino B, Pizzuti A, Dallapiccola B. Grouping of Multiple-Lentigines/LEOPARD and Noonan Syndromes on the PTPN11 Gene. Am J Hum Genet. 2002;71:389–394. doi: 10.1086/341528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Legius E, Schrander-Stumpel C, Schollen E, Pulles-Heintzberger C, Gewillig M, Fryns JP. PTPN11 mutations in LEOPARD syndrome. J Med Genet. 2002;39:571–574. doi: 10.1136/jmg.39.8.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pandit B, Sarkozy A, Pennacchio LA, Carta C, Oishi K, Martinelli S, Pogna EA, Schackwitz W, Ustaszewska A, Landstrom A, et al. Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat Genet. 2007;39:1007–1012. doi: 10.1038/ng2073. [DOI] [PubMed] [Google Scholar]
- 30.Sarkozy A, Carta C, Moretti S, Zampino G, Digilio MC, Pantaleoni F, Scioletti AP, Esposito G, Cordeddu V, Lepri F, et al. Germline BRAF mutations in Noonan, LEOPARD, and cardiofaciocutaneous syndromes: molecular diversity and associated phenotypic spectrum. Hum Mutat. 2009;30:695–702. doi: 10.1002/humu.20955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koudova M, Seemanova E, Zenker M. Novel BRAF mutation in a patient with LEOPARD syndrome and normal intelligence. Eur J Med Genet. 2009;52:337–340. doi: 10.1016/j.ejmg.2009.04.006. [DOI] [PubMed] [Google Scholar]
- 32.Gorlin RJ, Anderson RC, Moller JH. The leopard (multiple lentigines) syndrome revisited. Birth Defects Orig Artic Ser. 1971;7:110–115. [PubMed] [Google Scholar]
- 33.Digilio MC, Sarkozy A, de Zorzi A, Pacileo G, Limongelli G, Mingarelli R, Calabro R, Marino B, Dallapiccola B. LEOPARD syndrome: clinical diagnosis in the first year of life. Am J Med Genet A. 2006;140A:740–674. doi: 10.1002/ajmg.a.31156. [DOI] [PubMed] [Google Scholar]
- 34.Voron DA, Hatfield HH, Kalkhoff MD. Multiple lentigines syndrome: case report and review of the literature. Am J Med. 1976;60:447–456. doi: 10.1016/0002-9343(76)90764-6. [DOI] [PubMed] [Google Scholar]
- 35.Gorlin JR, Cohen MM, Levn LS. Leopard syndrome. In: Gorlin JR, Cohen MM, Levn LS, editors. Syndromes of the head and neck. Oxford University Press; New York: 1990. pp. 461–464. [Google Scholar]
- 36.Somerville J, Bonham-Carter RE. The heart in lentiginosis. Br Heart J. 1972;34:58–66. doi: 10.1136/hrt.34.1.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sarkozy A, Digilio MC, Zampino G, Dallapiccola B, Tartaglia M, Gelb BD. LEOPARD Syndrome: Clinical Aspects and Molecular Pathogenesis. Noonan Syndrome and Related Disorders. In: Zenker M, editor. Monogr Hum Genet. Vol. 17. Karger; Basel: 2009. pp. 55–65. [Google Scholar]
- 38.Mazzanti L, Cacciari E, Cicognani A, Bergamaschi R, Scarano E, Forabosco A. Noonan-like syndrome with loose anagen hair: a new syndrome? Am J Med Genet A. 118A:279–286. doi: 10.1002/ajmg.a.10923. [DOI] [PubMed] [Google Scholar]
- 39.Cordeddu V, Di Schiavi E, Pennacchio LA, Ma’ayan A, Sarkozy A, Fodale V, Cecchetti S, Cardinale A, Martin J, Schackwitz W, et al. Mutation of SHOC2 promotes aberrant protein N-myristoylation and causes Noonan-like syndrome with loose anagen hair. Nat Genet. 2009;41:1022–1026. doi: 10.1038/ng.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cantatore-Francis JL, Orlow SJ. Practical guidelines for evaluation of loose anagen hair syndrome. Arch Dermatol. 2009;145:1123–1128. doi: 10.1001/archdermatol.2009.220. [DOI] [PubMed] [Google Scholar]
- 41.Tosti A, Piraccini BM. Loose anagen hair syndrome and loose anagen hair. Arch Dermatol. 2002;138:521–522. doi: 10.1001/archderm.138.4.521. [DOI] [PubMed] [Google Scholar]
- 42.Cohen MM, Jr, Gorlin RJ. Noonan-like/multiple giant cell lesion syndrome. Am J Med Genet. 1991;40:159–166. doi: 10.1002/ajmg.1320400208. [DOI] [PubMed] [Google Scholar]
- 43.Tartaglia M, Kalidas K, Shaw A, Song X, Musat DL, van der Burgt I, Brunner HG, Bertola DR, Crosby A, Ion A, et al. PTPN11 mutations in Noonan syndrome: molecular spectrum, genotype-phenotype correlation, and phenotypic heterogeneity. Am J Hum Genet. 2002;70:1555–1563. doi: 10.1086/340847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sarkozy A, Obregon MG, Conti E, Esposito G, Mingarelli R, Pizzuti A, Dallapiccola B. A novel PTPN11 gene mutation bridges Noonan syndrome, multiple lentigines/LEOPARD syndrome and Noonan-like/multiple giant cell lesion syndrome. Eur J Hum Genet. 2004;12:1069–1072. doi: 10.1038/sj.ejhg.5201290. [DOI] [PubMed] [Google Scholar]
- 45.Jafarov T, Ferimazova N, Reichenberger E. Noonan-like syndrome mutations in PTPN11 in patients diagnosed with cherubism. Clin Genet. 2005;68:190–191. doi: 10.1111/j.1399-0004.2005.00475.x. [DOI] [PubMed] [Google Scholar]
- 46.Lee JC, Tartaglia M, Gelb BD, Fridrich K, Sachs S, Stratakis CA, Muenke M, Robey PG, Collins MT, Slavotinek A. Phenotypic and genotypic characterization of Noonan-like/multiple giant cell lesion syndrome. J Med Genet. 2005;42:e11. doi: 10.1136/jmg.2004.024091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mascheroni E, Digilio MC, Cortis E, Devito R, Sarkozy A, Capolino R, Dallapiccola B, Ugazio AG. Pigmented villonodular synovitis in a patient with Noonan syndrome and SOS1 gene mutation. Am J Med Genet A. 2008;146A:2966–2967. doi: 10.1002/ajmg.a.32538. [DOI] [PubMed] [Google Scholar]
- 48.Beneteau C, Cavé H, Moncla A, Dorison N, Munnich A, Verloes A, Leheup B. SOS1 and PTPN11 mutations in five cases of Noonan syndrome with multiple giant cell lesions. Eur J Hum Genet. 2009;17:1216–1221. doi: 10.1038/ejhg.2009.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hanna N, Parfait B, Talaat IM, Vidaud M, Elsedfy HH. SOS1: a new player in the Noonan-like/multiple giant cell lesion syndrome. Clin Genet. 2009;75:568–571. doi: 10.1111/j.1399-0004.2009.01149.x. [DOI] [PubMed] [Google Scholar]
- 50.Neumann TE, Allanson J, Kavamura I, Kerr B, Neri G, Noonan J, Cordeddu V, Gibson K, Tzschach A, Krüger G, et al. Multiple giant cell lesions in patients with Noonan syndrome and cardio-facio-cutaneous syndrome. Eur J Hum Genet. 2009;17:420–425. doi: 10.1038/ejhg.2008.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reynolds JF, Neri G, Herrmann JP, Blumberg B, Coldwell JG, Miles PV, Opitz JM. New multiple congenital anomalies/mental retardation syndrome with cardio-facio-cutaneous involvement--the CFC syndrome. Am J Med Genet. 1986;25:413–427. doi: 10.1002/ajmg.1320250303. [DOI] [PubMed] [Google Scholar]
- 52.Roberts A, Allanson J, Jadico SK, Kavamura MI, Noonan J, Opitz JM, Young T, Neri G. The cardiofaciocutaneous syndrome. J Med Genet. 2006;43:833–842. doi: 10.1136/jmg.2006.042796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fryer AE, Holt PJ, Hughes HE. The cardio-facio-cutaneous (CFC) syndrome and Noonan syndrome: Are they the same? Am J Med Genet. 1991;38:548–551. doi: 10.1002/ajmg.1320380410. [DOI] [PubMed] [Google Scholar]
- 54.Neri G, Zollino M, Reynolds JF. The Noonan-CFC controversy. Am J Med Genet. 1991;39:367–370. doi: 10.1002/ajmg.1320390323. [DOI] [PubMed] [Google Scholar]
- 55.Wieczorek D, Majewski F, Gillessen-Kaesbach G. Cardio-facio-cutaneous (CFC) syndrome-a distinct entity? Report of three patients demonstrating the diagnostic difficulties in delineation of CFC syndrome. Clin Genet. 1997;52:37–46. doi: 10.1111/j.1399-0004.1997.tb02512.x. [DOI] [PubMed] [Google Scholar]
- 56.Niihori T, Aoki Y, Narumi Y, Neri G, Cavé H, Verloes A, Okamoto N, Hennekam RC, Gillessen-Kaesbach G, Wieczorek D, et al. Germline KRAS and BRAF mutations in cardio-facio-cutaneous syndrome. Nat Genet. 2006;38:294–296. doi: 10.1038/ng1749. [DOI] [PubMed] [Google Scholar]
- 57.Rodriguez-Viciana P, Tetsu O, Tidyman WE, Estep AL, Conger BA, Cruz MS, McCormick F, Rauen KA. Germline mutations in genes within the MAPK pathway cause cardio-facio-cutaneous syndrome. Science. 311:1287–1290. doi: 10.1126/science.1124642. [DOI] [PubMed] [Google Scholar]
- 58.Narumi Y, Aoki Y, Niihori T, Neri G, Cavé H, Verloes A, Nava C, Kavamura MI, Okamoto N, Kurosawa K, et al. Molecular and clinical characterization of cardio-facio-cutaneous (CFC) syndrome: overlapping clinical manifestations with Costello syndrome. Am J Med Genet A. 2006;143A:799–807. doi: 10.1002/ajmg.a.31658. [DOI] [PubMed] [Google Scholar]
- 59.Nava C, Hanna N, Michot C, Pereira S, Pouvreau N, Niihori T, Aoki Y, Matsubara Y, Arveiler B, Lacombe D, et al. Cardio-facio-cutaneous and Noonan syndromes due to mutations in the RAS/MAPK signalling pathway: genotype phenotype relationships and overlap with Costello syndrome. J Med Genet. 2007;44:763–771. doi: 10.1136/jmg.2007.050450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schulz AL, Albrecht B, Arici C, van der Burgt I, Buske A, Gillessen-Kaesbach G, Heller R, Horn D, Hübner CA, Korenke GC, et al. Mutation and phenotypic spectrum in patients with cardio-facio-cutaneous and Costello syndrome. Clin Genet. 2008;73:62–70. doi: 10.1111/j.1399-0004.2007.00931.x. [DOI] [PubMed] [Google Scholar]
- 61.Costello JM. A new syndrome. NZ Med J. 1971;74:397. [Google Scholar]
- 62.Costello JM. A new syndrome: mental subnormality and nasal papillomata. Aust Paediatr J. 1977;13:114–118. doi: 10.1111/j.1440-1754.1977.tb01135.x. [DOI] [PubMed] [Google Scholar]
- 63.Zampino G, Mastroiacovo P, Ricci R, Zollino M, Segni G, Martini-Neri ME, Neri G. Costello syndrome: further clinical delineation, natural history, genetic definition, and nosology. Am J Med Genet. 1993;47:176–183. doi: 10.1002/ajmg.1320470210. [DOI] [PubMed] [Google Scholar]
- 64.Hennekam RC. Costello syndrome: an overview. Am J Med Genet C Semin Med Genet. 2003;117C:42–48. doi: 10.1002/ajmg.c.10019. [DOI] [PubMed] [Google Scholar]
- 65.Gripp KW. Tumor predisposition in Costello syndrome. Am J Med Genet C Semin Med Genet. 2005;137C:72–77. doi: 10.1002/ajmg.c.30065. [DOI] [PubMed] [Google Scholar]
- 66.Aoki Y, Niihori T, Kawame H, Kurosawa K, Ohashi H, Tanaka Y, Filocamo M, Kato K, Suzuki Y, Kure S, Matsubara Y. Germline mutations in HRAS proto-oncogene cause Costello syndrome. Nat Genet. 37:1038–1040. doi: 10.1038/ng1641. [DOI] [PubMed] [Google Scholar]
- 67.Gripp KW, Lin AE, Stabley DL, Nicholson L, Scott CI, Jr, Doyle D, Aoki Y, Matsubara Y, Zackai EH, Lapunzina P, et al. HRAS mutation analysis in Costello syndrome: genotype and phenotype correlation. Am J Med Genet A. 2006;140A:1–7. doi: 10.1002/ajmg.a.31047. [DOI] [PubMed] [Google Scholar]
- 68.Estep AL, Tidyman WE, Teitell MA, Cotter PD, Rauen RA. HRAS mutations in Costello syndrome: detection of constitutional activating mutations in codon 12 and 13 and loss of wild-type allele in malignancy. Am J Med Genet A. 2006;140A:8–16. doi: 10.1002/ajmg.a.31078. [DOI] [PubMed] [Google Scholar]
- 69.Kerr B, Delrue MA, Sigaudy S, Perveen R, Marche M, Burgelin I, Stef M, Tang B, Eden OB, O’Sullivan J, et al. Genotype-phenotype correlation in Costello syndrome: HRAS mutation analysis in 43 cases. J Med Genet. 2006;43:401–405. doi: 10.1136/jmg.2005.040352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zampino G, Pantaleoni F, Carta C, Cobellis G, Vasta I, Neri C, Pogna EA, De Feo E, Delogu A, Sarkozy A, et al. Diversity, parental germline origin, and phenotypic spectrum of de novo HRAS missense changes in Costello syndrome. Hum Mutat. 2007;28:265–272. doi: 10.1002/humu.20431. [DOI] [PubMed] [Google Scholar]
- 71.Friedman JM, Birch PH. Type 1 neurofibromatosis: a descriptive analysis of the disorder in 1,728 patients. Am J Med Genet. 1997;70:138–143. doi: 10.1002/(sici)1096-8628(19970516)70:2<138::aid-ajmg7>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 72.Williams VC, Lucas J, Babcock MA, Gutmann DH, Korf B, Maria BL. Neurofibromatosis type 1 revisited. Pediatrics. 2009;123:124–33. doi: 10.1542/peds.2007-3204. [DOI] [PubMed] [Google Scholar]
- 73.National Institutes of Health Consensus Development Conference Statement. Neurofibromatosis. Arch Neurol. 1988;45:575–578. [PubMed] [Google Scholar]
- 74.Shen MH, Harper PS, Upadhyaya M. Molecular genetics of neurofibromatosis type 1 (NF1) J Med Genet. 1996;33:2–17. doi: 10.1136/jmg.33.1.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fernel RE. Neurofibromatosis 1. Eur J Hum Genet. 2007;15:131–138. doi: 10.1038/sj.ejhg.5201676. [DOI] [PubMed] [Google Scholar]
- 76.Lee M-J, Stephenson DA. Recent developments in neurofibromatosis type 1. Curr Opin Neurol. 2007;20:135–141. doi: 10.1097/WCO.0b013e3280895da8. [DOI] [PubMed] [Google Scholar]
- 77.Allanson JE, Hall JG, Van Allen MI. Noonan phenotype associated with neurofibromatosis. Am J Med Genet. 1985;21:457–462. doi: 10.1002/ajmg.1320210307. [DOI] [PubMed] [Google Scholar]
- 78.Opitz JM, Weaver DD. The neurofibromatosis-Noonan syndrome. Am J Med Genet. 1985;21:477–490. doi: 10.1002/ajmg.1320210310. [DOI] [PubMed] [Google Scholar]
- 79.Carey JC. Neurofibromatosis-Noonan syndrome. Am J Med Genet. 75:263–264. doi: 10.1002/(sici)1096-8628(19980123)75:3<263::aid-ajmg7>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 80.Baralle D, Mattocks C, Kalidas K, Elmslie F, Whittaker J, Lees M, Ragge N, Patton MA, Winter RM, ffrench-Constant C. Different mutations in the NF1 gene are associated with neurofibromatosis-Noonan syndrome (NFNS) Am J Med Genet. 2003;119A:1–8. doi: 10.1002/ajmg.a.20023. [DOI] [PubMed] [Google Scholar]
- 81.De Luca A, Bottillo I, Sarkozy A, Carta C, Neri C, Bellacchio E, Schirinzi A, Conti E, Zampino G, Battaglia A, et al. NF1 gene mutations represent the major molecular event underlying neurofibromatosis-Noonan syndrome. Am J Hum Genet. 2005;77:1092–1101. doi: 10.1086/498454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hüffmeier U, Zenker M, Hoyer J, Fahsold R, Rauch A. A variable combination of features of Noonan syndrome and neurofibromatosis type I are caused by mutations in the NF1 gene. Am J Med Genet A. 2006;140A:2749–2756. doi: 10.1002/ajmg.a.31547. [DOI] [PubMed] [Google Scholar]
- 83.Bertola DR, Pereira AC, Passetti F, de Oliveira PS, Messiaen L, Gelb BD, Kim CA, Krieger JE. Neurofibromatosis-Noonan syndrome: Molecular evidence of the concurrence of both disorders in a patient. Am J Med Genet A. 2005;136:242–245. doi: 10.1002/ajmg.a.30813. [DOI] [PubMed] [Google Scholar]
- 84.Thiel C, Wilken M, Zenker M, Sticht H, Fahsold R, Gusek-Schneider GC, Rauch A. Independent NF1 and PTPN11 mutations in a family with neurofibromatosis-Noonan syndrome. Am J Med Genet A. 149A:1263–1267. doi: 10.1002/ajmg.a.32837. [DOI] [PubMed] [Google Scholar]
- 85.Nyström AM, Ekvall S, Allanson J, Edeby C, Elinder M, Holmström G, Bondeson ML, Annerén G. Noonan syndrome and neurofibromatosis type I in a family with a novel mutation in NF1. Clin Genet. 2009;76:524–534. doi: 10.1111/j.1399-0004.2009.01233.x. [DOI] [PubMed] [Google Scholar]
- 86.Watson GH. Pulmonary stenosis, cafe-au-lait spots, and dull intelligence. Arch Dis Child. 1967;42:303–307. doi: 10.1136/adc.42.223.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Partington MW, Burggraf GW, Fay JE, Frontini E. Pulmonary stenosis, cafe au lait spots and dull intelligence: the Watson syndrome revisited. Proc Greenwood Genet Center. 1985;4:105. [Google Scholar]
- 88.Allanson JE, Upadhyaya M, Watson GH, Partington M, Harper P, Huson S. Molecular linkage analysis of Watson syndrome. Am J Hum Genet. 1989;45(suppl):A38. [Google Scholar]
- 89.Allanson JE, Upadhyaya M, Watson GH, Partington M, MacKenzie A, Lahey D, MacLeod H, Sarfarazi M, Broadhead W, Harper PS, Huson SM. Watson syndrome: is it a subtype of type 1 neurofibromatosis? J Med Genet. 1991;28:752–756. doi: 10.1136/jmg.28.11.752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Upadhyaya M, Sarfarazi M, Huson S, Broadhead W, Allanson J, Fryer A, Harper PS. Linkage of Watson’s syndrome to chromosome 17 markers. J Med Genet. 1990;27:209. [Google Scholar]
- 91.Upadhyaya M, Shen M, Cherryson A, Farnham J, Maynard J, Huson SM, Harper PS. Analysis of mutations at the neurofibromatosis 1 (NF1) locus. Hum Mol Genet. 1992;1:735–740. doi: 10.1093/hmg/1.9.735. [DOI] [PubMed] [Google Scholar]
- 92.Tassabehji M, Strachan T, Sharland M, Colley A, Donnai D, Harris R, Thakker N. Tandem duplication within a neurofibromatosis type I (NF1) gene exon in a family with features of Watson syndrome and Noonan syndrome. Am J Hum Genet. 1993;53:90–95. [PMC free article] [PubMed] [Google Scholar]
- 93.Brems H, Chmara M, Sahbatou M, Denayer E, Taniguchi K, Kato R, Somers R, Messiaen L, De Schepper S, Fryns JP, et al. Germline loss-of-function mutations in SPRED1 cause a neurofibromatosis 1-like phenotype. Nat Genet. 2007;39:1120–1126. doi: 10.1038/ng2113. [DOI] [PubMed] [Google Scholar]
- 94.Pasmant E, Sabbagh A, Hanna N, Masliah-Planchon J, Jolly E, Goussard P, Ballerini P, Cartault F, Barbarot S, Landman-Parker J, et al. SPRED1 germline mutations causes a neurofibromatosis type 1 overlapping phenotype. J Med Genet. 2009;46:425–430. doi: 10.1136/jmg.2008.065243. [DOI] [PubMed] [Google Scholar]
- 95.Spurlock G, Bennett E, Chuzhanova N, Thomas N, Jim HP, Side L, Davies S, Haan E, Kerr B, Huson SM, Upadhyaya M. SPRED1 mutations (Legius syndrome): another clinically useful genotype for dissecting the neurofibromatosis type 1 phenotype. J Med Genet. 2009;46:431–437. doi: 10.1136/jmg.2008.065474. [DOI] [PubMed] [Google Scholar]
- 96.Messiaen L, Yao S, Brems H, Callens T, Sathienkijkanchai A, Denayer E, Spencer E, Arn P, Babovic-Vuksanovic D, Bay C, et al. Clinical and mutational spectrum of neurofibromatosis type 1-like syndrome. JAMA. 2009;302:2111–2118. doi: 10.1001/jama.2009.1663. [DOI] [PubMed] [Google Scholar]
- 97.Neel BG, Gu H, Pao L. The ‘Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem Sci. 2003;28:284–293. doi: 10.1016/S0968-0004(03)00091-4. [DOI] [PubMed] [Google Scholar]
- 98.Tartaglia M, Niemeyer CM, Shannon KM, Loh ML. SHP-2 and myeloid malignancies. Curr Opin Hematol. 2004;11:44–50. doi: 10.1097/00062752-200401000-00007. [DOI] [PubMed] [Google Scholar]
- 99.Dance M, Montagner A, Salles JP, Yart A, Raynal P. The molecular functions of Shp2 in the Ras/Mitogen-activated protein kinase (ERK1/2) pathway. Cell Signal. 2008;20:453–459. doi: 10.1016/j.cellsig.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 100.Xu D, Qu CK. Protein tyrosine phosphatases in the JAK/STAT pathway. Front Biosci. 2008;13:4925–4932. doi: 10.2741/3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hof P, Pluskey S, Dhe-Paganon S, Eck MJ, Shoelson SE. Crystal structure of the tyrosine phosphatase shp-2. Cell. 1998;92:441–450. doi: 10.1016/s0092-8674(00)80938-1. [DOI] [PubMed] [Google Scholar]
- 102.Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, Kremer H, van der Burgt I, Crosby AH, Ion A, Jeffery S, et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet. 2001;29:465–468. doi: 10.1038/ng772. [DOI] [PubMed] [Google Scholar]
- 103.Zenker M, Buheitel G, Rauch R, Koenig R, Bosse K, Kress W, Tietze HU, Doerr HG, Hofbeck M, Singer H, et al. Genotype-phenotype correlations in Noonan syndrome. J Pediatr. 2004;144:368–374. doi: 10.1016/j.jpeds.2003.11.032. [DOI] [PubMed] [Google Scholar]
- 104.Keilhack H, David FS, McGregor M, Cantley LC, Neel BG. Diverse biochemical properties of shp2 mutants. Implications for disease phenotypes. J Biol Chem. 2005;280:30984–30993. doi: 10.1074/jbc.M504699200. [DOI] [PubMed] [Google Scholar]
- 105.Tartaglia M, Martinelli S, Stella L, Bocchinfuso G, Flex E, Cordeddu V, Zampino G, van der Burgt I, Palleschi A, Petrucci TC, et al. Diversity and functional consequences of germline and somatic PTPN11 mutations in human disease. Am J Hum Genet. 2006;78:279–290. doi: 10.1086/499925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Martinelli S, Torreri P, Tinti M, Stella L, Bocchinfuso G, Flex E, Grottesi A, Ceccarini M, Palleschi A, Cesareni G, et al. Diverse driving forces underlie the invariant occurrence of the T42A, E139D, I282V and T468M SHP2 amino acid substitutions causing Noonan and LEOPARD syndromes. Hum Mol Genet. 2008;17:2018–2029. doi: 10.1093/hmg/ddn099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hanna N, Montagner A, Lee WH, Miteva M, Vidal M, Vidaud M, Parfait B, Raynal P. Reduced phosphatase activity of SHP-2 in LEOPARD syndrome: consequences for PI3K binding on Gab1. FEBS Lett. 580:2477–2482. doi: 10.1016/j.febslet.2006.03.088. [DOI] [PubMed] [Google Scholar]
- 108.Kontaridis MI, Swanson KD, David FS, Barford D, Neel BG. Ptpn11 (shp2) mutations in LEOPARD syndrome have dominant negative, not activating, effects. J Biol Chem. 281:6785–6792. doi: 10.1074/jbc.M513068200. [DOI] [PubMed] [Google Scholar]
- 109.Oishi K, Zhang H, Gault WJ, Wang CJ, Tan CC, Kim IK, Ying H, Rahman T, Pica N, Tartaglia M, Mlodzik M, Gelb BD. Phosphatase-defective LEOPARD syndrome mutations in PTPN11 gene have gain-of-function effects during Drosophila development. Hum Mol Genet. 18:193–201. doi: 10.1093/hmg/ddn336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Edouard T, Combier JP, Nédélec A, Bel-Vialar S, Métrich M, Conte-Auriol F, Lyonnet S, Parfait B, Tauber M, Salles JP, et al. Functional effects of PTPN11 (SHP2) mutations causing LEOPARD syndrome on epidermal growth factor-induced phosphoinositide 3-kinase/AKT/glycogen synthase kinase 3beta signaling. Mol Cell Biol. 2010;30:2498–2507. doi: 10.1128/MCB.00646-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Niemeyer CM, Kratz CP. Paediatric myelodysplastic syndromes and juvenile myelomonocytic leukaemia: molecular classification and treatment options. Br J Haematol. 140:610–624. doi: 10.1111/j.1365-2141.2007.06958.x. [DOI] [PubMed] [Google Scholar]
- 112.Loh ML, Vattikuti S, Schubbert S, Reynolds MG, Carlson E, Lieuw KH, Cheng JW, Lee CM, Stokoe D, Bonifas JM, et al. Mutations in PTPN11 implicate the SHP-2 phosphatase in leukemogenesis. Blood. 2004;103:2325–2331. doi: 10.1182/blood-2003-09-3287. [DOI] [PubMed] [Google Scholar]
- 113.Tartaglia M, Martinelli S, Cazzaniga G, Cordeddu V, Iavarone I, Spinelli M, Palmi C, Carta C, Pession A, Aricò M, et al. Genetic evidence for lineage- and differentiation stage-related contribution of somatic PTPN11 mutations to leukemogenesis in childhood acute leukemia. Blood. 2004;104:307–313. doi: 10.1182/blood-2003-11-3876. [DOI] [PubMed] [Google Scholar]
- 114.Tartaglia M, Martinelli S, Iavarone I, Cazzaniga G, Spinelli M, Giarin E, Petrangeli V, Carta C, Masetti R, Arico M, et al. Somatic PTPN11 mutations in childhood acute myeloid leukaemia. Br J Haematol. 2005;129:333–339. doi: 10.1111/j.1365-2141.2005.05457.x. [DOI] [PubMed] [Google Scholar]
- 115.Loh ML, Reynolds MG, Vattikuti S, Gerbing RB, Alonzo TA, Carlson E, Cheng JW, Lee CM, Lange BJ, Meshinchi S. PTPN11 mutations in pediatric patients with acute myeloid leukemia: Results from the children’s cancer group. Leukemia. 2004;18:1831–1834. doi: 10.1038/sj.leu.2403492. [DOI] [PubMed] [Google Scholar]
- 116.Chan RJ, Leedy MB, Munugalavadla V, Voorhorst CS, Li Y, Yu M, Kapur R. Human somatic PTPN11 mutations induce hematopoietic-cell hypersensitivity to granulocyte-macrophage colony-stimulating factor. Blood. 2005;105:3737–3742. doi: 10.1182/blood-2004-10-4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Schubbert S, Lieuw K, Rowe SL, Lee CM, Li X, Loh ML, Clapp DW, Shannon KM. Functional analysis of leukemia-associated PTPN11 mutations in primary hematopoietic cells. Blood. 2005;106:311–317. doi: 10.1182/blood-2004-11-4207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Shchelochkov OA, Patel A, Weissenberger GM, Chinault AC, Wiszniewska J, Fernandes PH, Eng C, Kukolich MK, Sutton VR. Duplication of chromosome band 12q24.11q24.23 results in apparent Noonan syndrome. Am J Med Genet A. 2008;146A:1042–1048. doi: 10.1002/ajmg.a.32215. [DOI] [PubMed] [Google Scholar]
- 119.Graham JM, Jr, Kramer N, Bejjani BA, Thiel CT, Carta C, Neri G, Tartaglia M, Zenker M. Genomic duplication of PTPN11 is an uncommon cause of Noonan syndrome. Am J Med Genet A. 2009;149A:2122–2128. doi: 10.1002/ajmg.a.32992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Quilliam LA, Khosravi-Far R, Huff SY, Der CJ. Guanine nucleotide exchange factors: activators of the Ras superfamily of proteins. Bioessays. 1995;17:395–404. doi: 10.1002/bies.950170507. [DOI] [PubMed] [Google Scholar]
- 121.Quilliam LA, Rebhun JF, Castro AF. A growing family of guanine nucleotide exchange factors is responsible for activation of Ras-family GTPases. Prog Nucleic Acid Res Mol Biol. 71:391–444. doi: 10.1016/s0079-6603(02)71047-7. [DOI] [PubMed] [Google Scholar]
- 122.Nimnual A, Bar-Sagi D. The two hats of SOS. Sci STKE. 2002;145:PE36. doi: 10.1126/stke.2002.145.pe36. [DOI] [PubMed] [Google Scholar]
- 123.Roberts AE, Araki T, Swanson KD, Montgomery KT, Schiripo TA, Joshi VA, Li L, Yassin Y, Tamburino AM, Neel BG, Kucherlapati RS. Germline gain-of-function mutations in SOS1 cause Noonan syndrome. Nat Genet. 2007;39:70–74. doi: 10.1038/ng1926. [DOI] [PubMed] [Google Scholar]
- 124.Tartaglia M, Pennacchio LA, Zhao C, Yadav KK, Fodale V, Sarkozy A, Pandit B, Oishi K, Martinelli S, Schackwitz W. Gain-of-function SOS1 mutations cause a distinctive form of Noonan syndrome. Nat Genet. 2007;39:75–79. doi: 10.1038/ng1939. [DOI] [PubMed] [Google Scholar]
- 125.Chardin P, Camonis JH, Gale NW, van Aelst L, Schlessinger J, Wigler MH, Bar-Sagi D. Human Sos1: a guanine nucleotide exchange factor for Ras that binds to GRB2. Science. 260:1338–1343. doi: 10.1126/science.8493579. [DOI] [PubMed] [Google Scholar]
- 126.Nimnual AS, Yatsula BA, Bar-Sagi D. Coupling of Ras and Rac guanosine triphosphatases through the Ras exchanger Sos. Science. 1998;279:560–563. doi: 10.1126/science.279.5350.560. [DOI] [PubMed] [Google Scholar]
- 127.Chen RH, Corbalan-Garcia S, Bar-Sagi D. The role of the PH domain in the signal-dependent membrane targeting of Sos. EMBO J. 16:1351–1359. doi: 10.1093/emboj/16.6.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sondermann H, Nagar B, Bar-Sagi D, Kuriyan J. Computational docking and solution x-ray scattering predict a membrane-interacting role for the histone domain of the Ras activator Son of sevenless. Proc Natl Acad Sci USA. 2005;102:16632–16637. doi: 10.1073/pnas.0508315102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zenker M, Horn D, Wieczorek D, Allanson J, Pauli S, van der Burgt I, Doerr HG, Gaspar H, Hofbeck M, Gillessen-Kaesbach G, et al. SOS1 is the second most common Noonan gene but plays no major role in cardio-facio-cutaneous syndrome. J Med Genet. 2007;44:651–656. doi: 10.1136/jmg.2007.051276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Narumi Y, Aoki Y, Niihori T, Neri G, Cavé H, Verloes A, Nava C, Kavamura MI, Okamoto N, Kurosawa K, et al. Molecular and clinical characterization of cardio-facio-cutaneous (CFC) syndrome: overlapping clinical manifestations with Costello syndrome. Am J Med Genet A. 2007;143A:799–807. doi: 10.1002/ajmg.a.31658. [DOI] [PubMed] [Google Scholar]
- 131.Nyström AM, Ekvall S, Berglund E, Björkqvist M, Braathen G, Duchen K, Enell H, Holmberg E, Holmlund U, Olsson-Engman M, et al. Noonan and cardio-facio-cutaneous syndromes: two clinically and genetically overlapping disorders. J Med Genet. 2008;45:500–506. doi: 10.1136/jmg.2008.057653. [DOI] [PubMed] [Google Scholar]
- 132.Margarit SM, Sondermann H, Hall BE, Nagar B, Hoelz A, Pirruccello M, Bar-Sagi D, Kuriyan J. Structural evidence for feedback activation by Ras.GTP of the Ras-specific nucleotide exchange factor SOS. Cell. 2003;112:685–695. doi: 10.1016/s0092-8674(03)00149-1. [DOI] [PubMed] [Google Scholar]
- 133.Sondermann H, Soisson SM, Boykevisch S, Yang SS, Bar-Sagi D, Kuriyan J. Structural analysis of autoinhibition in the Ras activator Son of sevenless. Cell. 2004;119:393–405. doi: 10.1016/j.cell.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 134.Kratz CP, Niemeyer CM, Thomas C, Bauhuber S, Matejas V, Bergsträsser E, Flotho C, Flores NJ, Haas O, Hasle H, et al. Mutation analysis of Son of Sevenless in juvenile myelomonocytic leukemia. Leukemia. 2007;21:1108–1109. doi: 10.1038/sj.leu.2404620. [DOI] [PubMed] [Google Scholar]
- 135.Swanson KD, Winter JM, Reis M, Bentires-Alj M, Greulich H, Grewal R, Hruban RH, Yeo CJ, Yassin Y, Iartchouk O, et al. SOS1 mutations are rare in human malignancies: implications for Noonan Syndrome patients. Genes Chromosomes Cancer. 2008;47:253–259. doi: 10.1002/gcc.20527. [DOI] [PubMed] [Google Scholar]
- 136.Wennerberg K, Rossman KL, Der CJ. The Ras superfamily at a glance. J Cell Sci. 2005;118:843–846. doi: 10.1242/jcs.01660. [DOI] [PubMed] [Google Scholar]
- 137.Barbacid M. Ras genes. Annu Rev Biochem. 1987;56:779–827. doi: 10.1146/annurev.bi.56.070187.004023. [DOI] [PubMed] [Google Scholar]
- 138.Adjei AA. Blocking oncogenic Ras signaling for cancer therapy. J Natl Cancer Inst. 2001;93:1062–1074. doi: 10.1093/jnci/93.14.1062. [DOI] [PubMed] [Google Scholar]
- 139.Clanton DJ, Hattori S, Shih TY. Mutations of the Ras gene product p21 that abolish guanine nucleotide binding. Proc Natl Acad Sci USA. 1986;83:5076–5080. doi: 10.1073/pnas.83.14.5076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Der CJ, Pan BT, Cooper GM. RasH mutants deficient in GTP binding. Mol Cell Biol. 6:3291–3294. doi: 10.1128/mcb.6.9.3291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Walter M, Clark SG, Levinson AD. The oncogenic activation of human p21ras by a novel mechanism. Science. 1986;233:649–652. doi: 10.1126/science.3487832. [DOI] [PubMed] [Google Scholar]
- 142.Denayer E, Parret A, Chmara M, Schubbert S, Vogels A, Devriendt K, Frijns JP, Rybin V, de Ravel TJ, Shannon K, et al. Mutation analysis in Costello syndrome: functional and structural characterization of the HRAS p.Lys117Arg mutation. Hum Mutat. 2008;29:232–239. doi: 10.1002/humu.20616. [DOI] [PubMed] [Google Scholar]
- 143.Gremer L, De Luca A, Merbitz-Zahradnik T, Dallapiccola B, Morlot S, Tartaglia M, Kutsche K, Ahmadian MR, Rosenberger G. Duplication of Glu37 in the switch I region of HRAS impairs effector/GAP binding and underlies Costello syndrome by promoting enhanced growth factor-dependent MAPK and AKT activation. Hum Mol Genet. 2010;19:790–802. doi: 10.1093/hmg/ddp548. [DOI] [PubMed] [Google Scholar]
- 144.Seeburg PH, Colby WW, Capon DJ, Goeddel DV, Levinson AD. Biological properties of human c-Ha-ras1 genes mutated at codon 12. Nature. 1984;312:71–75. doi: 10.1038/312071a0. [DOI] [PubMed] [Google Scholar]
- 145.van der Burgt I, Kupsky W, Stassou S, Nadroo A, Barroso C, Diem A, Kratz CP, Dvorsky R, Ahmadian MR, Zenker M. Myopathy caused by HRAS germline mutations: implications for disturbed myogenic differentiation in the presence of constitutive HRas activation. J Med Genet. 44:459–462. doi: 10.1136/jmg.2007.049270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Carta C, Pantaleoni F, Bocchinfuso G, Stella L, Vasta I, Sarkozy A, Digilio C, Palleschi A. Germline missense mutations affecting KRAS isoform b are associated with a severe noonan syndrome phenotype. Am J Hum Genet. 79:129–135. doi: 10.1086/504394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Schubbert S, Zenker M, Rowe SL, Böll S, Klein C, Bollag G, van der Burgt I, Musante L, Kalscheuer V, Wehner LE, et al. Germline KRAS mutations cause Noonan syndrome. Nat Genet. 2006;38:331–336. doi: 10.1038/ng1748. [DOI] [PubMed] [Google Scholar]
- 148.Zenker M, Lehmann K, Schulz AL, Barth H, Hansmann D, Koenig R, Korinthenberg R, Kreiss-Nachtsheim M, Meinecke P, Morlot S, et al. Expansion of the genotypic and phenotypic spectrum in patients with KRAS germline mutations. J Med Genet. 2007;44:131–135. doi: 10.1136/jmg.2006.046300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kratz CP, Zampino G, Kriek M, Kant SG, Leoni C, Pantaleoni F, Oudesluys-Murphy AM, Di Rocco C, Kloska SP, Tartaglia M, Zenker M. Craniosynostosis in patients with Noonan syndrome caused by germline KRAS mutations. Am J Med Genet. 149A:1036–1040. doi: 10.1002/ajmg.a.32786. [DOI] [PubMed] [Google Scholar]
- 150.Friday BB, Adjei AA. K-ras as a target for cancer therapy. Biochim Biophys Acta. 2005;1756:127–144. doi: 10.1016/j.bbcan.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 151.Johnson L, Greenbaum D, Cichowski K, Mercer K, Murphy E, Schmitt E, Bronson RT, Umanoff H, Edelmann W, Kucherlapati R, Jacks T. K-ras is an essential gene in the mouse with partial functional overlap with N-ras. Genes Dev. 1997;11:2468–2481. doi: 10.1101/gad.11.19.2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Koera K, Nakamura K, Nakao K, Miyoshi J, Toyoshima K, Hatta T, Otani H, Aiba A, Katsuki M. K-ras is essential for the development of the mouse embryo. Oncogene. 1997;15:1151–1159. doi: 10.1038/sj.onc.1201284. [DOI] [PubMed] [Google Scholar]
- 153.Plowman SJ, Williamson DJ, O’Sullivan MJ, Doig J, Ritchie AM, Harrison DJ, Melton DW, Arends MJ, Hooper ML, Patek CE. While K-ras is essential for mouse development, expression of the K-ras 4a splice variant is dispensable. Mol Cell Biol. 2003;23:9245–9250. doi: 10.1128/MCB.23.24.9245-9250.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Schubbert S, Bollag G, Lyubynska N, Nguyen H, Kratz CP, Zenker M, Niemeyer CM, Molven A, Shannon K. Biochemical and functional characterization of germ line KRAS mutations. Mol Cell Biol. 2007;27:7765–7770. doi: 10.1128/MCB.00965-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Cirstea IC, Kutsche K, Dvorsky R, Gremer L, Carta C, Horn D, Roberts AE, Lepri F, Merbitz-Zahradnik T, König R, et al. A restricted spectrum of NRAS mutations causes Noonan syndrome. Nat Genet. 2010;42:27–29. doi: 10.1038/ng.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Trovó-Marqui AB, Tajara EH. Neurofibromin: a general outlook. Clin Genet. 2006;70:1–13. doi: 10.1111/j.1399-0004.2006.00639.x. [DOI] [PubMed] [Google Scholar]
- 157.The I, Hannigan GE, Cowley GS, Reginald S, Zhong Y, Gusella JF, Hariharan IK, Bernards A. Rescue of a Drosophila NF1 mutant phenotype by protein kinase A. Science. 1997;276:791–794. doi: 10.1126/science.276.5313.791. [DOI] [PubMed] [Google Scholar]
- 158.Xu H, Gutmann DH. Mutations in the GAP-related domain impair the ability of neurofibromin to associate with microtubules. Brain Res. 1997;759:149–152. doi: 10.1016/s0006-8993(97)00328-4. [DOI] [PubMed] [Google Scholar]
- 159.Tong J, Hannan F, Zhu Y, Bernards A, Zhong Y. Neurofibromin regulates G protein-stimulated adenylyl cyclase activity. Nat Neurosci. 2002;5:95–96. doi: 10.1038/nn792. [DOI] [PubMed] [Google Scholar]
- 160.Dasgupta B, Dugan LL, Gutmann DH. The neurofibromatosis 1 gene product neurofibromin regulates pituitary adenylate cyclase-activating polypeptide-mediated signaling in astrocytes. J Neurosci. 2003;23:8949–8954. doi: 10.1523/JNEUROSCI.23-26-08949.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Vandenbroucke I, Van Oostveldt P, Coene E, De Paepe A, Messiaen L. Neurofibromin is actively transported to the nucleus. FEBS Lett. 2004;560:98–102. doi: 10.1016/S0014-5793(04)00078-X. [DOI] [PubMed] [Google Scholar]
- 162.Dorschner MO, Sybert VP, Weaver M, Pletcher BA, Stephens K. NF1 microdeletion breakpoints are clustered at flanking repetitive sequences. Hum Mol Genet. 2000;9:35–46. doi: 10.1093/hmg/9.1.35. [DOI] [PubMed] [Google Scholar]
- 163.Riva P, Corrado L, Natacci F, Castorina P, Wu B-L, Schneider GH, Clementi M, Tenconi R, Korf BR, Larizza L. NF1 microdeletion syndrome: refined FISH characterization of sporadic and familial deletions with locus-specific probes. Am J Hum Genet. 2000;66:100–109. doi: 10.1086/302709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Jenne DE, Tinschert S, Reimann H, Lasinger W, Thiel G, Hameister H, Kehrer-Sawatzki H. Molecular characterization and gene content of breakpoint boundaries in patients with neurofibromatosis type 1 with 17q11.2 microdeletions. Am J Hum Genet. 2001;69:516–527. doi: 10.1086/323043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Sawada S, Florell S, Purandare SM, Ota M, Stephens K, Viskochil D. Identification of NF1 mutations in both alleles of a dermal neurofibroma. Nat Genet. 1996;14:110–112. doi: 10.1038/ng0996-110. [DOI] [PubMed] [Google Scholar]
- 166.Serra E, Puig S, Otero D, Gaona A, Kruyer H, Ars E, Estivill X, Lazaro C. Confirmation of a double-hit model for the NF1 gene in benign neurofibromas. Am J Hum Genet. 1997;61:512–519. doi: 10.1086/515504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Side L, Taylor B, Cayouette M, Conner E, Thompson P, Luce M, Shannon K. Homozygous inactivation of the NF1 gene in bone marrow cells from children with neurofibromatosis type 1 and malignant myeloid disorders. New Eng J Med. 1997;336:1713–1720. doi: 10.1056/NEJM199706123362404. [DOI] [PubMed] [Google Scholar]
- 168.Bundschu K, Walter U, Schuh K. Getting a first clue about SPRED functions. Bioessays. 2007;29:897–907. doi: 10.1002/bies.20632. [DOI] [PubMed] [Google Scholar]
- 169.Wakioka T, Sasaki A, Kato R, Shouda T, Matsumoto A, Miyoshi K, Tsuneoka M, Komiya S, Baron R, Yoshimura A. Spred is a Sprouty-related suppressor of Ras signalling. Nature. 2001;412:647–651. doi: 10.1038/35088082. [DOI] [PubMed] [Google Scholar]
- 170.King JA, Straffon AF, D’Abaco GM, Poon CL, Smith CM, Buchert M, Corcoran NM, Hall NE, Callus BA, Sarcevic B, et al. Distinct requirements for the Sprouty domain for functional activity of Spred proteins. Biochem J. 2005;388:445–454. doi: 10.1042/BJ20041284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.De Schepper S, Maertens O, Callens T, Naeyaert JM, Lambert J, Messiaen L. Somatic mutation analysis in NF1 café au lait spots reveals two NF1 hits in the melanocytes. J Invest Dermatol. 2008;128:1050–1053. doi: 10.1038/sj.jid.5701095. [DOI] [PubMed] [Google Scholar]
- 172.Wellbrock C, Karasarides M, Marais R. The RAF proteins take centre stage. Nat. Rev. Mol Cell Biol. 2004;5:875–885. doi: 10.1038/nrm1498. [DOI] [PubMed] [Google Scholar]
- 173.Schreck R, Rapp UR. Raf kinases: oncogenesis and drug discovery. In J Cancer. 2006;119:2261–2271. doi: 10.1002/ijc.22144. [DOI] [PubMed] [Google Scholar]
- 174.Pritchard CA, Bolin L, Slattery R, Murray R, McMahon M. Post-natal lethality and neurological and gastrointestinal defects in mice with targeted disruption of the A-Raf protein kinase gene. Curr Biol. 1996;6:614–617. doi: 10.1016/s0960-9822(02)00548-1. [DOI] [PubMed] [Google Scholar]
- 175.Wojnowski L, Zimmer AM, Beck TW, Hahn H, Bernal R, Rapp UR, Zimmer A. Endothelial apoptosis in Braf-deficient mice. Nat Genet. 1997;16:293–297. doi: 10.1038/ng0797-293. [DOI] [PubMed] [Google Scholar]
- 176.Mikula M, Schreiber M, Husak Z, Kucerova L, Rüth J, Wieser R, Zatloukal K, Beug H, Wagner EF, Baccarini M. Embryonic lethality and fetal liver apoptosis in mice lacking the c-raf-1 gene. EMBO J. 2001;20:1952–1962. doi: 10.1093/emboj/20.8.1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Razzaque MA, Nishizawa T, Komoike Y, Yagi H, Furutani M, Amo R, Kamisago M, Momma K, Katayama H, Nakagawa N. Germline gain-of-function mutations in RAF1 cause Noonan syndrome. Nat Genet. 2007;39:1013–1017. doi: 10.1038/ng2078. [DOI] [PubMed] [Google Scholar]
- 178.Nyström AM, Ekvall S, Berglund E, Björkqvist M, Braathen G, Duchen K, Enell H, Holmberg E, Holmlund U, Olsson-Engman M, et al. Noonan and cardio-facio-cutaneous syndromes: two clinically and genetically overlapping disorders. J Med Genet. 2008;45:500–506. doi: 10.1136/jmg.2008.057653. [DOI] [PubMed] [Google Scholar]
- 179.Armour CM, Allanson JE. Further delineation of cardio-facio-cutaneous syndrome: clinical features of 38 individuals with proven mutations. J Med Genet. 2008;45:249–254. doi: 10.1136/jmg.2007.054460. [DOI] [PubMed] [Google Scholar]
- 180.Dhomen N, Marais R. New insight into BRAF mutations in cancer. Curr Opin Genet Dev. 2007;17:31–39. doi: 10.1016/j.gde.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 181.Selfors LM, Schutzman JL, Borland CZ, Stern MJ. soc-2 encodes a leucine-rich repeat protein implicated in fibroblast growth factor receptor signaling. Proc Natl Acad Sci USA. 1998;95:6903–6908. doi: 10.1073/pnas.95.12.6903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Sieburth DS, Sun Q, Han M. SUR-8, a conserved Ras-binding protein with leucine-rich repeats, positively regulates Ras-mediated signaling in C. elegans. Cell. 1998;94:119–130. doi: 10.1016/s0092-8674(00)81227-1. [DOI] [PubMed] [Google Scholar]
- 183.Li W, Han M, Guan KL. The leucine-rich repeat protein SUR-8 enhances MAP kinase activation and forms a complex with Ras and Raf. Genes Dev. 2000;14:895–900. [PMC free article] [PubMed] [Google Scholar]
- 184.Rodriguez-Viciana P, Oses-Prieto J, Burlinqame A, Fried M, McCormick F. A phosphatase holoenzyme comprised of Shoc2/Sur8 and the catalytic subunit of PP1 functions as an M-Ras effector to modulate Raf activity. Mol Cell. 2006;22:217–30. doi: 10.1016/j.molcel.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 185.Matsunaga-Udagawa R, Fujita Y, Yoshiki S, Terai K, Kamioka Y, Kiyokawa E, Yugi K, Aoki K, Matsuda M. The scaffold protein Shoc2/SUR-8 accelerates the interaction of Ras and Raf. J Biol Chem. 2010;285:7818–7826. doi: 10.1074/jbc.M109.053975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Boutin JA. Myristoylation. Cell Signal. 1997;9:15–35. doi: 10.1016/s0898-6568(96)00100-3. [DOI] [PubMed] [Google Scholar]
- 187.Farazi TA, Waksman G, Gordon JI. The biology and enzymology of protein N-myristoylation. J Biol Chem. 2001;276:39501–39504. doi: 10.1074/jbc.R100042200. [DOI] [PubMed] [Google Scholar]
- 188.Resh MD. Trafficking and signaling by fatty-acylated and prenylated proteins. Nat Chem Biol. 2006;2:584–590. doi: 10.1038/nchembio834. [DOI] [PubMed] [Google Scholar]
- 189.Zheng CF, Guan KL. Properties of MEKs: the kinases that phosphorylate and activate the extracellular signal-regulated kinases. J Biol Chem. 1993;268:23933–23939. [PubMed] [Google Scholar]
- 190.Giroux S, Tremblay M, Bernard D, Cardin-Girard JF, Aubry S, Larouche L, Rousseau S, Huot J, Landry J, Jeannotte L, Charron J. Embryonic death of Mek1-deficient mice reveals a role for this kinase in angiogenesis in the labyrinthine region of the placenta. Curr Biol. 1999;9:369–372. doi: 10.1016/s0960-9822(99)80164-x. [DOI] [PubMed] [Google Scholar]
- 191.Bélanger LF, Roy S, Tremblay M, Brott B, Steff AM, Mourad W, Hugo P, Erikson R, Charron J. Mek2 is dispensable for mouse growth and development. Mol Cell Biol. 2003;23:4778–4787. doi: 10.1128/MCB.23.14.4778-4787.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Cowley S, Paterson H, Kemp P, Marshall CJ. Activation of MAP kinase kinase is necessary and sufficient for PC12 differentiation and for transformation of NIH 3T3 cells. Cell. 1994;77:841–852. doi: 10.1016/0092-8674(94)90133-3. [DOI] [PubMed] [Google Scholar]
- 193.Mansour SJ, Matten WT, Hermann AS, Candia JM, Rong S, Fukasawa K, Vande Woude GF, Ahn NG. Transformation of mammalian cells by constitutively active MAP kinase kinase. Science. 1994;265:966–970. doi: 10.1126/science.8052857. [DOI] [PubMed] [Google Scholar]
- 194.Dentici ML, Sarkozy A, Pantaleoni F, Carta C, Lepri F, Ferese R, Cordeddu V, Martinelli S, Briuglia S, Digilio MC, et al. Spectrum of MEK1 and MEK2 gene mutations in cardio-facio-cutaneous syndrome and genotype-phenotype correlations. Eur J Hum Genet. 2009;17:733–740. doi: 10.1038/ejhg.2008.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Rauen KA, Tidyman WE, Estep AL, Sampath S, Peltier HM, Bale SJ, Lacassie Y. Molecular and functional analysis of a novel MEK2 mutation in cardio-facio-cutaneous syndrome: transmission through four generations. Am J Med Genet A. 2010;152A:807–814. doi: 10.1002/ajmg.a.33342. [DOI] [PMC free article] [PubMed] [Google Scholar]