

Abstract
Background
Polymorphism of the dopamine transporter genotype (DAT1) confers a small but significant susceptibility to Attention Deficit Hyperactivity Disorder (ADHD). We examined whether the volume of the head of caudate, a striatal structure with high DAT expression that is important for inhibitory function, differs by DAT1 in a children diagnosed with the disorder relative to age and IQ matched controls.
Method
Volume of the head of caudate was delineated in the right and left hemisphere and compared between 7–13 year old children with and without ADHD (Combined type) who were carriers of two (10/10) or one (9/10) copy of the 10-repeat DAT1 allele.
Results
Caudate volumes were overall smaller 10/10 than 9/10 children, particularly in the left than right hemisphere. While DAT1 effects did not vary by ADHD diagnosis, overall caudate volumes were smaller in ADHD relative to control children.
Conclusions
Altered caudate development associated with 10-repeat homozygosity of DAT1 may contribute susceptibility to ADHD.
Keywords: Brain, structure, VNTR, polymorphism, striatal
INTRODUCTION
Molecular genetic studies of the dopaminergic neurotransmitter system have suggested targets for elucidating the pathophysiology of Attention Deficit Hyperactivity Disorder (ADHD), one of the most common disorders of childhood. One promising target is the dopamine transporter (DAT) that regulates synaptic dopamine by reuptake following release (Madras et al., 2005). DAT is expressed most abundantly in the striatum where as the primary mechanism of dopamine clearance, it is a key determinant of synaptic dopamine levels. DAT is the target of methylphenidate, a common treatment for ADHD, which increases striatal dopamine particularly in the caudate by inhibiting DAT (Volkow et al., 2002). Molecular genetic studies have found a small [<4% (Waldman et al., 1998)] but significant association with ADHD of a variable number of tandem repeat sequences (VNTR) in the 3’-untranslated region (UTR) of the gene (SLC6A3) coding for the dopamine transporter (DAT1) such that ADHD was more prevalent in homozygous carriers of the 10 repeat allele [first reported by (Cook et al., 1995)]. Meta-analyses have confirmed this association although with low (1.2) odds ratio and effects sizes varied across studies suggesting that the association is influenced by moderating factors (Gizer et al., 2009). Thus, etiology of ADHD is posited to include genetic variation in striatal expression of DAT.
Homozygosity of the 10-repeat DAT1 allele may mediate susceptibility to ADHD via functional and structural brain characteristics associated with DAT that are shared with ADHD individuals. Functionally, there is some evidence indicating that ADHD and 10-repeat homozygous individuals display higher availability of DAT in the caudate. Several ligand-based imaging studies found that striatal DAT availability was higher in ADHD relative to control subjects [reviewed in (Spencer et al., 2005)]. Further, greater striatal DAT availability was also observed in 10-repeat homozygotes relative to heterozygotes with diagnosis of ADHD (Cheon et al., 2005) as well as without the diagnosis (Heinz et al., 2000). Further, in vitro studies have found that greater DAT expression was associated with the 10-repeat allele (Fuke et al., 2001; Mill et al., 2002; VanNess et al., 2005). However, in vivo studies of unaffected individuals have failed to replicate the finding, showing reduced (Jacobsen et al., 2000; van de Giessen et al., 2009; van Dyck et al., 2005), or no different (Krause et al., 2006; Martinez et al., 2001) striatal DAT availability in 10-repeat homozygotes relative to heterozygotes.
Structurally, there is initial evidence demonstrating that ADHD and 10-repeat homozygosity share reductions in volume of the caudate. Numerous structural imaging studies have shown that caudate volume was smaller in subjects with ADHD relative to controls [see review by (Seidman et al., 2005) and meta-analysis by (Valera et al., 2007)]. A recent meta-analysis, however, reported that volumes of globus pallidus and putamen but not caudate were reduced in ADHD (Ellison-Wright et al., 2008). Caudate volume was smaller in 10-repeat homozygous relative to heterozygous subjects including ADHD, their unaffected siblings, and controls (Durston et al., 2005). DAT1 effects did not extend to the prefrontal cortex that has extensive striatal projections but minimal DAT expression. Lack of DAT1 difference in prefrontal cortex was replicated in another study, although this study did not examine the caudate (Shaw et al., 2007). DAT1 effects did not interact statistically with ADHD diagnosis in both studies. Independent converging effects on caudate anatomy of DAT1 polymorphism and ADHD suggest that alteration in caudate anatomy may mediate susceptibility to ADHD.
In the present study, we sought confirming evidence for effects of DAT1 VNTR on the head of caudate (referred to as caudate henceforth) volume in children aged 7–13 years. While it serves to replicate findings of Durston et al., (2005), the present study also seeks to extend those findings in one important way: In the study by Durston et al., wide variation in developmental stages among the sample (age range 8–18 years) may have prevented the detection of a DAT1 X Diagnosis interaction on caudate volumes. Age is an important factor because developmental trajectories of caudate volume converge in adolescence such that volume was reduced in ADHD relative to controls in childhood but not in adolescence (Castellanos et al., 2002). By restricting age to preadolescence in the present study, a period when caudate volumetric differences are present between ADHD and controls, our design optimized detection of an interaction between DAT1 and ADHD diagnosis. ADHD children in the present study were diagnosed as Combined-type and treated with methylphenidate for at least 6 months. Based upon Durston et al.’s finding, we hypothesized that caudate volumes would be smaller in ADHD relative to control children and in 10/10 relative to 9/10 children. If supported, the present study would provide an important replication of Durston et al’s important first report of this finding. Furthermore, we also hypothesized that these variables would interact statistically, with the smallest caudate volume in 10/10 ADHD children. It is possible that we may not be able to detect this interaction, despite the restricted age range as our sample is modest in size and ethnically diverse; this may yield higher other genetic variation relative to Durston et al.’s study in all Dutch children. Nevertheless, this study provides an opportunity to test for a DAT1 X Diagnosis interaction.
METHODS AND MATERIALS
Subjects
Twenty-six typically developing children (controls) and thirty-three children with ADHD (Combined type) who were being chronically treated with methylphenidate for at least 6 months, aged 7–13 years were recruited from the Washington DC area through advertisements in the newsletter and on-hold phone messages at Children’s National Medical Center and were paid for participation. Parental consent and children’s assent were obtained according to guidelines of the Georgetown University Institutional Review Board.
Demographic information is presented in Table 1. All participants with ADHD provided documentation from the diagnosing clinician (either a physician or a psychologist) which was reviewed by a clinical psychologist (LK) to confirm DSM-IV criteria were met (i.e., presence of 6 out of 9 symptoms of inattention and 6 out of 9 symptoms of hyperactivity/impulsivity in more than one setting, present before age 7 years and causing impairment). All participants were being treated with methylphenidate during and prior to participation in the study; parents reported that they were satisfied with their child’s current medication. Further, an ADHD Rating Scale-IV (DuPaul et al., 1998) was completed by a parent for each child. In order to screen for co-morbid psychiatric disorders, the Behavioral Assessment System for Children [BASC (Reynolds and Kamphaus, 1998)] was administered for each child. Exclusion criteria included: (a) Full Scale IQ below 85 as measured by the Wechsler Abbreviated Scale of Intelligence (WASI); (b) Woodcock-Johnson Letter Word ID or Word Attack standard scores below 85; and (c) Parental report of neurological (e.g., epilepsy) disorders; (d) Mood disorders in both groups screened by the BASC; (e) Siblings with ADHD diagnosis in control children. IQ and reading measures were assessed on medication. IQ and reading disability cutoffs ensured children’s ability to comprehend and follow instructions and assent procedures associated with MRI scanning.
Table 1.
Mean (standard deviation in parenthesis) and Range for demographic variables. Neither diagnosis nor DAT1 groups differed statistically on any variable (ps > .08) except BASC and ADHD Rating scale scores which were higher in ADHD relative to controls (ps < .001); these did not differ by genotype within each group (ps > .15).
| ADHD 10/10 | ADHD 9/10 | Control 10/10 | Control 9/10 | |
|---|---|---|---|---|
| N | 18 | 15 | 16 | 10 |
| Gender (males) | 14 | 10 | 10 | 7 |
| Age (in years) | 10.5 (1.6) | 10.5 (1.8) | 10.8 (1.5) | 10.3 (1.5) |
| Dosage (mg/kg) | .81 (.40) R: .46–2.06 |
.93 (.47) R: .37–1.89 |
N/A | N/A |
| Full Scale IQ | 111.1 (12.3) R: 92–133 |
112.7 (13.9) R: 87–142 |
111.5 (5.9)* R: 103–129 |
111.1 (11.7) R: 89–130 |
| Woodcock-Johnson Letter Word ID |
108.1 (10.5) R: 91–128 |
108.5 (12.9) R: 91–139 |
114.8 (8.5) R: 99–132 |
108.4 (9.4) R: 91–122 |
| Woodcock-Johnson Word Attack |
103.7 (7.8) R: 93–117 |
104.7 (10.9) R: 89–129 |
111.2 (9.6) R: 96–127 |
103.7 (10.3) R: 90–126 |
| BASC Hyperactivity (T score) | 67.5 (15.4) * R: 36–92 |
65.2 (17.2) R: 46–105 |
40.9 (6.9) R: 30–55 |
46.3 (11.9) R: 33–69 |
| BASC Attention Problems (T score) |
66.1 (9.2) * R: 50–82 |
67.7 (7.3) R: 53–79 |
45.7 (8.1) R: 32–58 |
50.5 (9.6) 39–71 |
| ADHD Rating Scale Hyperactivity |
16.2 (6.4) R: 7–26 |
15.8 (7.7) R: 3–27 |
1.0 (0.9) ** R: 0–3 |
2.7 (3.1) R: 0–11 |
| ADHD Rating Scale Inattention |
17.9 (4.6) R: 11–26 |
18.9 (6.7) R: 10–27 |
3.4 (2.8) ** R: 0–8 |
6.2 (4.5) R: 1–15 |
Missing data for one child.
Missing data from three children. ADHD Rating Scale range: 0–27. BASC – Behavioral Assessment System for Children
Of the controls, ten children (7 boys) were heterozygous for the 10-repeat allele (termed 9/10) and sixteen children (10 boys) were homozygous for the 10-repeat allele (termed 10/10). Of the children with ADHD, fifteen children (10 boys) were 9/10 and twenty (14 boys) children were 10/10. Children homozygous for the 9-repeat allele (termed 9/9) were excluded from analysis due to low sample size (n = 4). The control group comprised 8% Alaskan Native, 4% Asian, 15% African-American, 8% Hispanic, 11% no report, 46% Caucasian, and 8% Caucasian/Asian. The ADHD group comprised 3% Asian, 12 % African-American, 6 % Hispanic, 6% no report, 61% Caucasian, 6% Caucasian/Asian, and 6% Caucasian/African-American. Gender composition and age did not differ among the four subgroups (χ2 = 1.01, p = .80; χ2 = 173, p = .31) and allelic distribution did not differ between ADHD and control groups (χ2 = .29, p = .59). Further, the four subgroups did not differ in age and Full-scale IQ (ps > .89). ADHD children had significantly higher hyperactivity and inattention scores than controls (ps < .001) but the two genotype ADHD groups (i.e., 10/10 and 9/10) did not differ in severity of symptoms (ps > .58) assessed by the ADHD rating scale and BASC. Current dosage did not differ between genotype groups (p = .72, range: .22–2.06 mg/kg).
Imaging Procedure
A Siemens Trio 3T MRI scanner (Erlangen, Germany) was used to acquire a high resolution sagittal T1-weighted structural scan using a 3D MPRAGE sequence with a scan time of 6:51 min and the following parameters: TR = 1600 ms, TE =4.4 ms, 256 × 256-mm FOV, 160-mm slab with 1-mm-thick slices, 256 × 256 × 160 matrix (effective resolution of 1.0 mm3), 1 excitation and a 15° flip angle. Subjects watched a film or television series of their choice during the scan. Head motion was minimized by small foam cushions placed on the sides of the subject’s head.
Data Analysis
Images were inspected by two trained experimenters for artifacts due to motion (e.g., ghosting) and poor contrast between grey and white matter boundaries; all images included in this study were without these artifacts. Using SPM2 (Wellcome Department of Cognitive Neurology, London), raw images were processed using the optimized voxel-based morphometry methods outlined by Ashburner and colleagues (Ashburner and Friston, 2000; Mechelli et al., 2005). Images were first segmented into grey matter, white matter and CSF. The grey matter segment was then normalized to the grey matter template derived from the Montreal Neurological Institute T1 structural template. Use of an adult template for normalization for this age range has been validated in several studies (Burgund et al., 2002; Muzik et al., 2000). These normalization parameters were then applied to the raw image to normalize the whole brain. Normalized whole brain images from each child were averaged to create a single mean brain image for the ADHD and control groups separately. Two regions of interest (ROIs) were drawn manually on these two mean images, for right and left caudate using FSL (Centre for Functional Magnetic Resonance Imaging of the Brain, University of Oxford, London, UK). The boundary of the head of the caudate was traced and included part of the globus pallidus, as it could not be resolved (see Figure 1). These template ROIs were then transformed into native space for each child using the inverse of the spatial normalization parameters. These native space ROIs were then overlaid on each subject’s whole brain images applying a voxel intensity threshold to control for boundary effects of normalization. Using the Duvernoy Atlas (Duvernoy, 1999) as a guide, each ROI was then inspected collectively by two experimenters blind to subject identity, who edited the boundaries in all three dimensions using a paint brush size of 1 voxel (1 mm), to be consistent with the area of the head of caudate in each subject. The medial boundary was the lateral border of the lateral ventricle and the lateral boundary was the medial aspect of the internal capsule. This normalization procedure, modeled after Semrud-Clikeman et al. (Semrud-Clikeman et al., 2006), controlled for global differences in cerebral volume and differences in dimensions in brain alignment during image acquisition. For each subject, grey matter volumes of the right and left caudate ROI and of the whole brain, and white matter volume for the whole brain were measured using FSL (Smith et al., 2004; Woolrich et al., 2009). One-third of the brains were selected randomly for re-measuring with ROI tracing done by another rater. Interrater reliability between the original ROIs and the re-measured ones was .88.
Figure 1.
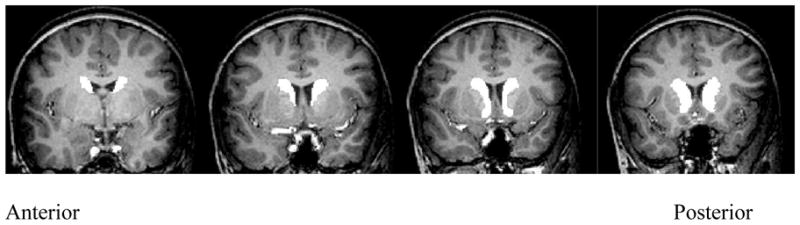
Sample caudate region of interest
Genotyping
DNA was extracted from whole blood with a PureGene kit from 10 ml of whole blood or taken from cheek swab or Oragene saliva kits (DNA Genotek Inc., Ottawa, Ontario, Canada). Genotyping was performed in the following manner. PCR was carried out in a 10 ul volume containing 50 ng of genomic DNA, 0.5 uM of each primer 5′-NED-TGTGGTGTAGGGAACGGCCTGAG-3′ and 5′-(GTTTCTT)CTTCCTGGAGGTCACGGCTCAAGG-3′, 800 uM of each dNTP (dATP, dCTP, dGTP, dTTP), 1 x EXT PCR buffer (including 1.5 mM MgCl2), GC melt, and 0.3 units DyNAzyme EXT polymerase. Samples were amplified on a 9700 thermal cycler with an initial 12-minute step to heat-activate the enzyme, 45 cycles consisting of a denaturation step of 95 degrees C for 30 sec., an annealing step of 68 degrees C for 45 sec., and an extension step of 72 degrees C for 3 min. Products were injected onto an ABI 3730XL multi-capillary array genetic analyzer. Alleles were called with GeneMapper software, blind to all phenotypic information.
RESULTS
Total whole brain grey and white matter, and caudate volumes for the left and right hemisphere were computed for each subject. First, we determined whether mean whole brain grey and white matter volumes differed by DAT1 (10/10, 9/10) and Diagnosis (ADHD, Controls) in separate factorial analyses of variance (ANOVAs). Second, we determined whether caudate volumes differed by DAT1 and Diagnosis and Hemisphere (left, right) in a mixed ANOVA including age and Total Cerebral Volume (TCV, whole brain grey + white brain white) as covariates. In this ANOVA, DAT1 and Diagnosis were between-subjects variables and Hemisphere was a within-subjects variable.
For both whole brain grey (ADHD – 9/10: Mean = 705 cm3, SD = 54 cm3; 10/10: Mean = 714 cm3, SD = 61 cm3; Control 9/10: Mean = 690 cm3, SD = 50 cm3; 10/10:Mean = 715 cm3, SD = 53 cm3) and white matter (ADHD - 9/10: Mean = 358 cm3, SD = 46 cm3; 10/10:Mean = 355 cm3, SD = 34 cm3; Control – 9/10: Mean = 354 cm3, SD = 42 cm3; 10/10:Mean = 361 cm3, SD = 43 cm3), main effects of Diagnosis and DAT1 and their interaction were not significant (ps > .24). Thus, there were no global brain differences between the groups. Analysis of caudate volumes revealed a main effect of Diagnosis indicating that caudate volumes were smaller in ADHD than control children, F (1, 53) = 6.61, p = .01, η2 = .11 (see Figure 2). Further, there was a main effect of DAT1 indicating that caudate volumes were smaller in children with 10/10 than 9/10 genotype, F (1, 53) = 7.00, p = .01, η2 = .11. The Diagnosis X DAT1 interaction was not significant (p = .97) indicating that genotypic differences did not vary between ADHD and control groups. The DAT1 X Hemisphere interaction, however, was significant, F (1, 53) = 4.07, p = .05, η2 = .07. Breaking down this interaction with post-hoc t-tests revealed that caudate volumes were smaller in the left than right hemisphere in 10/10 children, t(33) = 3.12, p = .01, d = .33, but not in 9/10 children (p = .84). Further, 10/10 children had smaller caudate volumes than the 9/10 children in the left (t (57) = 2.51, p = .02, d = .67) but not the right (p = .21) hemisphere (see Figure 3). No other interactions were significant (ps > .10). Further, caudate volumes did not correlate with symptom measures (BASC Inattention, Hyperactivity) in any group or in the whole sample (ps > .29) or with age (ps > .36).
Figure 2.

Mean (and standard error) volumes (in mm3) of the left and right caudate for the ADHD (dark) and Control (light) groups. Caudate volumes were smaller in ADHD (M = 4935 mm3, SD = 552 mm3) than control (M = 5267 mm3, SD = 685 mm3) children, F (1, 53) = 6.61, p = .01, η2 = .11.
Figure 3.

Mean (and standard error) volumes (in mm3) of the left and right caudate for the 10/10 (dark) and 9/10 (light) DAT1 groups. Caudate volumes were smaller in 10/10 (M = 4952 mm3, SD = 619 mm3) than 9/10 (M = 5257 mm3, SD = 615 mm3) children, F (1, 53) = 7.00, p = .01, η2 = .11. The DAT1 X Hemisphere interaction was significant, F (1, 53) = 4.07, p = .05, η2 = .07 (10/10 Left Caudate: M = 4850 mm3, SD = 575 mm3; 10/10 Right Caudate: M = 5054 mm3, SD = 653 mm3; 9/10 Left Caudate: M = 5250 mm3, SD = 645 mm3; 9/10 Right Caudate: M = 5264 mm3, SD = 597 mm3).
DISCUSSION
We examined whether volume of the head of caudate differed by DAT1 polymorphism in preadolescent children with and without diagnosis of ADHD. Results showed that while there were no differences in global grey and white matter by diagnosis or DAT1 group, caudate volumes were smaller in 10-repeat homozygotes relative to heterozygotes, particularly in the left relative to the right hemisphere. Further, ADHD children had smaller caudate volumes than control children. No other interactions with DAT1, ADHD diagnosis, or hemisphere reached significance.
The present study not only replicates but also extends findings from Durston et al. (2005), the first and only study to examine caudate volumetric differences by DAT1 and ADHD diagnosis, in three important ways. First, our finding of smaller caudate volume in 10-repeat homozygotes relative to heterozygotes and lack of statistical interaction with ADHD diagnosis supports findings of Durston et al. (2005). Lack of a DAT1 X Diagnosis interaction in the present study, in which subjects’ age was restricted to late childhood and diagnosis to one subtype of ADHD, indicates that neither of these factors prevented detection of an interaction in Durston et al.’s study. In the context of molecular genetic findings associating 10-repeat homozygosity with prevalence of ADHD [meta-analysis by (Yang et al., 2007)], similar effects of ADHD and 10-repeat homozygosity on caudate volume suggest shared structural processes determining neuronal and synaptic density, influencing phenotypic expression. Specifically, relative to heterozygotes, homozygous 10-repeat unaffected children had higher hyperactivity (Mill et al., 2005) and poor inhibitory task performance [TEA-Ch Opposite Worlds task (Cornish et al., 2005); errors of commission on the Continuous Performance Test (Loo et al., 2003)], properties of the ADHD phenotype (Vaidya and Stollstorff, 2008). In light of the importance of the caudate to inhibitory function, the present findings suggest that effects of 10-repeat homozygosity on striatal structural development may play a role in translating risk for ADHD. However, additional genetic or environmental factors, and their interaction, are likely to also contribute because a preliminary functional imaging study found reduced left caudate activation during response inhibition in 10-repeat homozygotes relative to heterozygotes with ADHD and their unaffected siblings, but not in controls (Durston et al., 2008). Control sample size in that study was very small (9-carriers = 4, 10/10=5) and thus, these findings need to be replicated in future studies. Nevertheless, together these findings suggest that the effects of DAT1 VNTR on caudate physiology and anatomy are important properties for identifying vulnerability to ADHD.
Second, our finding of smaller caudate volume in ADHD relative to control children supports results of Durston et al., (2005) as well as numerous other studies [see review by (Seidman et al., 2005) and meta-analysis by (Valera et al., 2007) but see (Ellison-Wright et al., 2008)]. The caudate is a part of the nigrostriatal dopamine pathway and thalamo-cortical projections linking prefrontal cortex to the striatum, circuitry that is important for inhibitory control of motor action, a key domain of impairment in ADHD. Indeed, caudate volume was associated with performance on response inhibition tasks in ADHD and control children (Casey et al., 1997). Caudate functional impairment in ADHD subjects is evident on numerous functional imaging studies that showed reduced activation during inhibitory control relative to control subjects [recently reviewed in (Bush et al., 2005; Vaidya and Stollstorff, 2008)]. Together, these findings have contributed to the view that the caudate is a principal site of pathophysiology in ADHD.
Third, we found a hemispheric difference in the DAT1 effect on caudate volume that was not observed in Durston et al.’s study (2005). As Durston et al’s study included a larger sample than ours and also the fact that the present hemispheric difference was a small effect, this finding ought to be interpreted with caution. Caudate volume was smaller in the left than right hemisphere in the 10-repeat homozygotes relative to heterozygotes. The finding of smaller left caudate volume in 10/10 children sheds some light on past volumetric findings of inconsistent hemispheric differences in ADHD as well as control children (summarized in Table 2). They suggest that the composition of DAT1 allele status in the sample (whether majority were 10-repeat homozygotes or heterozygotes) is likely to influence the nature of observed caudate asymmetry. Localization of the effect of DAT1 to the left hemisphere in the present study is consistent with the DAT1 functional imaging findings discussed earlier (Durston et al., 2008) in which left caudate activation was reduced in 10/10 relative to 9-carriers with ADHD and their unaffected siblings.
Table 2.
Hemispheric differences in caudate size in ADHD and control children.
| Study | ADHD/Controls | Caudate Findings |
|---|---|---|
| Hynd et al. 1993 | 11/11 | L caudate larger than R caudate in controls. Reversed in ADHD (L<R) |
| Castellanos et al. 1994 | 50/48 | L caudate smaller than R caudate in controls. Not significant in ADHD |
| Castellanos et al. 1996 | 57/55 | L caudate smaller than R caudate in controls. Not significant in ADHD |
| Filipek et al. 1997 | 15/15 | L caudate smaller in ADHD compared to controls, but not R caudate |
| Mataro et al. 1997 | 11/19 | R caudate larger in ADHD compared to controls, but not L caudate |
| Semrud-Clikeman et al. 2000 | 10/11 | L caudate smaller in ADHD compared to controls, but not R caudate. |
| Pineda et al. 2002 | 30/15 | L caudate larger than R caudate in ADHD |
The present findings suggest that individual differences in DAT gene expression influence structural brain development. Twin studies have shown that individual differences in brain morphology are highly heritable indicating that shared genes influence brain structural development (Bartley et al., 1997). Dopaminergic genes, in particular, may exert early influence on brain development in light of the trophic role of dopamine in neurogenesis, division, migration, and differentiation (Nieoullon, 2002). Alternatively, DAT1 structural differences may reflect plasticity in dopamine receptor actions due to transporter differences. Plasticity of dopaminergic structures (e.g., increased caudate volume) is evident following prolonged exposure to antipsychotic medication that attenuates dopamine levels, and has been attributed to compensatory receptor regulation (Chakos et al., 1995; Chakos et al., 1994). Differences due to DAT1 in the present study could not be attributed to prolonged exposure to methylphenidate because they were not selective to ADHD children. It is important to note that other genetic polymorphisms interact with DAT1 to influence caudate anatomy. A recent study in healthy adults found that caudate volume was reduced in 10-repeat homozygotes relative to 9-carriers carrying the GT variant of the D2 receptor genotype that is associated with reduced presynaptic expression, whereas the DAT1 differences were in the opposite direction (10/10 > 9 carriers) for the GG variant (Bertolino et al., 2009). Furthermore, interactive effects of DAT1 and the catechol-O-methyltransferase (COMT) genotypes have been observed in prefrontal activation during working memory (Bertolino et al., 2008). Unlike the interaction with DRD2, DAT1 does not appear to interact with DRD4 polymorphism with respect to caudate and prefrontal volumes (Durston et al., 2005; Shaw et al., 2007). Regarding DAT1, an in vitro study showed that single nucleotide polymorphisms, in addition to the VNTR, also influence gene expression (Miller and Madras, 2002). Thus, many other sources of genetic variation are present in our and past studies, and perhaps reduced statistical power to detect significant interaction with ADHD diagnosis.
In sum, the present findings replicate Durston et al’s past finding that the DAT1 VNTR influences volume of the caudate, in a sample that is relatively homogeneous with respect to childhood age and exposure to methylphenidate. It is important to note that an important limitation of our study is the relatively small sample size. This coupled with high genetic diversity that is likely to stem from an ethnically diverse sample, could have reduced power to detect interaction with diagnosis or raised the likelihood of Type 2 error. Thus, the finding of a lack of an interaction must be viewed with caution. Further, while all children were subtyped as Combined-type upon initial clinical diagnoses, they were in long-term treatment at the time of testing and thus, severity of symptoms varied among the sample as suggested by the wide range of rating scores. Nonetheless, our results concur with Durston et al’s who included a larger sample than ours. Replication studies are important for advancing the burgeoning field of imaging genetics, generally, and our understanding of striatal pathophysiology in ADHD, particularly. The present finding of smaller caudate volume in carriers of two copies of the 10-repeat allele, a genotype that confers small but significant risk for ADHD, suggests that alteration in striatal structural characteristics is an etiological risk factor for ADHD.
Acknowledgments
This work was supported by NIMH grants #MH065395 to CJV and #MH70564 to MS.
References
- Ashburner J, Friston KJ. Voxel-based morphometry--the methods. Neuroimage. 2000;11(6 Pt 1):805–21. doi: 10.1006/nimg.2000.0582. [DOI] [PubMed] [Google Scholar]
- Bartley AJ, Jones DW, Weinberger DR. Genetic variability of human brain size and cortical gyral patterns. Brain. 1997;120 ( Pt 2):257–69. doi: 10.1093/brain/120.2.257. [DOI] [PubMed] [Google Scholar]
- Bertolino A, Di Giorgio A, Blasi G, Sambataro F, Caforio G, Sinibaldi L, Latorre V, Rampino A, Taurisano P, Fazio L, et al. Epistasis between dopamine regulating genes identifies a nonlinear response of the human hippocampus during memory tasks. Biol Psychiatry. 2008;64(3):226–34. doi: 10.1016/j.biopsych.2008.02.001. [DOI] [PubMed] [Google Scholar]
- Bertolino A, Fazio L, Di Giorgio A, Blasi G, Romano R, Taurisano P, Caforio G, Sinibaldi L, Ursini G, Popolizio T, et al. Genetically determined interaction between the dopamine transporter and the D2 receptor on prefrontostriatal activity and volume in humans. J Neurosci. 2009;29(4):1224–34. doi: 10.1523/JNEUROSCI.4858-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgund ED, Kang HC, Kelly JE, Buckner RL, Snyder AZ, Petersen SE, Schlaggar BL. The feasibility of a common stereotactic space for children and adults in fMRI studies of development. Neuroimage. 2002;17(1):184–200. doi: 10.1006/nimg.2002.1174. [DOI] [PubMed] [Google Scholar]
- Bush G, Valera EM, Seidman LJ. Functional neuroimaging of attention-deficit/hyperactivity disorder: a review and suggested future directions. Biol Psychiatry. 2005;57(11):1273–84. doi: 10.1016/j.biopsych.2005.01.034. [DOI] [PubMed] [Google Scholar]
- Casey BJ, Castellanos FX, Giedd JN, Marsh WL, Hamburger SD, Schubert AB, Vauss YC, Vaituzis AC, Dickstein DP, Sarfatti SE, et al. Implication of right frontostriatal circuitry in response inhibition and attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 1997;36(3):374–83. doi: 10.1097/00004583-199703000-00016. [DOI] [PubMed] [Google Scholar]
- Castellanos FX, Lee PP, Sharp W, Jeffries NO, Greenstein DK, Clasen LS, Blumenthal JD, James RS, Ebens CL, Walter JM, et al. Developmental trajectories of brain volume abnormalities in children and adolescents with attention-deficit/hyperactivity disorder. Jama. 2002;288(14):1740–8. doi: 10.1001/jama.288.14.1740. [DOI] [PubMed] [Google Scholar]
- Chakos MH, Lieberman JA, Alvir J, Bilder R, Ashtari M. Caudate nuclei volumes in schizophrenic patients treated with typical antipsychotics or clozapine. Lancet. 1995;345(8947):456–7. doi: 10.1016/s0140-6736(95)90441-7. [DOI] [PubMed] [Google Scholar]
- Chakos MH, Lieberman JA, Bilder RM, Borenstein M, Lerner G, Bogerts B, Wu H, Kinon B, Ashtari M. Increase in caudate nuclei volumes of first-episode schizophrenic patients taking antipsychotic drugs. Am J Psychiatry. 1994;151(10):1430–6. doi: 10.1176/ajp.151.10.1430. [DOI] [PubMed] [Google Scholar]
- Cheon KA, Ryu YH, Kim JW, Cho DY. The homozygosity for 10-repeat allele at dopamine transporter gene and dopamine transporter density in Korean children with attention deficit hyperactivity disorder: relating to treatment response to methylphenidate. Eur Neuropsychopharmacol. 2005;15(1):95–101. doi: 10.1016/j.euroneuro.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Cook EH, Jr, Stein MA, Krasowski MD, Cox NJ, Olkon DM, Kieffer JE, Leventhal BL. Association of attention-deficit disorder and the dopamine transporter gene. Am J Hum Genet. 1995;56(4):993–8. [PMC free article] [PubMed] [Google Scholar]
- Cornish KM, Manly T, Savage R, Swanson J, Morisano D, Butler N, Grant C, Cross G, Bentley L, Hollis CP. Association of the dopamine transporter (DAT1) 10/10-repeat genotype with ADHD symptoms and response inhibition in a general population sample. Mol Psychiatry. 2005;10(7):686–98. doi: 10.1038/sj.mp.4001641. [DOI] [PubMed] [Google Scholar]
- DuPaul GJ, Power TJ, Anastopoulos AD, Reid R. ADHD- Rating Scales DSM-IV for parents and teachers. New York: Guilford; 1998. [Google Scholar]
- Durston S, Fossella JA, Casey BJ, Hulshoff Pol HE, Galvan A, Schnack HG, Steenhuis MP, Minderaa RB, Buitelaar JK, Kahn RS, et al. Differential effects of DRD4 and DAT1 genotype on fronto-striatal gray matter volumes in a sample of subjects with attention deficit hyperactivity disorder, their unaffected siblings, and controls. Mol Psychiatry. 2005;10(7):678–85. doi: 10.1038/sj.mp.4001649. [DOI] [PubMed] [Google Scholar]
- Durston S, Fossella JA, Mulder MJ, Casey BJ, Ziermans TB, Vessaz MN, Van Engeland H. Dopamine transporter genotype conveys familial risk of attention-deficit/hyperactivity disorder through striatal activation. J Am Acad Child Adolesc Psychiatry. 2008;47(1):61–7. doi: 10.1097/chi.0b013e31815a5f17. [DOI] [PubMed] [Google Scholar]
- Ellison-Wright I, Ellison-Wright Z, Bullmore E. Structural brain change in Attention Deficit Hyperactivity Disorder identified by meta-analysis. BMC Psychiatry. 2008;8:51. doi: 10.1186/1471-244X-8-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuke S, Suo S, Takahashi N, Koike H, Sasagawa N, Ishiura S. The VNTR polymorphism of the human dopamine transporter (DAT1) gene affects gene expression. Pharmacogenomics J. 2001;1(2):152–6. doi: 10.1038/sj.tpj.6500026. [DOI] [PubMed] [Google Scholar]
- Gizer IR, Ficks C, Waldman ID. Candidate gene studies of ADHD: a meta-analytic review. Hum Genet. 2009;126(1):51–90. doi: 10.1007/s00439-009-0694-x. [DOI] [PubMed] [Google Scholar]
- Heinz A, Goldman D, Jones DW, Palmour R, Hommer D, Gorey JG, Lee KS, Linnoila M, Weinberger DR. Genotype influences in vivo dopamine transporter availability in human striatum. Neuropsychopharmacology. 2000;22(2):133–9. doi: 10.1016/S0893-133X(99)00099-8. [DOI] [PubMed] [Google Scholar]
- Jacobsen LK, Staley JK, Zoghbi SS, Seibyl JP, Kosten TR, Innis RB, Gelernter J. Prediction of dopamine transporter binding availability by genotype: a preliminary report. Am J Psychiatry. 2000;157(10):1700–3. doi: 10.1176/appi.ajp.157.10.1700. [DOI] [PubMed] [Google Scholar]
- Krause J, Dresel SH, Krause KH, La Fougere C, Zill P, Ackenheil M. Striatal dopamine transporter availability and DAT-1 gene in adults with ADHD: no higher DAT availability in patients with homozygosity for the 10-repeat allele. World J Biol Psychiatry. 2006;7(3):152–7. doi: 10.1080/15622970500518444. [DOI] [PubMed] [Google Scholar]
- Loo SK, Specter E, Smolen A, Hopfer C, Teale PD, Reite ML. Functional effects of the DAT1 polymorphism on EEG measures in ADHD. J Am Acad Child Adolesc Psychiatry. 2003;42(8):986–93. doi: 10.1097/01.CHI.0000046890.27264.88. [DOI] [PubMed] [Google Scholar]
- Madras BK, Miller GM, Fischman AJ. The dopamine transporter and attention-deficit/hyperactivity disorder. Biol Psychiatry. 2005;57(11):1397–409. doi: 10.1016/j.biopsych.2004.10.011. [DOI] [PubMed] [Google Scholar]
- Martinez D, Gelernter J, Abi-Dargham A, van Dyck CH, Kegeles L, Innis RB, Laruelle M. The variable number of tandem repeats polymorphism of the dopamine transporter gene is not associated with significant change in dopamine transporter phenotype in humans. Neuropsychopharmacology. 2001;24(5):553–60. doi: 10.1016/S0893-133X(00)00216-5. [DOI] [PubMed] [Google Scholar]
- Mechelli A, Price CJ, Friston KJ, Ashburner J. Voxel-Based Morphometry of the Human Brain: Methods and Applications. Current Medical Imaging Reviews. 2005;1(1):105–113. [Google Scholar]
- Mill J, Asherson P, Browes C, D'Souza U, Craig I. Expression of the dopamine transporter gene is regulated by the 3′ UTR VNTR: Evidence from brain and lymphocytes using quantitative RT-PCR. Am J Med Genet. 2002;114(8):975–9. doi: 10.1002/ajmg.b.10948. [DOI] [PubMed] [Google Scholar]
- Mill J, Xu X, Ronald A, Curran S, Price T, Knight J, Craig I, Sham P, Plomin R, Asherson P. Quantitative trait locus analysis of candidate gene alleles associated with attention deficit hyperactivity disorder (ADHD) in five genes: DRD4, DAT1, DRD5, SNAP-25, and 5HT1B. Am J Med Genet B Neuropsychiatr Genet. 2005;133(1):68–73. doi: 10.1002/ajmg.b.30107. [DOI] [PubMed] [Google Scholar]
- Miller GM, Madras BK. Polymorphisms in the 3′-untranslated region of human and monkey dopamine transporter genes affect reporter gene expression. Mol Psychiatry. 2002;7(1):44–55. doi: 10.1038/sj.mp.4000921. [DOI] [PubMed] [Google Scholar]
- Muzik O, Chugani DC, Juhasz C, Shen C, Chugani HT. Statistical parametric mapping: assessment of application in children. Neuroimage. 2000;12(5):538–49. doi: 10.1006/nimg.2000.0651. [DOI] [PubMed] [Google Scholar]
- Nieoullon A. Dopamine and the regulation of cognition and attention. Prog Neurobiol. 2002;67(1):53–83. doi: 10.1016/s0301-0082(02)00011-4. [DOI] [PubMed] [Google Scholar]
- Reynolds CR, Kamphaus RW, editors. Behavior Assessment System for Children. Circle Pines, MN: American Guidance Service; 1998. [Google Scholar]
- Seidman LJ, Valera EM, Makris N. Structural brain imaging of attention-deficit/hyperactivity disorder. Biol Psychiatry. 2005;57(11):1263–72. doi: 10.1016/j.biopsych.2004.11.019. [DOI] [PubMed] [Google Scholar]
- Semrud-Clikeman M, Pliszka SR, Lancaster J, Liotti M. Volumetric MRI differences in treatment-naive vs chronically treated children with ADHD. Neurology. 2006;67(6):1023–7. doi: 10.1212/01.wnl.0000237385.84037.3c. [DOI] [PubMed] [Google Scholar]
- Shaw P, Gornick M, Lerch J, Addington A, Seal J, Greenstein D, Sharp W, Evans A, Giedd JN, Castellanos FX, et al. Polymorphisms of the dopamine D4 receptor, clinical outcome, and cortical structure in attention-deficit/hyperactivity disorder. Arch Gen Psychiatry. 2007;64(8):921–31. doi: 10.1001/archpsyc.64.8.921. [DOI] [PubMed] [Google Scholar]
- Smith SM, Jenkinson M, Woolrich MW, Beckmann CF, Behrens TE, Johansen-Berg H, Bannister PR, De Luca M, Drobnjak I, Flitney DE, et al. Advances in functional and structural MR image analysis and implementation as FSL. Neuroimage. 2004;23(Suppl 1):S208–19. doi: 10.1016/j.neuroimage.2004.07.051. [DOI] [PubMed] [Google Scholar]
- Spencer TJ, Biederman J, Madras BK, Faraone SV, Dougherty DD, Bonab AA, Fischman AJ. In vivo neuroreceptor imaging in attention-deficit/hyperactivity disorder: a focus on the dopamine transporter. Biol Psychiatry. 2005;57(11):1293–300. doi: 10.1016/j.biopsych.2005.03.036. [DOI] [PubMed] [Google Scholar]
- Vaidya CJ, Stollstorff M. Cognitive neuroscience of Attention Deficit Hyperactivity Disorder: current status and working hypotheses. Dev Disabil Res Rev. 2008;14(4):261–7. doi: 10.1002/ddrr.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valera EM, Faraone SV, Murray KE, Seidman LJ. Meta-analysis of structural imaging findings in attention-deficit/hyperactivity disorder. Biol Psychiatry. 2007;61(12):1361–9. doi: 10.1016/j.biopsych.2006.06.011. [DOI] [PubMed] [Google Scholar]
- van de Giessen EM, de Win MM, Tanck MW, van den Brink W, Baas F, Booij J. Striatal dopmaine transporter availability associated with polymorphisms in the dopamine transporter gene SLC6A3. Journal of Nuclear Medicine. 2009;50(1):45–52. doi: 10.2967/jnumed.108.053652. [DOI] [PubMed] [Google Scholar]
- van Dyck CH, Malison RT, Jacobsen LK, Seibyl JP, Staley JK, Laruelle M, Baldwin RM, Innis RB, Gelernter J. Increased dopamine transporter availability associated with the 9-repeat allele of the SLC6A3 gene. J Nucl Med. 2005;46(5):745–51. [PubMed] [Google Scholar]
- VanNess SH, Owens MJ, Kilts CD. The variable number of tandem repeats element in DAT1 regulates in vitro dopamine transporter density. BMC Genet. 2005;6:55. doi: 10.1186/1471-2156-6-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Fowler JS, Logan J, Franceschi D, Maynard L, Ding YS, Gatley SJ, Gifford A, Zhu W, et al. Relationship between blockade of dopamine transporters by oral methylphenidate and the increases in extracellular dopamine: therapeutic implications. Synapse. 2002;43(3):181–7. doi: 10.1002/syn.10038. [DOI] [PubMed] [Google Scholar]
- Waldman ID, Rowe DC, Abramowitz A, Kozel ST, Mohr JH, Sherman SL, Cleveland HH, Sanders ML, Gard JM, Stever C. Association and linkage of the dopamine transporter gene and attention-deficit hyperactivity disorder in children: heterogeneity owing to diagnostic subtype and severity. Am J Hum Genet. 1998;63(6):1767–76. doi: 10.1086/302132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolrich MW, Jbabdi S, Patenaude B, Chappell M, Makni S, Behrens T, Beckmann C, Jenkinson M, Smith SM. Bayesian analysis of neuroimaging data in FSL. Neuroimage. 2009;45(1 Suppl):S173–86. doi: 10.1016/j.neuroimage.2008.10.055. [DOI] [PubMed] [Google Scholar]
- Yang B, Chan RC, Jing J, Li T, Sham P, Chen RY. A meta-analysis of association studies between the 10-repeat allele of a VNTR polymorphism in the 3′-UTR of dopamine transporter gene and attention deficit hyperactivity disorder. Am J Med Genet B Neuropsychiatr Genet. 2007;144B(4):541–50. doi: 10.1002/ajmg.b.30453. [DOI] [PubMed] [Google Scholar]