

Abstract
Hypoxia-inducible factor 1 (HIF-1) mediates adaptive responses to reduced oxygen availability by regulating gene expression. A critical cell-autonomous adaptive response to chronic hypoxia controlled by HIF-1 is reduced mitochondrial mass and/or metabolism. Exposure of HIF-1-deficient fibroblasts to chronic hypoxia results in cell death due to excessive levels of reactive oxygen species (ROS). HIF-1 reduces ROS production under hypoxic conditions by multiple mechanisms including: a subunit switch in cytochrome c oxidase from the COX4-1 to COX4-2 regulatory subunit that increases the efficiency of complex IV; induction of pyruvate dehydrogenase kinase 1, which shunts pyruvate away from the mitochondria; induction of BNIP3, which triggers mitochondrial selective autophagy; and induction of microRNA-210, which blocks assembly of Fe/S clusters that are required for oxidative phosphorylation. HIF-1 is also required for ischemic preconditioning and this effect may be due in part to its induction of CD73, the enzyme that produces adenosine. HIF-1-dependent regulation of mitochondrial metabolism may also contribute to the protective effects of ischemic preconditioning.
1. Introduction
The story of life on Earth is a tale of oxygen production and utilization. Approximately 3 billion years ago, primitive single-celled organisms evolved the capacity for photosynthesis, a biochemical process in which photons of solar energy are captured by chlorophyll and used to power the reaction of CO2 and H2O to form glucose and O2. The subsequent rise in the atmospheric O2 concentration over the next billion years set the stage for the ascendance of organisms with the capacity for respiration, a process that consumes glucose and O2 and generates CO2, H2O, and energy in the form of ATP. Some of these single-celled organisms eventually took up residence within the cytoplasm of other cells and devoted all of their effort to energy production as mitochondria. Compared to the conversion of glucose to lactate by glycolysis, the complete oxidation of glucose by respiration provided such a large increase in energy production that it made possible the evolution of multicellular organisms. Among metazoan organisms, the progressive increase in body size during evolution was accompanied by progressively more complex anatomic structures that function to ensure the adequate delivery of O2 to all cells, ultimately resulting in the sophisticated circulatory and respiratory systems of vertebrates.
All metazoan cells can sense and respond to reduced O2 availability (hypoxia). Adaptive responses to hypoxia can be cell autonomous, such as the alterations in mitochondrial metabolism that are described below, or non-cell-autonomous, such as changes in tissue vascularization (reviewed in ref. 1). Primary responses to hypoxia need to be distinguished from secondary responses to sequelae of hypoxia, such as the adaptive responses to ATP depletion that are mediated by AMP kinase (reviewed in ref 2). In contrast, recent data suggest that O2 and redox homeostasis are inextricably linked and that changes in oxygenation are inevitably associated with changes in the levels of reactive oxygen species (ROS), as will be discussed below.
2. HIF-1 Regulates Oxygen Homeostasis in All Metazoan Species
A key regulator of the developmental and physiological networks required for the maintenance of O2 homeostasis is hypoxia-inducible factor 1 (HIF-1). HIF-1 is a heterodimeric transcription factor that is composed of an O2-regulated HIF-1α subunit and a constitutively expressed HIF-1β subunit [3,4]. HIF-1 regulates the expression of hundreds of genes through several major mechanisms. First, HIF-1 binds directly to hypoxia response elements, which are cis-acting DNA sequences located within target genes [5]. The binding of HIF-1 results in the recruitment of co-activator proteins that activate gene transcription (Fig. 1A). Only rarely does HIF-1 binding result in transcriptional repression [6]. Instead, HIF-1 represses gene expression by indirect mechanisms, which are described below. Second, among the genes activated by HIF-1 are many that encode transcription factors [7], which when synthesized can bind to and regulate (either positively or negatively) secondary batteries of target genes (Fig. 1B). Third, another group of HIF-1 target genes encode members of the Jumonji domain family of histone demethylases [8,9], which regulate gene expression by modifying chromatin structure (Fig. 1C). Fourth, HIF-1 can activate the transcription of genes encoding microRNAs [10], which bind to specific mRNA molecules and either block their translation or mediate their degradation (Fig. 1D). Fifth, the isolated HIF-1α subunit can bind to other transcription factors [11,12] and inhibit (Fig. 1E) or potentiate (Fig. 1F) their activity.
Fig. 1.
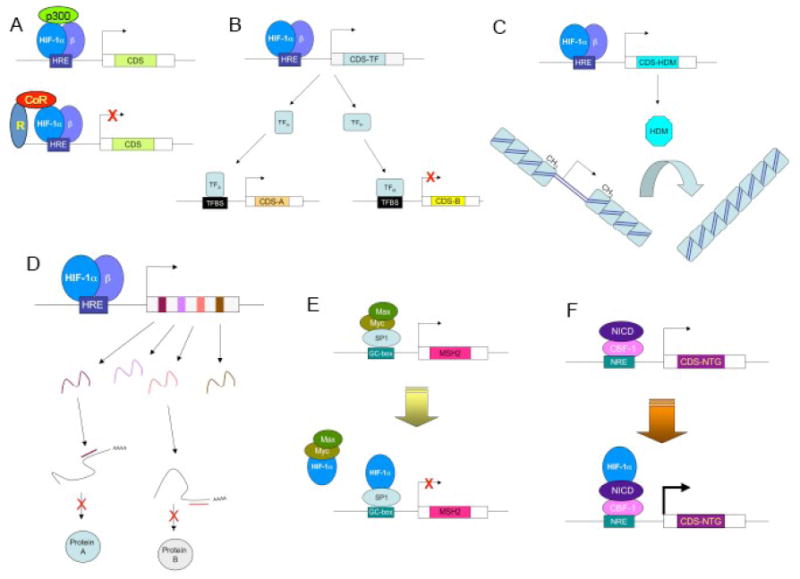
Mechanisms by which HIF-1 regulates gene expression. (A) Top: HIF-1 binds directly to target genes at a cis-acting hypoxia response element (HRE) and recruits coactivator proteins such as p300 to increase gene transcription. The coding sequence (CDS) of these HIF-1 target genes encode proteins that mediate adaptive responses to hypoxia. Bottom: There are only several rare examples in which HIF-1 binding leads to transcriptional repression. (B) HIF-1 binds directly to and transactivates a gene that encodes a transcription factor (TF), which either activates (TFA) or represses (TFB) the expression of secondary target genes by binding to its cognate transcription factor binding site (TFBS). (C) HIF-1 activates the transcription of genes encoding proteins with histone demethylase (HDM) activity. Demethylation of histones alters chromatin structure, which either decreases (as illustrated) or increases the expression of genes within the modified chromatin. (D) HIF-1 activates transcription of genes encoding microRNAs that bind to mRNAs and either block their translation or induce their degradation. (E) HIF-1a binds to the transcription factor SP1 and blocks activation of the gene encoding the mismatch DNA repair protein MSH2, thereby functioning as a co-repressor. (F) HIF-1a binds to the Notch intracellular domain (NICD) and potentiates transcriptional activation of Notch target genes, thereby functioning as a co-activator. (Adapted from ref. 50.)
HIF-1α and HIF-1β are present in all metazoan species, including the simple roundworm Caenorhabitis elegans [13], which consists of ~103 cells and has no specialized systems for O2 delivery. The fruit fly Drosophila melanogaster evolved tracheal tubes, which conduct air into the interior of the body from which it diffuses to surrounding cells. In vertebrates, the development of the circulatory and respiratory systems was accompanied by the appearance of HIF-2α, which is also O2-regulated and heterodimerizes with HIF-1β [14] but is only expressed in a restricted number of cell types [15], whereas HIF-1α and HIF-1β are expressed in all human and mouse tissues [16]. In Drosophila, the ubiquitiously expressed HIF-1α ortholog is designated Similar [17] and the paralogous gene that is expressed specifically in tracheal tubes is designated Trachealess [18].
3. HIF-1 Activity is Regulated by Oxygen
In the presence of O2, HIF-1α and HIF-2α are subjected to hydroxylation by prolyl-4-hydroxylase domain proteins (PHDs) that use O2 and α-ketoglutarate as substrates and generate CO2 and succinate as by-products [19]. Prolyl hydroxylation is required for binding of the von Hipple-Lindau protein, which recruits a ubiquitin-protein ligase that targets HIF-1α and HIF-2α for proteasomal degradation (Fig. 2). Under hypoxic conditions, the rate of hydroxylation declines and the non-hydroxylated proteins accumulate. HIF-1α transactivation domain function is also O2-regulated [20,21]. Factor inhibiting HIF-1 (FIH-1) represses transactivation domain function [22] by hydroxylating asparagine residue 803 in HIF-1α, thereby blocking the binding of the co-activators p300 and CBP [23].
Fig. 2.
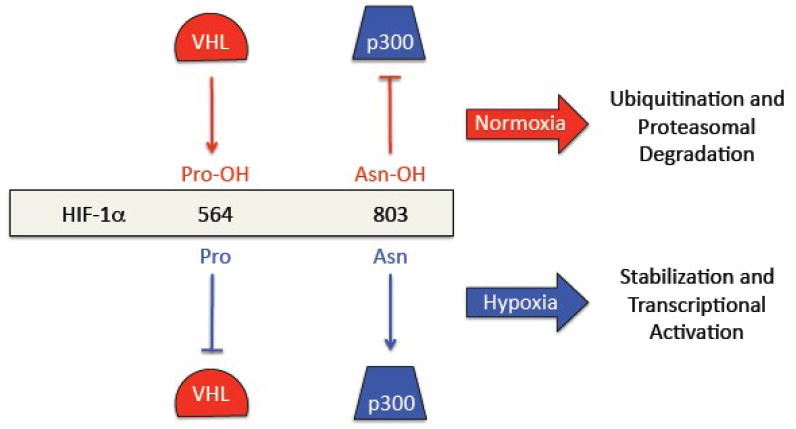
Negative regulation of HIF-1 activity by oxygen. Top: In the presence of O2: prolyl hydroxylation of HIF-1a leads to binding of the von Hippel-Lindau protein (VHL), which recruits a ubiquitin protein-ligase that targets HIF-1a for proteasomal degradation; and asparaginyl hydroxylation of HIF-1a blocks the binding of the coactivator protein p300. Bottom: Under hypoxic conditions, the hydroxylation reactions are inhibited, leading to decreased VHL binding and protein stabilization as well as increased p300 binding and transcriptional activation.
When cells are acutely exposed to hypoxic conditions, the generation of ROS at complex III of the mitochondrial electron transport chain (ETC) increases and is required for the induction of HIF-1α protein levels [24]. More than a decade after these observations were first made, the precise mechanism by which hypoxia increases ROS generation and by which ROS induces HIF-1α accumulation remain unknown. However, the prolyl and asparaginyl hydroxylases contain Fe2+ in their active site and oxidation to Fe3+ would block their catalytic activity. Since O2 is a substrate for the hydroxylation reaction, anoxia also results in a loss of enzyme activity. However, the concentration at which O2 becomes limiting for prolyl or asparaginyl hydroxylase activity in vivo is not known.
3. HIF-1 Regulates the Balance Between Oxidative and Glycolytic Metabolism
All metazoan organisms depend on mitochondrial respiration as the primary mechanism for generating sufficient amounts of ATP to maintain cellular and systemic homeostasis. Respiration, in turn, is dependent on an adequate supply of O2 to serve as the final electron acceptor in the ETC. In this process, electrons are transferred from complex I (or complex II) to complex III, then to complex IV, and finally to O2, which is reduced to water. This orderly transfer of electrons generates a proton gradient across the inner mitochondrial membrane that is used to drive the synthesis of ATP. At each step of this process, some electrons combine with O2 prematurely, resulting in the production of superoxide anion, which is reduced to hydrogen peroxide through the activity of mitochondrial superoxide dismutase. The efficiency of electron transport appears to be optimized to the physiological range of O2 concentrations, such that ATP is produced without the production of excess superoxide, hydrogen peroxide, and other ROS at levels that would result in the increased oxidation of cellular macromolecules and subsequent cellular dysfunction or death. In contrast, when O2 levels are acutely increased or decreased, an imbalance between O2 and electron flow occurs, which results in increased ROS production.
When cells such as mouse embryo fibroblasts (MEFs) are acutely subjected to hypoxia, e.g. a reduction of ambient O2 concentration from 20% to 0.5-1%, ROS levels increase within minutes. However, when MEFs are maintained at 0.5-1% O2 for several days, ROS levels actually decrease relative to cells that are maintained at 20% O2 [25,26]. When these experiments were performed with MEFs that are homozygous for a knockout (KO) allele at the locus encoding HIF-1α, chronic hypoxia causes ROS levels to increase rather than decrease, resulting in cell death within several days. Treatment of the cells with a free radical scavenger significantly increases the survival of the KO MEFs.
Remarkably, HIF-1a KO MEFs cultured at 1% O2 have higher ATP levels than wild-type MEFs cultured at 20% O2, demonstrating that O2 is not limiting for oxidative phosphorylation under these conditions [26]. However, continued high rates of respiration of KO cells under hypoxic conditions are associated with the production of lethal levels of ROS. Thus, the primary adaptive significance of the HIF-1-dependent switch from oxidative to glycolytic metabolism under hypoxic conditions is not maintenance of ATP levels (this is the purview of AMP kinase) but rather the maintenance of redox homeostasis.
MEFs require HIF-1 activity to make two critical metabolic adaptations to chronic hypoxia. First, HIF-1 activates the gene encoding pyruvate dehydrogenase (PDH) kinase 1 (PDK1), which phosphorylates and inactivates the catalytic subunit of PDH, the enzyme that converts pyruvate to acetyl coenzyme A (AcCoA) for entry into the mitochondrial tricarboxylic acid (TCA) cycle [25]. Second, HIF-1 activates the gene encoding BNIP3, a member of the Bcl-2 family of mitochondrial proteins, which triggers selective mitochondrial autophagy [26]. Interference with the induction of either of these proteins in hypoxic cells results in increased ROS production and increased cell death. Overexpression of either PDK1 or BNIP3 rescues HIF-1α-null MEFs. By shunting pyruvate away from the mitochondria, PDK1 decreases flux through the ETC and thereby counteracts the reduced efficiency of electron transport under hypoxic conditions, which would otherwise increase ROS production. PDK1 functions cooperatively with the product of another HIF-1 target gene, LDHA [27], which converts pyruvate to lactate, thereby further reducing available substrate for the PDH reaction.
PDK1 effectively reduces flux through the TCA cycle and thereby reduces flux through the ETC in cells that primarily utilize glucose as a substrate for oxidative phosphorylation. However, PDK1 is predicted to have little effect on ROS generation in cells that utilize fatty acid oxidation as their source of AcCoA. Hence another strategy to reduce ROS generation under hypoxic conditions is selective mitochondrial autophagy [26]. MEFs reduce their mitochondrial mass and O2 consumption by >50% after only two days at 1% O2. BNIP3 competes with Beclin-1 for binding to Bcl-2, thereby freeing Beclin-1 to activate autophagy. Using short hairpin RNAs to knockdown expression of BNIP3, Beclin-1, or Atg5 (another component of the autophagy machinery) phenocopied HIF-1α-null cells by preventing hypoxia-induced reductions in mitochondrial mass and O2 consumption as a result of failure to induce autophagy [26]. HIF-1-regulated expression of BNIP3L also contributes to hypoxia-induced autophagy [28]. Remarkably, mice heterozygous for the HIF-1α KO allele have a significantly increased ratio of mitochondrial:nuclear DNA in their lungs (even though this is the organ that is exposed to the highest O2 concentrations), indicating that HIF-1 regulates mitochondrial mass under physiological conditions in vivo [26]. In contrast to the selective mitochondrial autophagy that is induced in response to hypoxia as described above, autophagy (of unspecified cellular components) induced by anoxia does not require HIF-1, BNIP3, or BNIP3L, but is instead regulated by AMP kinase [29].
Another demonstration that HIF-1 regulates mitochondrial ROS production over the physiological range of O2 concentrations was the finding that cells express the COX4-1 regulatory subunit of cytochrome c oxidase (ETC complex IV) under aerobic conditions but switch to the COX4-2 subunit under hypoxic conditions. HIF-1 activates transcription of the genes encoding COX4-2 and LON, a mitochondrial protease that is required for degradation of COX4-1 [30]. It appears that the subunit switch is designed to optimize COX activity under hypoxic conditions. These results suggest that under conditions of chronic hypoxia, ETC complex IV can also become a source of increased ROS production if the COX4 subunit switch does not occur.
Most recently, another mechanism has been reported by which HIF-1 can block both mitochondrial respiration and ETC activity. HIF-1 activates the transcription of the microRNA miR-210 in many cell types [10,31] and miR-210 was shown to block expression of the iron-sulfur cluster assembly proteins ISCU1/2, which are required for the function of the TCA cycle enzyme aconitase and ETC complex I [32].
The multiplicity of HIF-1-mediated mechanisms identified so far by which cells regulate mitochondrial metabolism in response to changes in cellular O2 concentration (Fig. 3) suggests that this is a critical adaptive response to hypoxia. The fundamental nature of this physiological response is underscored by the fact that yeast also switch COX4 subunits in an O2-dependent manner but do so by an entirely different molecular mechanism [33], since yeast do not have a HIF-1α homologue. Thus, it appears that by convergent evolution both unicellular and multicellular eukaryotes possess mechanisms by which they modulate mitochondrial metabolism to maintain redox homeostasis despite changes in O2 availability. Indeed, it is the balance between energy, oxygen, and redox homeostasis that represents the key to life with oxygen.
Fig. 3.
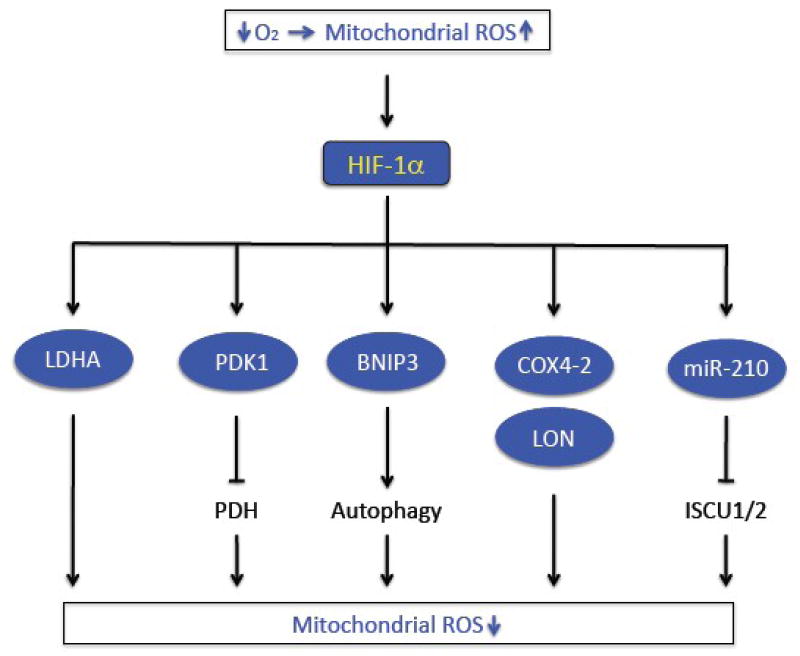
Regulation of mitochondrial metabolism by HIF-1. Acute hypoxia leads to increased mitochondrial generation of reactive oxygen species (ROS). Decreased O2 and increased ROS levels lead to decreased HIF-1a hydroxylation (see Fig. 2) and increased HIF-1-dependent transcription of genes encoding proteins (LDHA, PDK1, BNIP3, COX4-2, LON) or microRNA (miR-210) that reduce mitochondrial respiration and ROS production.
5. HIF-1 is a Critical Mediator of Hypoxic and Ischemic Cardiac Preconditioning
Exposure of the heart to one or more brief (~5-min) episodes of ischemia protects against myocardial cell death following a prolonged (~30-min) episode of ischemia, a phenomenon known as ischemic preconditioning [34]. Preconditioning results in two phases of protection, an acute or early phase immediately following the preconditioning stimulus and lasting no more than several hours and a delayed or late phase starting ~24 hours after the preconditioning stimulus and lasting for several days [35]. Perfusion of the heart with hypoxic blood [36] or adenosine analogues [37] is sufficient to induce acute preconditioning. Chronic exposure of rats to ambient hypoxia (3 weeks at 10% O2) also protects the heart against ischemia-reperfusion injury [38].
Exposure of mice to alternating cycles of ambient hypoxia (6% O2 for 6 min) and reoxygenation (21% O2 for 6 min) for 1 h induces delayed cardiac preconditioning and this effect is lost in Hif1a+/- mice that are heterozygous for the HIF-1α KO allele [39]. This protective effect appears to be mediated at least in part by the expression (in the kidney) and secretion into the bloodstream of the hormone erythropoietin (EPO), which is the product of the first identified HIF-1 target gene [3]. Direct administration of EPO into isolated perfused hearts results in acute protection against ischemia-reperfusion injury [40].
When isolated perfused hearts from wild type mice are subjected to two cycles of global no-flow ischemia (5 min) and reperfusion (5 min) as an acute preconditioning stimulus, they are protected against subsequent prolonged ischemia (30 min) and reperfusion, whereas this protection is completely lost in hearts from Hif1a+/- mice [41]. In contrast, hearts from Hif1a+/- mice are protected against ischemia-reperfusion injury when adenosine perfusion is used as the acute preconditioning stimulus, indicating a specific defect in responding to ischemia [41]. Inhibition of HIF-1a expression by transduction of small interfering RNAs into the heart also blocks ischemic preconditioning [42].
Adenosine levels increase in hearts subjected to ischemic preconditioning and pretreatment with adenosine receptor antagonists blocks the protective effect of ischemic preconditioning [43], whereas treatment with adenosine receptor agonists mimics ischemia as a preconditioning stimulus [35,43]. Adenosine is formed from extracellular ATP through the activity of two enzymes: Ectonucleoside triphosphate diphosphohydrolase 1 (ENTPD1, also known as CD39) converts ATP to ADP and then to AMP; and ecto-5’-nucleotidase (NT5E, also known as CD73) converts AMP to adenosine. CD73 expression is induced in hypoxic cells and this involves direct binding of HIF-1 to the Nt5e gene [44]. CD73 KO mice lack ischemic preconditioning [45]. Remarkably, hearts from Hif1a+/- mice are protected against ischemia-reperfusion injury by adenosine perfusion [41]. Taken together, these results suggest that production of adenosine during ischemic preconditioning is dependent on normal levels of HIF-1 and that loss of HIF-1 activity leads to insufficient production of adenosine, which in turn leads to the loss of preconditioning. Adenosine administration allows the defect in HIF-1-dependent adenosine production to be bypassed, thereby resulting in the rescue of ischemic Hif1a+/- hearts.
6. Does Regulation of Mitochondria by HIF-1 also Contribute to Preconditioning?
As described above, a major role of HIF-1 is to prevent excess mitochondrial ROS production under hypoxic conditions, suggesting that HIF-1 activation during ischemic preconditioning might protect the heart by inhibiting mitochondrial metabolism. Yet, as in the case of the adenosine pathway, the known mechanisms by which HIF-1 regulates mitochondrial respiration require transcriptional activation of target genes and the subsequent synthesis of the target gene products. However, a preconditioning protocol consisting of two cycles of 5-min ischemia/5-min reperfusion followed by a 30-min ischemic insult provides less than one hour for a protective response to be put in play before reperfusion occurs. Is this enough time?
An alternative model is that HIF-1a enters the mitochondria and directly alters mitochondrial metabolism, presumably by a mechanism that is independent of its known roles as a transcription factor. This proposal is not as radical as it seems, because an increasing number of transcription factors have been found in the mitochondria, including p53 [46], glucocorticoid receptor [47], and STAT3, which promotes oxidative phosphorylation by an undetermined mechanism [48]. In support of this model, a recent study found HIF-1a in the mitochondria of cultured cardiac myocytes subjected to a “preconditioning stimulus” consisting of 4 cycles of 1-hour hypoxia and 1-hour reoxygenation [49]. No specific effects of HIF-1a on mitochondrial metabolism were reported. Since this cell culture model is of limited relevance to the study of ischemic cardiac preconditioning, further studies are required to more rigorously test the intriguing possibility that HIF-1a mediates preconditioning via direct effects on mitochondrial metabolism.
Acknowledgments
Work in the author’s laboratory is supported by Public Health Service grants from NCI, NHLBI, and NIGMS. G.L.S. is the C. Michael Armstrong Professor at The Johns Hopkins University School of Medicine.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rey S, Semenza GL. Hypoxia-inducible factor 1-dependent mechanisms of vascularization and vascular remodeling. Cardiovasc Res. 2010;86:236–242. doi: 10.1093/cvr/cvq045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fogarty S, Hardie DG. Development of protein kinase activators: AMPK as a target in metabolic disorders and cancer. Biochim Biophys Acta. 2010;1804:581–591. doi: 10.1016/j.bbapap.2009.09.012. [DOI] [PubMed] [Google Scholar]
- 3.Wang GL, Semenza GL. Purification and characterization of hypoxia-inducible factor 1. J Biol Chem. 1995;270:1230–1237. doi: 10.1074/jbc.270.3.1230. [DOI] [PubMed] [Google Scholar]
- 4.Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA. 1995;92:5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Semenza GL, Wang GL. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol. 1992;12:5447–5454. doi: 10.1128/mcb.12.12.5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mole DR, Blancher C, Copley RR, Pollard PJ, Gleadle JM, Ragoussis J, Ratcliffe PJ. Genome-wide association of hypoxia-inducible factor (HIF)-1α and HIF-2α DNA binding with expression profiling of hypoxia-inducible transcripts. J Biol Chem. 2009;284:16767–16775. doi: 10.1074/jbc.M901790200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Manalo DJ, Rowan A, Lavoie T, Natarajan L, Kelly BD, Ye SQ, Garcia JG, Semenza GL. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood. 2005;105:659–669. doi: 10.1182/blood-2004-07-2958. [DOI] [PubMed] [Google Scholar]
- 8.Beyer S, Kristensen MM, Jensen KS, Johansen JV, Staller P. The histone demethylases JMJD1A and JMJD2B are transcriptional targets of hypoxia-inducible factor HIF. J Biol Chem. 2008;283:36542–36552. doi: 10.1074/jbc.M804578200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pollard PJ, Loenarz C, Mole DR, McDonough MA, Gleadle JM, Schofield CJ, Ratcliffe PJ. Regulation of Jumonji-domain-containing histone demethylases by hypoxia-13 inducible factor (HIF)-1α. Biochem J. 2008;416:387–394. doi: 10.1042/BJ20081238. [DOI] [PubMed] [Google Scholar]
- 10.Kulshreshtha R, Ferracin M, Wojcik SE, Garzon R, Alder H, Agosto-Perez FJ, Davuluri R, Liu CG, Croce CM, Negrini M, Calin GA, Ivan M. A microRNA signature of hypoxia. Mol Cell Biol. 2007;27:1859–1867. doi: 10.1128/MCB.01395-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koshiji M, To KK, Hammer S, Kumamoto K, Harris AL, Modrich P, Huang LE. HIF-1α induces genetic instability by transcriptionally downregulating MutSα expression. Mol Cell. 2005;17:793–803. doi: 10.1016/j.molcel.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 12.Gustafsson MV, Zheng X, Pereira T, Gradin K, Jin S, Lundkvist J, Ruas JL, Poellinger L, Lendahl U, Bondesson M. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev Cell. 2005;9:617–628. doi: 10.1016/j.devcel.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 13.Jiang H, Guo R, Powell-Coffman JA. The Caenorhabditis elegans hif-1 gene encodes a bHLH-PAS protein that is required for adaptation to hypoxia. Proc Natl Acad Sci USA. 2001;98:7916–7921. doi: 10.1073/pnas.141234698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wiesener MS, Turley H, Allen WE, Willam C, Eckhardt KU, Talks KL, Wood SM, Gatter KC, Harris AL, Pugh CW, Ratcliffe PJ, Maxwell PH. Induction of endothelial PAS domain protein-1 by hypoxia: characterization and comparison with hypoxia-inducible factor-1α. Blood. 1998;92:2260–2268. [PubMed] [Google Scholar]
- 15.Flamme I, Fröhlich T, von Reutern TM, Kappel A, Damert A, Risau W. HRF, a putative basic helix-loop-helix-PAS-domain transcription factor is closely related to hypoxia-inducible factor-1α and developmentally expressed in blood vessels. Mech Dev. 1997;63:51–60. doi: 10.1016/s0925-4773(97)00674-6. [DOI] [PubMed] [Google Scholar]
- 16.Wiener CM, Booth G, Semenza GL. In vivo expression of mRNAs encoding hypoxia-inducible factor 1. Biochem Biophys Res Commun. 1996;225:485–488. doi: 10.1006/bbrc.1996.1199. [DOI] [PubMed] [Google Scholar]
- 17.Bacon NC, Wappner P, O’Rourke JF, Bartlett SM, Shilo B, Pugh CW, Ratcliffe PJ. Regulation of the Drosophila bHLH-PAS protein Sima by hypoxia: functional evidence for homology with mammalian HIF-1α. Biochem Biophys Res Commun. 1998;249:811–816. doi: 10.1006/bbrc.1998.9234. [DOI] [PubMed] [Google Scholar]
- 18.Wilk R, Weizman I, Shilo BZ. trachealess encodes a bHLH-PAS protein that is an inducer of tracheal cell fates in Drosophila. Genes Dev. 1996;10:93–102. doi: 10.1101/gad.10.1.93. [DOI] [PubMed] [Google Scholar]
- 19.Kaelin WG, Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30:393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 20.Jiang BH, Zheng JZ, Leung SW, Roe R, Semenza GL. Transactivation and inhibitory domains of hypoxia-inducible factor 1α: Modulation of transcriptional activity by oxygen tension. J Biol Chem. 1997;272:19253–19260. doi: 10.1074/jbc.272.31.19253. [DOI] [PubMed] [Google Scholar]
- 21.Pugh CW, O’Rourke JF, Nagao M, Gleadle JM, Ratcliffe PJ. Activation of hypoxia-inducible factor-1: definition of regulatory domains within the α subunit. J Biol Chem. 1997;272:11205–11214. doi: 10.1074/jbc.272.17.11205. [DOI] [PubMed] [Google Scholar]
- 22.Mahon PC, Hirota K, Semenza GL. FIH-1: a novel protein that interacts with HIF-1α and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 2001;15:2675–2686. doi: 10.1101/gad.924501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002;16:1466–1471. doi: 10.1101/gad.991402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chandel NS, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM, Schumacker PT. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1α during hypoxia: a mechanism of O2 sensing. J Biol Chem. 2000;275:25130–25138. doi: 10.1074/jbc.M001914200. [DOI] [PubMed] [Google Scholar]
- 25.Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177–185. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 26.Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, Gonzalez FJ, Semenza GL. Mitochondrial autophagy is a HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283:10892–10903. doi: 10.1074/jbc.M800102200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 27.Semenza GL, Jiang BH, Leung SW, Passantino R, Concordet JP, Maire P, Giallongo A. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J Biol Chem. 1996;271:32529–32537. doi: 10.1074/jbc.271.51.32529. [DOI] [PubMed] [Google Scholar]
- 28.Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouysségur J, Mazure NM. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol. 2009;29:2570–2581. doi: 10.1128/MCB.00166-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Papandreou I, Lim AL, Laderoute K, Denko NC. Hypoxia signals autophagy in tumor cells via AMPK activity, independent of HIF-1, BNIP3, and BNIP3L. Cell Death Differ. 2008;15:1572–1581. doi: 10.1038/cdd.2008.84. [DOI] [PubMed] [Google Scholar]
- 30.Fukuda R, Zhang H, Kim JW, Shimoda L, Dang CV, Semenza GL. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell. 2007;129:111–122. doi: 10.1016/j.cell.2007.01.047. [DOI] [PubMed] [Google Scholar]
- 31.Corn PG. Hypoxic regulation of miR-210: shrinking targets expand HIF-1’s influence. Cancer Biol Ther. 2008;7:265–267. doi: 10.4161/cbt.7.2.5745. [DOI] [PubMed] [Google Scholar]
- 32.Chan SY, Zhang YY, Hemann C, Mahoney CE, Zweier JL, Loscalzo J. MicroRNA-210 controls mitochondrial metabolism during hypoxia by repressing the iron-sulfur cluster assembly proteins ISCU1/2. Cell Metab. 2009;10:273–284. doi: 10.1016/j.cmet.2009.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burke PV, Poyton RO. Structure/function of oxygen-regulated isoforms in cytochrome c oxidase. J Exp Biol. 1998;201:1163–1175. doi: 10.1242/jeb.201.8.1163. [DOI] [PubMed] [Google Scholar]
- 34.Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–1136. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- 35.Marber MS, Latchman DS, Walker JM, Yellon DM. Cardiac stress protein elevation 24 hours after brief ischemia or heat stress is associated with resistance to myocardial infarction. Circulation. 1993;88:1264–1272. doi: 10.1161/01.cir.88.3.1264. [DOI] [PubMed] [Google Scholar]
- 36.Shizukuda Y, Mallet RT, Lee SC, Downey HF. Hypoxic preconditioning of ischemic canine myocardium. Cardiovasc Res. 1992;26:534–542. doi: 10.1093/cvr/26.5.534. [DOI] [PubMed] [Google Scholar]
- 37.Liu GS, Thornton J, Van Winkle DM, Stanley AW, Olsson RA, Downey JM. Protection against infarction afforded by preconditioning is mediated by A1 adenosine receptors in rabbit heart. Circulation. 1991;84:350–356. doi: 10.1161/01.cir.84.1.350. [DOI] [PubMed] [Google Scholar]
- 38.Tajima M, Katayose D, Bessho M, Isoyama S. Acute ischemic preconditioning and chronic hypoxia independently increase myocardial tolerance to ischemia. Cardiovasc Res. 1994;28:312–319. doi: 10.1093/cvr/28.3.312. [DOI] [PubMed] [Google Scholar]
- 39.Cai Z, Manalo DJ, Wei G, Rodriguez ER, Fox-Talbot K, Lu H, Zweier JL, Semenza GL. Hearts from rodents exposed to intermittent hypoxia or erythropoietin are protected against ischemia-reperfusion injury. Circulation. 2003;108:79–85. doi: 10.1161/01.CIR.0000078635.89229.8A. [DOI] [PubMed] [Google Scholar]
- 40.Cai Z, Semenza GL. Phosphatidylinositol-3-kinase signaling is required for erythropoietin-mediated acute protection against myocardial ischemia/reperfusion injury. Circulation. 2004;109:2050–2053. doi: 10.1161/01.CIR.0000127954.98131.23. [DOI] [PubMed] [Google Scholar]
- 41.Cai Z, Zhong H, Bosch-Marce M, Fox-Talbot K, Wang L, Wei C, Trush MA, Semenza GL. Complete loss of ischemic preconditioning-induced cardioprotection in mice with partial deficiency of HIF-1α. Cardiovasc Res. 2008;77:463–470. doi: 10.1093/cvr/cvm035. [DOI] [PubMed] [Google Scholar]
- 42.Eckle T, Köhler D, Lehmann R, El Kasmi K, Eltzschig HK. Hypoxia-inducible factor-1 is central to cardioprotection: a new paradigm for ischemic preconditioning. Circulation. 2008;118:166–175. doi: 10.1161/CIRCULATIONAHA.107.758516. [DOI] [PubMed] [Google Scholar]
- 43.Thornton JD, Liu GS, Olsson RA, Downey JM. Intravenous pretreatment with A1-selective adenosine analogues protects the heart against infarction. Circulation. 1992;85:659–665. doi: 10.1161/01.cir.85.2.659. [DOI] [PubMed] [Google Scholar]
- 44.Synnestvedt K, Furuta GT, Comerford KM, Louis N, Karhausen J, Eltzschig HK, Hansen KR, Thompson LF, Colgan SP. Ecto-5’-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J Clin Invest. 2002;110:993–1002. doi: 10.1172/JCI15337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eckle T, Krahn T, Grenz A, Köhler D, Mittelbronn M, Ledent C, Jacobson MA, Osswald H, Thompson LF, Unertl K, Eltzschig HK. Cardioprotection by ecto-5’-nucleotidase (CD73) and A2B adenosine receptors. Circulation. 2007;115:1581–1590. doi: 10.1161/CIRCULATIONAHA.106.669697. [DOI] [PubMed] [Google Scholar]
- 46.Marchenko ND, Zaika A, Moll UM. Death signal-induced localization of p53 protein to mitochondria, a potential role in apoptotic signaling. J Biol Chem. 2000;275:16202–16212. doi: 10.1074/jbc.275.21.16202. [DOI] [PubMed] [Google Scholar]
- 47.Psarra AM, Sekeris CE. Glucocorticoid receptors and other nuclear transcription factors in mitochondria and possible functions. Biochim Biophys Acta. 2009;1787:431–436. doi: 10.1016/j.bbabio.2008.11.011. [DOI] [PubMed] [Google Scholar]
- 48.Gough DJ, Corlett A, Schlessinger K, Wegrzyn J, Larner AC, Levy DE. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science. 2009;324:1713–1716. doi: 10.1126/science.1171721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rane S, He M, Sayed D, Vashistha H, Malhotra A, Sadoshima J, Vatner DE, Vatner SF, Abdellatif M. Downregulation of miR-199a derepresses hypoxia-inducible factor 1a and sirtuin 1 and recapitulates hypoxia preconditioning in cardiac myocytes. Circ Res. 2009;104:879–886. doi: 10.1161/CIRCRESAHA.108.193102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Semenza GL. Oxygen homeostasis. Systems Biol Med. 2010 doi: 10.1002/wsbm.69. [DOI] [PubMed] [Google Scholar]