

Abstract
Bipolar disorder (BPD) and schizophrenia (SZ) are severe psychiatric illnesses with a combined prevalence of 4%. A disturbance of energy metabolism is frequently observed in these disorders. Several pieces of evidence point to an underlying dysfunction of mitochondria: i) decreased mitochondrial respiration; (ii) changes in mitochondrial morphology; iii) increases in mitochondrial DNA (mtDNA) polymorphisms and in levels of mtDNA mutations; iv) downregulation of nuclear mRNA molecules and proteins involved in mitochondrial respiration; v) decreased high-energy phosphates and decreased pH in the brain; and vi) psychotic and affective symptoms, and cognitive decline in mitochondrial disorders. Furthermore, transgenic mice with mutated mitochondrial DNA polymerase show mood disorder-like phenotypes. In this review, we will discuss the genetic and physiological components of mitochondria and the evidence for mitochondrial abnormalities in BPD and SZ. We will furthermore describe the role of mitochondria during brain development and the effect of current drugs for mental illness on mitochondrial function. Understanding the role of mitochondria, both developmentally as well as in the ailing brain, is of critical importance to elucidate pathophysiological mechanisms in psychiatric disorders.
Introduction
Overlapping phenotypes between bipolar disorder and schizophrenia
Bipolar disorder (BPD) and schizophrenia (SZ), two disorders accompanied by psychosis, affect approximately 4% of the world population. It is widely accepted that a combination of genetic vulnerability and environmental factors lead to the manifestation of these illnesses (Burmeister et al., 2008; Karlsgodt et al., 2008; Lewis and Levitt, 2002). Both diseases have a neurodevelopmental component, with the onset of symptoms occurring most frequently during late adolescence or early adulthood (Maier et al., 2006). Because of the multi-factorial pathogenesis of BPD and SZ, the disease severity and rate of progression can vary greatly from patient to patient (Jablensky, 2006).
Schizophrenia and BPD-I share many overlapping phenotypes (Lin and Mitchell, 2008; Thaker, 2008). Evidence for shared genetic factors derives from pharmacology, pathology, family studies and DNA linkage analysis (Berrettini, 2003; Berrettini, 2000; Craddock et al., 2006; Heckers et al., 2002; Post, 1999). Among the overlapping traits are disturbances in energy metabolism and in mitochondrial function.
Evidence for disturbed mitochondrial function in bipolar disorder and schizophrenia
Abnormalities in energy metabolism were found in functional assays (Cavelier et al., 1995; Maurer et al., 2001; Regenold et al., 2009) and in magnetic resonance spectroscopy studies (Dager et al., 2004; Deicken et al., 1995b; Frey et al., 2007; Jensen et al., 2006; Kato et al., 1993; Kato et al., 1995; Ongur et al., 2009; Stork and Renshaw, 2005; Yurgelun-Todd et al., 1996). Gene and protein expression studies point to a decrease of factors and enzymes involved in ATP generation and storage (Iwamoto et al., 2005; Karry et al., 2004; Klushnik et al., 1991; Konradi et al., 2004; MacDonald et al., 2006; Middleton et al., 2002; Mill et al., 2008; Pennington et al., 2008; Washizuka et al., 2005; Washizuka et al., 2009), and linkage analysis showed an association of both disorders with genes involved in mitochondrial function (Kirk et al., 1999; Washizuka et al., 2003b; Washizuka et al., 2006; Xu et al., 2008; Zhang et al., 2009). Mitochondrial structural abnormalities have been reported in patients with BPD (Cataldo et al.), and both diseases are associated with mitochondrial DNA (mtDNA) mutations and polymorphisms (Amar et al., 2007; Kato et al., 1997; Kato et al., 2000; Kato, 2001; Munakata et al., 2004; Munakata et al., 2005; Quiroz et al., 2008; Rollins et al., 2009; Shao et al., 2008; Ueno et al., 2009). Conversely, bona fide mitochondrial diseases are frequently comorbid with psychotic symptoms and misdiagnosed as SZ or BPD (Campos et al., 2001; Fattal et al., 2006; Grover et al., 2006; Mancuso et al., 2008; Prayson and Wang, 1998). The latter observation makes a particularly strong case for a role of mitochondria in the clinical symptoms of psychosis, as the primary disease-causing event in mitochondrial disorders is the mtDNA mutation.
Mood stabilizers provide partial protection against mitochondrial toxicity, whereas first-generation antipsychotic drugs might have adverse effects on mitochondrial respiration (Bachmann et al., 2009; King et al., 2001; Maurer and Moller, 1997; Maurer et al., 2009; Pereira et al., 1992; Prince et al., 1997b; Sagara, 1998; Struewing et al., 2007; Valvassori et al.; Washizuka et al., 2009).
Mitochondrial pathology could be the consequence of genetic susceptibility (Brandon et al., 2005b; Ishizuka et al., 2006; James et al., 2004; Kvajo et al., 2008; Millar et al., 2005; Stoffel et al., 1996), secondary to dysregulation of other neurotransmitter systems (Ben-Shachar, 2002; Brenner-Lavie et al., 2009; Chen et al., 2008) or the results of environmental impacts such as exposure to toxins, famine, infections and substance abuse (Brown and Derkits; Brown and Yamamoto, 2003; Kroll, 2007; Kyle and Pichard, 2006), all of which are known risk factors for BPD and SZ.
Mitochondrial genetics
Mitochondria are cell organelles surrounded by a double membrane, each of which is a phospholipid bilayer. The intermembrane space separates both membranes (figure 1), and the mitochondrial matrix is the innermost area, surrounded by the double membrane. The matrix contains the enzymes for the citric acid cycle (tricarboxylic acid cycle, TCA), which supply the proton/electron donors NADH+H+ and FADH2 for the electron transport chain. The inner mitochondrial membrane is folded into cristae to provide increased surface area for the electron transport chain, a major component of the inner membrane. The electron transport chain pumps protons from the matrix into the intermembrane space, and produces energy in the form of ATP upon re-entry of protons into the matrix (figure 1).
Figure 1.
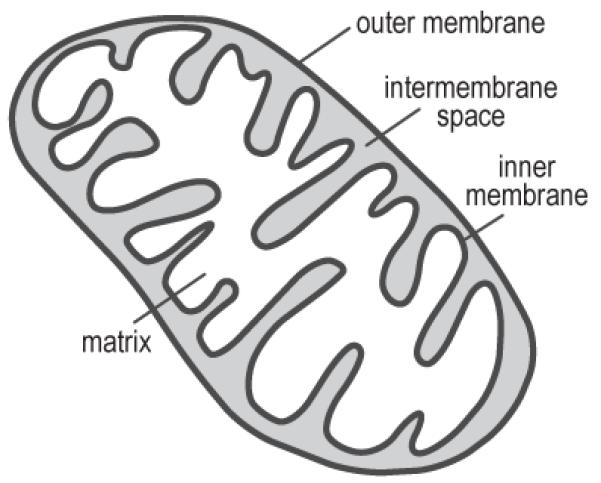
Mitochondrial anatomy: Mitochondria are intracellular organelles that contain both outer and inner membranes separated by an intermembrane space. The innermost mitochondrial compartment, the matrix, contains the enzymes necessary for tricarboxylic acid (TCA) cycle, while the inner membrane contains the components of the electron transport chain.
Mitochondria are thought to have originated from a symbiotic relationship between an anaerobic proto-eukaryotic cell that engulfed an aerobic bacterium. As such, mitochondria retain bacterial characteristics, including a circular, polycistronic DNA that lacks both introns and histones. The 16.6-kB mitochondrial genome (mtDNA) contains 37 genes: 22 transfer RNAs (tRNAs), two ribosomal RNAs (rRNAs), seven genes for proteins of the electron transport chain complex I, one complex III gene, three complex IV genes, and two complex V genes (Wallace, 2005). This leaves the majority of mitochondrial proteins encoded in the nuclear DNA. The two strands of mtDNA are referred to as the heavy strand, which contains the majority of mitochondrial genes, and the light strand, which contains the NADH dehydrogenase 6 (mt-ND6) gene and eight tRNA genes. Since mitochondrial genes contain no introns, the only noncoding region of mtDNA is the regulatory D-loop, which contains promoters for both mtDNA strands and the origin of replication for the heavy strand.
Mitochondrial DNA is organized into DNA-protein clusters called nucleoids, which are tethered to the inner mitochondrial membrane (Clay Montier et al., 2009; Iborra et al., 2004). By comparing the number of mtDNA-positive foci and the number of mtDNA copies in a human cell line, it was estimated that each nucleoid contained an average of eight mtDNA molecules (Iborra et al., 2004), though there is probably considerable variability. In addition to mtDNA, nucleoids contain protein elements essential to replication and transcription of the mitochondrial genome, such as mitochondrial transcription factors and the helicase Twinkle (Iborra et al., 2004), both of which are encoded in the nuclear DNA. The mechanism for mtDNA replication is not yet fully characterized, but it is thought that mitochondrial transcription factor A (TFAM) and DNA polymerase γ (POLG) bind in the D-loop at the origin of replication on the heavy strand. The heavy strand is then replicated until the origin of the light strand is reached, roughly 2/3 of the way through the DNA molecule. At this point, light strand replication begins in the opposite direction. Transcription of mitochondrial RNA is thought to occur in a similar manner as replication, and after synthesis of a polycistronic transcript, tRNAs fold and are cleaved, producing individual mRNAs for each gene, which are polyadenylated before translation by mitochondrial ribosomes (Wallace, 2005).
All cells are polyploid for mtDNA, and mtDNA content varies greatly among tissues (Frahm et al., 2005; McInerny et al., 2009; Pysh and Khan, 1972). Unlike nuclear DNA replication, mtDNA replication does not coordinate with the cell cycle. At some point, however, replication of the mitochondrial genome requires communication between mitochondria and the nucleus because the transcription factors required by mtDNA for replication, DNA polymerase and helicase, are nucleus-encoded. The complete mechanism by which mtDNA copy number is regulated is not yet known, but TFAM is thought to play a role in this process, as TFAM knockout mice show mtDNA depletion, resulting in deficient electron transport and ATP synthesis, and the development of myopathy (Silva et al., 2000; Wredenberg et al., 2002). TFAM overexpression, in contrast, results in increased expression of some mitochondrial genes, but does not seem to increase mitochondrial function above normal levels (Ekstrand et al., 2004).
Mitochondrial DNA copy number varies between brain regions: quantitative, real-time PCR (qPCR) against the mtDNA-encoded genes NADH dehydrogenase 1 (mt-ND1), mt-ND4, cytochrome B, and 12S rRNA revealed copy numbers between 2,000 and 5,000 per cell in the midbrain and striatum of rodents (McInerny et al., 2009). Copy number also varies depending on the region assayed (McInerny et al., 2009). The spinal cord, striatum, and cortex have mtDNA copy numbers of 10,000 or more per cell, the medulla possesses roughly 8,000 copies per cell, and the midbrain and cerebellum each have 4,000 copies or less per cell (Frahm et al., 2005; McInerny et al., 2009).
An ultrastructural analysis of mitochondria in neurons and glia across various regions of adult rat brains showed variations among regions and cell types in mitochondrial size and number, as well as density and length of cristae within mitochondria (Pysh and Khan, 1972). Astrocytes tended to have less dense cristae, but higher mitochondrial numbers and mitochondrial volume fraction than oligodendrocytes. Neurons had higher cristae packing than glia, as well as less variation in mitochondrial size across brain regions. The volume fraction of mitochondria in neurons was found to be lowest in cerebellar granule cells, which correlates with the observed lower cerebellar mtDNA copy number (Frahm et al., 2005; McInerny et al., 2009). Of all cell types, neurons had the highest mitochondrial volume fraction, which correlates with their high energy demands. Astrocytes contained a mitochondrial volume fraction that was just below that of neurons, which could be attributed to the role of astrocytes in signaling and metabolic support of neurons.
Mitochondrial physiology: oxidative phosphorylation, calcium buffering and apoptosis
A central function of mitochondria is to convert the chemical energy stored in sugars to high-energy phosphates, a usable form of cellular energy that is produced by the electron transport chain in the process of oxidative phosphorylation (OXPHOS), also known as mitochondrial respiration. Glycolysis in the cytoplasm breaks glucose down into pyruvate, which is further metabolized to NADH+H+ in the mitochondrial matrix by the TCA cycle (figure 2). NADH+H+ then transfers its high-energy electrons to NADH dehydrogenase, which is complex I of the electron transport chain (figure 2A, 3). The TCA cycle also produces succinate, which carries electrons to complex II (succinate dehydrogenase). The electrons are passed from complexes I or II to coenzyme Q, followed by complex III and cytochrome c (coenzyme Q: cytochrome c reductase), and complex IV (cytochrome c oxidase). Electrons from complex IV combine with O2 to produce H2O (figure 3). With each transfer of an electron from one carrier to the next, the electron releases energy, which is coupled to transport of hydrogen ions from the matrix to the intermembrane space, resulting in an electrochemical gradient across the inner mitochondrial membrane. Hydrogen ions pass back into the matrix through complex V (ATP synthase), an energy-producing event used to synthesize ATP from ADP and inorganic phosphate (Brandon et al., 2005a; Wallace, 1999; Wallace, 2005).
Figure 2.
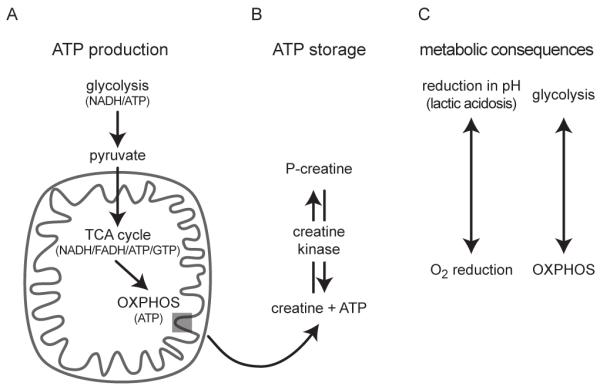
Energy metabolism and metabolic consequences: A) Glycolysis, a cytosolic process, breaks down glucose to pyruvate. The net yield of glycolysis is two molecules of ATP and two molecules of the electron donor NADH, which are shuttled into the mitochondria. Likewise, pyruvate crosses into the mitochondria and is converted acetyl-CoA, which enters the tricarboxylic acid (TCA) cycle. The conversion to acetyl-CoA produces NADH. The TCA cycle, then produces three molecules of NADH, one molecule of the electron donor FADH2, as well as one molecule of ATP or GTP. NADH and FADH2 donate electrons to the electron transport chain of the inner mitochondrial membrane, where ATP is synthesized during oxidative phosphorylation (OXPHOS). Each glucose molecule produces 36 ATP molecules when metabolized by OXPHOS. B) Creatine kinase uses ATP to phosphorylate creatine and produce phosphocreatine (P-creatine), a stable product for extended storage of ATP. During periods of high energy demand, creatine kinase acts as a phosphatase to retrieve ATP from P-creatine. C) metabolic shift from OXPHOS to glycolysis results in cellular acidification through lactic acidosis, a metabolization from pyruvate to lactate. Conversely, OXPHOS activity measured as reduction in O2.
Figure 3.
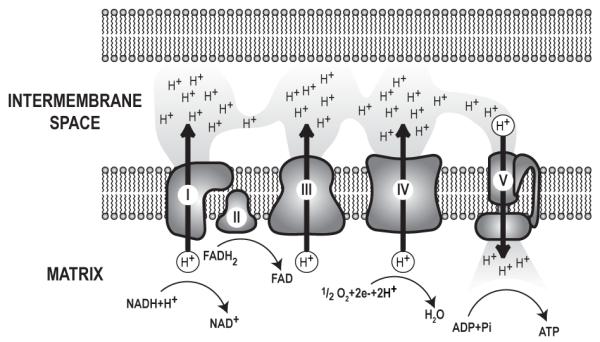
The electron transport chain: Electron transport chain complexes I-V are part of the inner mitochondrial membrane. NADH and FADH2 from the tricarboxylic acid (TCA) cycle donate electrons to complexes I and II, respectively. These electrons are transferred to complex III and complex IV, sequentially. With each transfer, the electrons release energy. Energy release from electrons is coupled to the movement of protons from the matrix to the intermembrane space, resulting in a proton gradient across the inner mitochondrial membrane. Upon reaching complex IV, electron pairs combine with ½ O2 and 2H+ to create H2O. Complex V, or ATP synthase, releases energy stored in the proton gradient by allowing proton flow into the matrix. Complex V couples this energy release to the synthesis of ATP from ADP and Pi. Each complex is assembled from a number of proteins. For example, complex I, the largest complex, has 39 proteins encoded in the nuclear DNA and 7 proteins encoded in the mtDNA, while complex II, the smallest complex, has 4 nuclear encoded proteins.
In addition to providing cellular energy, mitochondria form microdomains with calcium (Ca2+) influx sites to buffer cytosolic Ca2+ (Rizzuto and Pozzan, 2006). Ca2+ released into the cytosol from either external sources such as NMDA receptors or from the endoplasmic reticulum is taken up by mitochondria and released slowly, preventing high levels of cytosolic Ca2+ from inducing stress and excitotoxicity (Baron et al., 2003; Rizzuto and Pozzan, 2006). Ca2+ uptake into mitochondria stimulates enzymes of the TCA cycle and facilitates increased ATP synthesis through increased production of NADH+H+ (Orrenius et al., 2003). This is important since Ca2+ activates many cellular processes with high energy demands, and the clearance and storage of Ca2+ requires ATP-dependent pumps. Mitochondrial Ca2+ overload, however, can have detrimental effects on mitochondrial viability and ultimately causes cell death (Orrenius et al., 2003). Glucose deprivation, for example, can cause mitochondrial membrane depolarization and rises in intracellular Ca2+, increasing the cell’s susceptibility to excitotoxicity (Isaev et al., 2008; Stelmashook et al., 2009).
Mitochondria are pivotal for cellular resilience and the management of stress. Mitochondria contain proteins that prevent and facilitate apoptosis. Insults such as excess Ca2+ influx, decreased mitochondrial membrane potential, or DNA damage from oxidative stress can result in mitochondrial membrane permeabilization. Often, this event occurs through the opening of the mitochondrial permeability transition pore (mPTP), a protein complex spanning the inner and outer mitochondrial membranes. Though the identity of all mPTP components has not yet been fully determined, the complex is likely composed of the voltage-dependent anion channel (VDAC), cyclophilin D, and the adenine nucleotide translocase (Galluzzi et al., 2009). Opening of the mPTP results in release of intramitochondrial proteins such as cytochrome c and pro-caspases. In the cytosol, cytochrome c activates apoptotic protease activating factor 1 (Apaf1), which assembles into the apoptosome structure (figure 4). The apoptosome activates procaspase-9, an initiator caspase that is cleaved into caspase-9. Caspase-9 then activates executioner caspases such as caspase-3, which facilitate nuclear DNA cleavage and breakdown of the cytoskeleton and nuclear lamina, causing cells to assume an apoptotic, rounded morphology (Hotchkiss et al., 2009).
Figure 4.
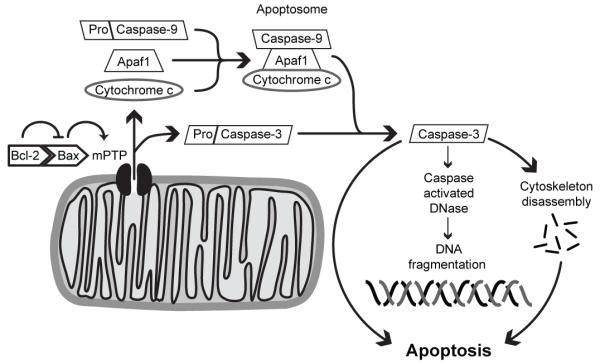
Mitochondria-mediated apoptosis: Pro-apoptotic proteins such as Bax promote opening of the mitochondrial permeability transition pore (mPTP). Anti-apoptotic proteins such as Bcl-2 bind and inhibit pro-apoptotic proteins. Once open, the mPTP causes release of intramitochondrial proteins such as cytochrome c and procaspase-3. In the cytosol, cytochrome c assembles with apoptotic protease activating factor 1 (Apaf1) and procaspase-9 into the apoptosome, where procaspase-9 is cleaved to yield active caspase-9. Caspase-9, then, facilitates cleavage-mediated activation of procaspase-3 to caspase-3, which in turn facilitates DNA fragmentation, cytoskeletal disassembly, and apoptotic cell death.
On the mitochondrial membrane are proteins of the Bcl-2 family, which promote or prevent apoptosis through protein-protein interactions. Pro-apoptotic members of the family, such as Bax and Bad, facilitate the opening of the mPTP (Galluzzi et al., 2009; Ouyang and Giffard, 2004). Anti-apoptotic proteins, such as Bcl-2 and Bcl-xL, prevent apoptosis by binding and inhibiting pro-apoptotic proteins (Chipuk and Green, 2008; Ouyang and Giffard, 2004), (figure 4). Overexpression of these anti-apoptotic proteins in mice enhances cell viability and resilience after insult, whereas knockout results in premature death (Einat et al., 2005; Murphy et al., 1996).
Apoptosis serves important developmental functions. During mammalian central nervous system development, an abundance of neurons is produced in proliferative zones surrounding the ventricles. These ventricular zones are the site of neurogenesis and supply neurons to the cortex, striatum, and hippocampus. Neurons are produced in excess and nearly half must be eliminated during early adolescence through mitochondria-mediated apoptotic mechanisms. Interestingly, many survival factors and growth cues that determine whether a neuron will survive or die are affected by lithium, a mood stabilizer used to treat BPD. These factors include BDNF, the Wnt pathway, the ERK/MAPK pathway, and the PLC/PIP2 pathway (Machado-Vieira et al., 2009).
The role of mitochondria in oxidative stress and DNA damage
Reactive oxygen species (ROS) are oxygen-containing free radicals created as byproducts of both physiological and abnormal electron transport. At baseline, roughly 1-5% of all oxygen consumed by the cell is converted to ROS (Lee and Wei, 2005), but ROS are produced in larger quantities after a mitochondrial insult, such as excess Ca2+ or exposure to mitochondrial toxins. ROS arise when electrons are prematurely released from complexes near the beginning of the electron transport chain. This results in the production of superoxide anions (O2·−). Excess ROS production can also occur during nutrient overload, when electron transport chain complexes are oversaturated with electrons and release electrons promiscuously (Russell et al., 2002; Wallace, 2005). Superoxide anions can be converted to H2O2 by superoxide dismutases, and to water by glutathione peroxidases or catalase (Wallace, 2005). When ROS are not neutralized, they damage proteins, lipids, and nucleic acids. Within the electron transport chain, the iron-sulfur (Fe-S) clusters that mediate electron transport in complexes I and III are most susceptible to oxidative damage (Wallace, 2005). Mitochondrial DNA is also particularly vulnerable to oxidative stress since it is located at the main site of ROS production and lacks protective histones (Wallace, 2005).
In addition to mtDNA damage due to ROS exposure, the lack of histones and the limited proofreading ability of POLG causes a higher mutation rate in mtDNA relative to nuclear DNA. Mitochondrial DNA mutations accumulate throughout a lifetime, while mtDNA levels and mitochondrial function tend to decline with age (Wallace, 2005). Although mtDNA levels increase with cell size, cells from older donors contained less mtDNA per picogram of protein than that of younger probands (Shmookler Reis and Goldstein, 1983).
Oxidative damage induces point mutations but can also result in large deletions of mtDNA. A 5 kB mtDNA deletion, termed the “common deletion”, spans the mt-ND3, mt-ND4, mt-ND5, cytochrome c oxidase III (mt-COIII), mt-ATPase6, and mt-ATPase8 genes, as well as several tRNA genes (Brandon et al., 2005a). This deletion is one of the more prevalent mtDNA alterations, but occurs at low rates in non-diseased tissues and tends to increase in prevalence with age (Brandon et al., 2005a; Fuke et al., 2008; McInerny et al., 2009). In some mitochondrial disorders, such as chronic progressive external opthalmoplegia and Kearns-Sayre Syndrome, this deletion reaches high levels. As a result, the cells with high levels of this common deletion produce more reactive oxygen species when stimulated with an H2O2 insult, resulting in reduced cell viability (Jou et al., 2005; Liu et al., 2007; Wang and Lu, 2009).
Mitochondrial diseases
Mutations in mtDNA result in debilitating diseases if they affect a large percentage of the mitochondrial population. Since nearly all mitochondria are inherited from the mother, diseases caused by mtDNA mutations exhibit a matrilineal inheritance pattern (Shao et al., 2008). Multiple mtDNA genotypes can exist within the same cell, a state termed heteroplasmy (figure 5). A one-celled embryo with both wild-type and mutant mtDNA copies can pass on an uneven proportion of mutant mtDNAs to either daughter cell, resulting in tissue-specific variations in the proportion of mutated mtDNA (figure 5). This condition is termed ‘mosaicism’ (Youssoufian and Pyeritz, 2002). When the proportion of mutated mtDNA passes a particular threshold, symptoms of mitochondrial pathology become evident. The symptoms are dependent on which tissue has the highest amount of mutated mitochondria as well as which tissue has the highest energy demand. Mutations of any gene in the mitochondrial genome could affect expression or function of electron transport chain proteins. Detrimental mitochondrial mutations impair the effectiveness of OXPHOS, and highly OXPHOS-reliant tissues such as the brain, pancreas, and muscle are more commonly affected (Wallace, 2005). Common symptoms of mitochondrial disease include seizure, blindness, diabetes, muscle weakness, and cardiomyopathy (DiMauro and Schon, 2008). While many of the mitochondrial disorder symptoms are physical deficiencies, psychiatric symptoms are frequently comorbid (Campos et al., 2001; Fattal et al., 2006; Grover et al., 2006; Mancuso et al., 2008; Prayson and Wang, 1998).
Figure 5.

Mitochondrial mutations are distributed in a mosaic pattern. During fission of mitochondria, mutant and wild-type mtDNAs can be distributed unevenly, A. Heteroplasmic oocytes contain a mixture of normal and mutated mitochondria, B. Mitochondria are randomly partitioned to the daughter cells during mitosis. The presence of tissue-specific differences in mutant mtDNAs is termed mosaicism.
Morphological and physiological changes in mitochondria in psychiatric diseases
Unlike most other tissues, the brain depends exclusively on glycolysis and OXPHOS to create ATP. Thus, any mitochondrial pathology will have the most pronounced effect on the brain.
Morphological changes of mitochondria have been observed in BPD and SZ in brain tissue as well as peripheral tissue. Mitochondria were significantly smaller in the prefrontal cortex (PFC) of BPD patients compared to normal controls. (Cataldo et al.). In the PFC of SZ patients, oligodendrocytes showed drastic reductions in mitochondrial numbers and volume (Uranova et al., 2001). SZ patients had also fewer mitochondria in the striatum than controls or individuals with other psychiatric diagnoses (Kung and Roberts, 1999).
In peripheral cells and cell lines, a number of pathological changes were observed. The cellular location of mitochondria was altered in lymphoblastoid and fibroblast cell lines of BPD patients (Cataldo et al.). Lymphocytes of BPD patients failed to respond to glucose deprivation while lymphocytes of normal controls responded with an upregulation of the elements of the electron transport chain (Naydenov et al., 2007). Mononuclear cells of SZ patients had enlarged mitochondria with fragmented cristae, which was independent of antipsychotic drug type used (Inuwa et al., 2005). In a similar study, activated lymphocytes in individuals with SZ had a lower mitochondrial density than controls (Uranova et al., 2007).
Abnormalities in mitochondrial function have been observed in both BPD and SZ. For example, SZ patients showed large reductions in cytochrome-c oxidase activity in the caudate nucleus, the frontal cortex and the temporal cortex (Cavelier et al., 1995; Maurer et al., 2001). Rodent studies suggest that this reduction is not caused by treatment (Prince et al., 1997a; Prince et al., 1997b). In the PFC of BPD patients, complex I activity was impaired and associated with increased protein oxidation and nitration (Andreazza et al., 2010). Impairment of OXPHOS activity causes accumulation of lactate and unprocessed glucose (figure 2A). In cerebrospinal fluid from BPD patients, lactate and glucose levels were increased, indicating a shift from mitochondrial respiration to glycolysis (Regenold et al., 2009). These changes were not observed in SZ patients in the same study, although peripheral blood mononuclear cells from first-onset, antipsychotic-naive SZ patients showed an increased expression of proteins associated with the glycolysis pathway after stimulation with staphylococcal enterotoxin (Herberth et al.). Dysregulation of the glycolysis pathway is in line with the increased prevalence of metabolic syndrome and insulin resistance observed in psychotic patients (Regenold et al., 2002). Although antipsychotic drugs can contribute to abnormal glucose metabolism (Newcomer, 2004), increased insulin resistance is a hallmark of psychotic disorders (Regenold et al., 2002).
The response to metabolic stress is impaired in BPD, and this impairment is closely linked to mitochondria (Naydenov et al., 2007). Peripheral blood mononuclear cells of BPD patients lacked the molecular response to glucose deprivation shown by cells of normal controls (Naydenov et al., 2007). Microarray analysis of the peripheral blood mononuclear cells revealed that controls upregulated expression of electron transport genes in response to glucose deprivation, while cells from BPD patients failed to show any changes in mitochondrial gene expression. Increased number of electron transport chain units can potentially improve ATP output by increasing the chance of contact with NADH+H+ and reducing the chance of NADH+H+ diffusion. BPD mitochondria fail to maintain ATP output, which could heighten their sensitivity to mild insults. (Plant et al., 2002)
Ca2+ homeostasis is impaired in both BPD and SZ. Since mitochondria buffer cytosolic Ca2+, impaired Ca2+ homeostasis has detrimental effects on mitochondrial function and viability (Orrenius et al., 2003). Elevated Ca2+ levels have been detected in peripheral cells from BPD and SZ patients (Dubovsky et al., 1992; Kusumi et al., 1992; Ripova et al., 1997). Lymphoblastoid cell lines derived from BPD patients show higher Ca2+ peaks after thapsigargin, thrombin- or lysophosphatidic acid-mediated stimulation (Dubovsky et al., 1992; Kato et al., 2003; Ripova et al., 1997; Wasserman et al., 2004).
Taken together, the data provide ample evidence for abnormalities in mitochondrial structure and function in BPD and SZ. The combination of elevated Ca2+ levels and decreased ATP production will cause continuous cellular stress and reduce the ability to appropriately respond to temporary peaks in stressful stimuli such as increased glutamate release during emotional situations and drug use, or decreased glucose levels during times of famine or deliberate starvation. These factors are known risk factors for psychotic episodes (Brown and Derkits; Brown and Yamamoto, 2003; Kroll, 2007; Kyle and Pichard, 2006)
Genetic evidence for abnormal mitochondrial function in BPD and SZ: Mutations and polymorphisms in mtDNA and nuclear DNA.
Large family studies have revealed increased rates of BPD and SZ within families, suggesting a genetic vulnerability (Gershon et al., 1982). While diseases caused by mtDNA mutations display maternal inheritance, evidence for an increase in maternal parent-of-origin effect in psychiatric disorders is at best weak (Kato et al., 1996; Lan et al., 2007; McMahon et al., 1995). In a group of 439 families affected by SZ, rates of maternal and paternal inheritance patterns were roughly equal, and 52% of the families investigated had ill relatives on both sides of the family (DeLisi et al., 2000). The prevalence of mental illness-associated mtDNA single nucleotide polymorphisms (SNPs) has also been investigated. Several groups have assessed the prevalence of particular mtDNA SNPs in disease populations relative to controls (Amar et al., 2007; Kato et al., 2000; Kato, 2001; Kazuno et al., 2009; Marchbanks et al., 2003; McMahon et al., 2000; Munakata et al., 2004; Munakata et al., 2005; Shao et al., 2008; Ueno et al., 2009). Most groups identified different SNPs across the mitochondrial genome as having disease associations, and no particular gene or region of mtDNA appears to be targeted. Participants’ ancestry may contribute to the variability, as sites with disease-associated SNPs in one haplogroup may fail to be polymorphic in another (Kato, 2001; Shao et al., 2008). The data suggest that mtDNA mutations in BPD and SZ might be somatic rather than inherited, indicating either an overall increased vulnerability of mtDNA, or a higher exposure to DNA-damaging factors.
The common deletion can be inherited maternally or acquired such as in response to oxidative stress. Significant increases in the prevalence of the common deletion were found in the cortex of BPD probands and suicide victims (Kato et al., 1997). In contrast, a similar study of frontal cortex brains from individuals with BPD or SZ failed to find an association with common deletion levels (Kakiuchi et al., 2005). The percentage of mtDNA that contains the common deletion is quite low, which might make it difficult to obtain consistent results (McInerny et al., 2009). Low common deletion levels also bring into question the impact of damaged DNA on brain function, since the prevailing intact mtDNA should be able to overcome the deleterious effects of mutant mtDNA (McInerny et al., 2009). However, due to mosaicism, deletion levels can vary even between adjacent tissues and the results might not accurately reflect a potential pathology.
Several disease-associated mitochondrial SNPs have been found to have functional implications. Patients with the 5178A>C polymorphism, which is associated with BPD, tended to have lower frontal lobe pH compared to patients with 5178A when assayed by MRS (Kato and Kato, 2000; Kato et al., 2000). The SZ- and BPD-associated SNP 3243A>G, which lies in a leucine-tRNA gene, hampers the tRNA’s aminoacylation, resulting in a compensatory upregulation of the leucyl-tRNA synthetase LARS2 that is seen both in cybrids containing 3243A>G and patient brains (Munakata et al., 2005). A polymorphism in the mt-ND1 gene which is associated with BPD, 3644T>C, causes decreased mitochondrial membrane potential and complex I activity (Munakata et al., 2004). The BPD-associated 10398A>G polymorphism has been linked to decreased mitochondrial matrix pH, and higher baseline and post-stimulation mitochondrial Ca2+ levels (Kato and Kato, 2000; Kato et al., 2003; Kazuno et al., 2006; Kazuno et al., 2008; Washizuka et al., 2003a). Interestingly, the latter two effects are tempered by the mood stabilizer valproic acid (VPA) (Kato et al., 2003; Kazuno et al., 2008). In addition to VPA-related alterations in Ca2+ responses, the 10398A>G SNP was found to be more prevalent in BPD patients that did not respond to lithium treatment (Washizuka et al., 2003a). An mtDNA variant observed in SZ, 12027T>C results in the change from isoleucine to threonine of the mt-ND4 subunit of NADH-ubiquinone reductase (Marchbanks et al., 2003).
The vast majority of genes encoding mitochondrial proteins are products of nuclear DNA and are subject to Mendelian inheritance patterns and epigenetic regulation. Nucleus-encoded mitochondrial genes are involved in OXPHOS, the TCA cycle, shuttle systems that control influx and efflux of molecules from mitochondria, and factors involved in mtDNA replication such as TFAM and POLG. A number of genetic risk factor for SZ and BPD are associated with mitochondria and mitochondrial function. Disease-associated mutations in nuclear complex I genes have been identified, for example, and could contribute to observed reductions in complex I mRNA and protein expression levels (Andreazza et al., 2010; Ben-Shachar and Karry, 2008; Karry et al., 2004; Washizuka et al., 2003b; Washizuka et al., 2005; Washizuka et al., 2006; Washizuka et al., 2009; Xu et al., 2008). Reductions in complex I protein expression have also been found to correlate with reduced complex I function as well as increased protein oxidation and nitrosylation (Andreazza et al., 2010).
Some nuclear genes associated with BPD and SZ are connected to mitochondria. DISC-1, which encodes disrupted in schizophrenia 1, was identified in a large Scottish pedigree as a susceptibility gene for mental illness including SZ and BPD. While the function of DISC-1 is not entirely known, it has been shown to interact with structural proteins that organize the cytoskeleton and synaptic areas of developing neurons (Ishizuka et al., 2006). Animal models of mutant DISC-1 have revealed that the protein localizes predominantly to mitochondria (James et al., 2004). Expression of truncated DISC-1 in cell lines was used to mimic the human DISC-1 mutation. Compared to full-length DISC-1, truncated DISC-1 showed increased nuclear localization (Brandon et al., 2005b; Millar et al., 2005). Overexpression of truncated DISC-1 isoforms induced abnormal mitochondrial morphologies, indicating changes in mitochondrial dynamics such as fission and fusion (Millar et al., 2005).
Another gene locus associated with SZ and BPD is G72/G30 (Bass et al., 2009; Boks et al., 2007; Chumakov et al., 2002; Hattori et al., 2003; Prata et al., 2008; Williams et al., 2006). While no gene product has yet been identified for G30, G72 (D-amino acid oxidase activator) encodes a protein that is expressed in cerebellum, hippocampus, amygdala, and cortex, including DLPFC (Boks et al., 2007; Kvajo et al., 2008; Lang et al., 2007; Otte et al., 2009). The G72 protein localizes to mitochondria, and its overexpression can alter mitochondrial morphology by enhancing mitochondrial fragmentation (Kvajo et al., 2008). While these changes do not appear to affect cell survival, they may play a role in dendritic arborization, as neurons overexpressing the G72 gene product exhibit increased dendritic branching (Kvajo et al., 2008).
Velocardiofacial syndrome has a high incidence of psychotic symptoms (Arinami, 2006; Hercher and Bruenner, 2008; Ousley et al., 2007). The disease is caused by a deletion of 22q11.2. A number of genes in the most commonly deleted area are important for mitochondrial function, such as the mitochondrial citrate transporter, the mitochondrial ribosomal protein MRPL40, proline dehydrogenase, thioredoxin reductase 2, and the zinc finger protein, ZDHHC8 (Maynard et al., 2008). Genetic studies of SZ have shown associations with these genes (Chen et al., 2004; Li et al., 2004; Liu et al., 2002). The research focus in the deletion region is mostly on the dopamine system (i.e. catechol-o-methyltransferase) and on genes involved in synaptic connectivity (Karayiorgou et al.), and detailed information on the contribution of mitochondria to the symptoms has not been collected yet.
While the evidence for a primary mitochondrial pathology in BPD and SZ is mixed, the evidence for psychotic and mood disorder symptoms in bona fide mitochondrial disorders is quite strong (Campos et al., 2001; Fattal et al., 2006; Grover et al., 2006; Mancuso et al., 2008; Prayson and Wang, 1998). The data demonstrate that mitochondria are important factors of consideration in psychiatric disorders, though they might not necessarily be the primary cause for these disorders.
Molecular evidence for abnormal mitochondrial function in BPD and SZ: distorted gene and protein levels
In studies of psychiatric patients, units of the electron transport chain have consistently shown reduced expression profiles. For example, the 24-kDa and 51-kDa subunits of complex I were significantly decreased in the PFC in SZ (Karry et al., 2004). In the same study, complex I subunits were also decreased in the striatum. In the cerebellum, a similar pattern of expression was observed, indicating that impairments in mitochondrial function may be widespread in individuals with these disorders. In a gene expression study of the hippocampus, an extensive downregulation of genes involved in the electron transport chain was observed in BPD (Konradi et al., 2004). Similar observations were made in the hippocampus in SZ (Altar et al., 2005), and a downregulation of TCA cycle genes was observed in the PFC (Middleton et al., 2002).
Although the argument has been made that only brain samples with decreased pH show decreased mitochondrial pathologies, and thus these samples should be removed (Vawter et al., 2006), one needs to consider that a decrease in mitochondrial respiration will cause a shift to glycolysis and thus a drop in pH (see figure 2A, 2C). Removing samples with lower pH removes all samples with mitochondrial pathologies, as has been well demonstrated (Vawter et al., 2006). Magnetic resonance spectroscopy studies of living patients circumvent the problem of low pH in postmortem samples, and further support the notion of a link between brain pH and altered energy metabolism (Dager et al., 2004; Kato et al., 1993; Kato et al., 1998). These studies are outlined further below.
Creatine kinase is an enzyme involved in the storage of high-energy phosphates produced by mitochondria (figure 2B). A decreased output of ATP might lead to a reduction in creatine kinase. It is thus not surprising that levels of creatine kinase mRNA were downregulated in the hippocampus and PFC in BPD and SZ (MacDonald et al., 2006), another indication of altered energy metabolism.
In lymphoblastoid cell lines, a complex I gene (NDUFV2) was downregulated in BPDI but not BPDII (Washizuka et al., 2009). Abnormalities were also found in peripheral blood mononuclear cells of BPD patients, which failed to respond to glucose deprivation stress (Naydenov et al., 2007).
Mitochondrial pathology may not be uniform across all brain regions and cell types. In contrast to the various studies reporting a downregulation of mitochondrial genes in the brain, complex I activity and expression of the 24- and 51-kDa iron-sulfur flavoprotein subunits were increased in platelets of SZ patients, with no change in BPD patients (Ben-Shachar et al., 1999; Dror et al., 2002). The degree of increase in complex I activity correlated directly with the severity of positive symptoms (Dror et al., 2002). Moreover, the parieto-occipital cortex showed increased mRNA levels of selected complex I genes in BPD (Ben-Shachar and Karry, 2008) and SZ (Karry et al., 2004).
One should not assume that disturbances in mitochondrial function might have the same behavioral cluster independent of tissue type or brain area. Firstly, mitochondrial mutations need to be present in brain areas involved in psychoses. Secondly, bioenergetic demands vary across brain areas and are linked to stressors such as Ca2+ and glutamate. Thus, the percentage of mutated mitochondria needed to cause problems varies. Neurons in some brain areas might be able to handle slight mitochondrial abnormalities while neurons in other brain areas cease to function properly, particularly when under increased energy demand. These differences might enable some behaviors to remain unaffected, while other behaviors become affected.
In vivo evidence of mitochondrial dysfunction in psychiatric diseases: imaging techniques
In vivo evidence for mitochondrial involvement in psychiatric illnesses derives from magnetic resonance spectroscopy (MRS), an imaging technique that allows visualization of energy-related metabolite levels and pH in the brain. Inefficient OXPHOS leads to accumulation of lactic acid from glycolysis and acidification of the tissue (figure 2C), as well as depletion of high-energy phosphates. The high-energy phosphates ATP and phosphocreatine (PCr) are detectable by MRS (figure 2). MRS can also measure levels of other metabolic markers such as phosphomonoesters (PMEs), which include the phospholipid precursors phosphoethanolamine and phosphocholine. PMEs are often used as markers of phospholipid synthesis levels in tissue (Kato et al., 1993); reductions in PMEs indicate lipid breakdown.
MRS enables the measurement of brain metabolic activity in alive, awake patients. Kato et al. found reduced frontal lobe pH in medicated and unmedicated BPD patients (Kato et al., 1998). Although the sample size was small, comparison of medicated to unmedicated patients revealed no significant difference in brain pH, suggesting that mood stabilizers do not affect brain pH. MRS was furthermore performed during euthymic and manic states (Kato et al., 1993). In the euthymic state, participants showed reduced frontal lobe pH and PME levels as compared to the manic state. The intra-individual differences between euthymia and mania point toward state-dependent mitochondrial abnormalities in BPD, rather than a predetermined pathology. These data were further supported by two-dimensional proton echo-planar spectroscopic imaging of medication-free BPD patients in a depressed or mixed-mood state, which showed a shift from oxidative phosphorylation toward glycolysis (Dager et al., 2004).
Reduced PME and increased phosphodiester (PDE) levels were observed in the frontal cortex in BPD and SZ patients, indicating increased lipid breakdown in the brain (Deicken et al., 1995b; Fujimoto et al., 1992; Klemm et al., 2001; Pettegrew et al., 1991; Stanley et al., 1994). BPD patients also exhibited a reduction and a left-right asymmetry in PCr levels that was not seen in controls, suggesting that BPD patients have abnormalities in either the production or maintenance of high-energy phosphates (Deicken et al., 1995b; Kato et al., 1994).
Likewise, SZ patients exhibit markers of increased lipid breakdown in frontal cortex, with increased PDEs in both frontal hemispheres, and reduced PMEs in left frontal cortex. The severity and degree of asymmetry of such abnormalities directly relates to performance on the PFC-reliant Wisconsin Card Sorting Test (Deicken et al., 1995c). In addition, increased PCr and decreased ATP levels in the right temporal lobe of SZ patients correlates with positive symptom severity (Deicken et al., 1995a).
The use of advanced imaging techniques has revealed metabolic deficiencies in psychiatric patients that are consistent with the mitochondrial pathology seen in postmortem studies. Although these studies cannot differentiate primary pathologies from secondary consequences, it is quite likely that mitochondrial pathologies are responsible for some of the symptoms observed in BPD and SZ.
Rodent studies provide evidence for a link of mitochondrial function with behavioral endophenotypes of psychiatric disorders
Of the many animal models of SZ and BPD, few have examined mitochondrial pathologies. Mice that have been genetically modified to express mutant POLG specifically in neurons displayed increased startle response and altered circadian behavior (Kasahara et al., 2006). These behaviors were ameliorated by lithium treatment as well as by electroconvulsive shock treatment (Kasahara et al., 2008). The results support the notion that mutations in mitochondria-related nuclear genes can reproduce behaviors seen in psychiatric illness. In the ventral hippocampal rodent model of SZ, reduced PFC complex I protein levels were observed after puberty, but not before—a time course consistent with that of the onset of SZ (Ben-Shachar et al., 2009).
Effects of mood stabilizers and antipsychotics on mitochondria
The predominant treatments for BPD and SZ are mood stabilizers and antipsychotic drugs. Most of these drugs were discovered serendipitously and, despite impressive efforts, their mechanism of action if only partly understood. The mood stabilizer VPA is metabolized in mitochondria, and many antipsychotic drugs or their metabolites are taken up into mitochondria (Inuwa et al., 2005; Li et al., 1991). In the human neuroblastoma cell line, SH-SY5Y, therapeutic doses of VPA and lithium induced increases in mitochondrial oxygen consumption and mitochondrial membrane potential. The drugs also normalized complex IV activity and caspase expression after a methamphetamine-induced insult (Bachmann et al., 2009). Lithium increased the activity of electron transport chain complexes I, II and III in homogenates of the human PFC, while a slight reduction was seen in complex IV activity at very high drug doses (Maurer et al., 2009). This indicates that mood stabilizers may alleviate some of the symptoms of BPD by improving mitochondrial function.
Lithium and VPA affect and stabilize intracellular Ca2+ dynamics (Kazuno et al., 2008; Quiroz et al.; Shalbuyeva et al., 2007; Wasserman et al., 2004). Rat brain mitochondria incubated in lithium-containing media maintained their membrane potential better after a Ca2+-mediated insult than mitochondria incubated in normal media (Shalbuyeva et al., 2007). Both drugs also have antiapoptotic effects. They are known to inhibit cytochrome c release from mitochondria and to increase expression of the anti-apoptotic gene Bcl-2 (Bachmann et al., 2009; Chen and Chuang, 1999; Michaelis et al., 2006; Shalbuyeva et al., 2007). In lymphoblastoid cell lines, VPA enhanced expression of the ETC complex I subunit NDUFV2 (Washizuka et al., 2009). In SH-SY5Y human neuroblastoma cell lines, lithium treatment attenuated rotenone-induced elevations in caspase-3 activity, and prevented the late stages of apoptosis (King et al., 2001; Shalbuyeva et al., 2007). Cerebellar granule cells exhibited reduced expression of pro-apoptotic proteins Bax and p53 after lithium treatment, and both lithium and VPA enhance cell viability and reduce apoptosis rates (Chen and Chuang, 1999; Michaelis et al., 2006). In addition, lithium and VPA raise expression of glutathione-S-transferase, which participates in reversal of oxidative damage (Bachmann et al., 2009; Wang et al., 2004).
VPA is a histone deacetylase inhibitor and facilitates gene expression via this epigenetic mechanism. Deacetylated histones inhibit transcription of the sequences to which they are bound. VPA increases acetylation of histones H3 and H4, an event associated with enhanced expression of brain-derived neurotrophic factor (BDNF) and glutamic acid decarboxylase 67 (GAD67) (Gavin et al., 2009; Sharma et al., 2006).
The first generation of antipsychotic drugs, “conventional antipsychotics”, are potent inhibitors of the dopamine D2 receptor (Seeman, 1987). “Atypical antipsychotic drugs” target other neurotransmitter systems, including serotonin, norepinephrine, and cannabinoids, and produce fewer extrapyramidal side effects (Carpenter and Koenig, 2008). Antipsychotic drugs do not improve the mitochondrial pathology observed in SZ, and some of these drugs inhibit complex I activity. The effects of typical and atypical antipsychotic drugs on mitochondrial electron transport chain complexes were examined in acute or chronic drug regimens of adult male rats (Prince et al., 1997a). Both haloperidol and fluphenazine reduced complex I activity in striatum, frontal cortex, hippocampus, and cerebellum. Similar results were observed when rats received chronic low doses of the mitochondrial toxin MPTP. Treatments with the atypical antipsychotic clozapine, however, did not alter complex I activity. Both typical and atypical antipsychotic drugs increased complex IV activity in frontal cortex, but only atypical antipsychotic drugs increased complex IV activity in the hippocampus. In rat liver, typical, but not atypical, antipsychotics inhibited complex I activity and facilitated activity of complex IV (Modica-Napolitano et al., 2003). Despite the increased complex IV activity seen with many antipsychotics, it appears that their inhibition of complex I results in an overall inhibition of maximal oxygen consumption rate after uncoupling and, more importantly, might reduce the ability to produce ATP. This suggests that antipsychotics might contribute to mitochondrial instability. Overall, studies that investigate the effects of antipsychotics and mood stabilizers on mitochondria are very important and provide insight into how the drugs might alleviate or contribute to the symptoms of BPD and SZ.
Concluding remarks
Neurons are uniquely dependent on mitochondria for energy production. However, mitochondria are not just important for ATP synthesis, but they are also involved in Ca2+ homeostasis, apoptosis, brain development, and brain function. A large number of studies have shown that the overall stability of mitochondria, the efficiency of OXPHOS, and the ability to buffer Ca2+ and neutralize ROS, are affected in BPD and SZ. While many of these disturbances could be secondary to lifestyle and drug treatment, evidence from animal and genetic studies indicates that in some cases mitochondrial pathology might be the primary cause.
Reduced mitochondrial viability and function causes decreased ATP production and reserves. Ion pumps need ATP to maintain the membrane potential in neurons. Proper information processing depends on the ability of the neuron to maintain a solid membrane potential with minimal leakage. One could imagine how a flaccid membrane potential could lead to (a) diminished information processing, i.e. not all information gets adequately propagated, or (b) chance depolarization leading to an erroneous signal. Furthermore, with little ATP reserves (such a PCr) stressful situations or periods of decreased glucose availability could cause severe malfunctions, explaining known risk factors for affective disorders and SZ (Brown and Derkits, 2010; Brown and Yamamoto, 2003; Kroll, 2007; Kyle and Pichard, 2006).
A question that arises is why are higher brain functions and behaviors predominantly affected and why are other metabolic and neuronal systems seemingly functioning normally? First, the energy demands of particular neuronal types might be responsible for their vulnerability. For example, in the hippocampus certain populations of GABA interneurons have higher activities of cytochrome C, indicating higher energy demands in these cells (Gulyas et al., 2006). Interestingly, markers for GABA neurons are dramatically reduced in the hippocampus in SZ and BPD (Benes et al., 1998; Heckers et al., 2002), and in BPD are correlated with a reduction in nuclear genes coding for the mitochondrial electron transport chain (Konradi et al., 2004). Thus, neurons with the highest energy demand are abnormal in BPD and SZ. Other neurons might have lower energy demands and less vulnerability to subtle mitochondrial abnormalities.
Second, different neuronal populations might respond differently to stressors such as glutamate. Populations with high sensitivity or high exposure to stressors would be particularly vulnerable during episodes of low ATP availability. Thirdly, minor problems in neuronal circuits might be more obvious in higher-level cognitive function and thought processing compared to less complex functions. Fourthly, some neuronal circuits or neurotransmitter systems might be able to compensate for cell stress and even cell loss. This is well known for the nigrostriatal pathway in Parkinson’s disease, where 70-80% of dopaminergic neurons are lost before the first motor symptoms emerge (Bernheimer et al., 1973). This level of neuroplasticity might be protective during mitochondrial damage, but does not seem to be available in cortical areas. Finally, in the case of mtDNA abnormalities, mosaicism could cause some brain areas to be affected but not others.
Any combination of the factors above can explain how a universal cell organelle might affect specific behaviors. Mitigating genetic and environmental factors might have some influence on which microcircuits are more protected while facilitating factors might cause cellular stress in other circuits. Thus, the genetic and external environments influence the clinical presentation and diagnosis.
No matter where in the sequence of disease-causing events mitochondria might rank, mitochondrial pathology could be an important factor in the manifestation of clinical symptoms. Thus, therapeutic approaches to strengthen mitochondrial function could significantly reduce symptoms and increase the quality of life of patients.
Acknowledgement
The work was supported by MH084131, MH74000 and MH67999. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding institute or the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Altar CA, Jurata LW, Charles V, Lemire A, Liu P, Bukhman Y, Young TA, Bullard J, Yokoe H, Webster MJ, Knable MB, Brockman JA. Deficient hippocampal neuron expression of proteasome, ubiquitin, and mitochondrial genes in multiple schizophrenia cohorts. Biol Psychiatry. 2005;58:85–96. doi: 10.1016/j.biopsych.2005.03.031. [DOI] [PubMed] [Google Scholar]
- Amar S, Shamir A, Ovadia O, Blanaru M, Reshef A, Kremer I, Rietschel M, Schulze TG, Maier W, Belmaker RH, Ebstein RP, Agam G, Mishmar D. Mitochondrial DNA HV lineage increases the susceptibility to schizophrenia among Israeli Arabs. Schizophr Res. 2007;94:354–358. doi: 10.1016/j.schres.2007.04.020. [DOI] [PubMed] [Google Scholar]
- Andreazza AC, Shao L, Wang JF, Young LT. Mitochondrial complex I activity and oxidative damage to mitochondrial proteins in the prefrontal cortex of patients with bipolar disorder. Arch Gen Psychiatry. 2010;67:360–368. doi: 10.1001/archgenpsychiatry.2010.22. [DOI] [PubMed] [Google Scholar]
- Arinami T. Analyses of the associations between the genes of 22q11 deletion syndrome and schizophrenia. J Hum Genet. 2006;51:1037–1045. doi: 10.1007/s10038-006-0058-5. [DOI] [PubMed] [Google Scholar]
- Bachmann RF, Wang Y, Yuan P, Zhou R, Li X, Alesci S, Du J, Manji HK. Common effects of lithium and valproate on mitochondrial functions: protection against methamphetamine-induced mitochondrial damage. Int J Neuropsychopharmacol. 2009;12:805–822. doi: 10.1017/S1461145708009802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron KT, Wang GJ, Padua RA, Campbell C, Thayer SA. NMDA-evoked consumption and recovery of mitochondrially targeted aequorin suggests increased Ca2+ uptake by a subset of mitochondria in hippocampal neurons. Brain Res. 2003;993:124–132. doi: 10.1016/j.brainres.2003.09.022. [DOI] [PubMed] [Google Scholar]
- Bass NJ, Datta SR, McQuillin A, Puri V, Choudhury K, Thirumalai S, Lawrence J, Quested D, Pimm J, Curtis D, Gurling HM. Evidence for the association of the DAOA (G72) gene with schizophrenia and bipolar disorder but not for the association of the DAO gene with schizophrenia. Behav Brain Funct. 2009;5:28. doi: 10.1186/1744-9081-5-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Shachar D, Zuk R, Gazawi H, Reshef A, Sheinkman A, Klein E. Increased mitochondrial complex I activity in platelets of schizophrenic patients. Int J Neuropsychopharmacol. 1999;2:245–253. doi: 10.1017/S1461145799001649. [DOI] [PubMed] [Google Scholar]
- Ben-Shachar D. Mitochondrial dysfunction in schizophrenia: a possible linkage to dopamine. J Neurochem. 2002;83:1241–1251. doi: 10.1046/j.1471-4159.2002.01263.x. [DOI] [PubMed] [Google Scholar]
- Ben-Shachar D, Karry R. Neuroanatomical pattern of mitochondrial complex I pathology varies between schizophrenia, bipolar disorder and major depression. PLoS One. 2008;3:e3676. doi: 10.1371/journal.pone.0003676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Shachar D, Nadri C, Karry R, Agam G. Mitochondrial complex I subunits are altered in rats with neonatal ventral hippocampal damage but not in rats exposed to oxygen restriction at neonatal age. J Mol Neurosci. 2009;38:143–151. doi: 10.1007/s12031-008-9144-9. [DOI] [PubMed] [Google Scholar]
- Benes FM, Kwok EW, Vincent SL, Todtenkopf MS. A reduction of nonpyramidal cells in sector CA2 of schizophrenics and manic depressives. Biol Psychiatry. 1998;44:88–97. doi: 10.1016/s0006-3223(98)00138-3. [DOI] [PubMed] [Google Scholar]
- Bernheimer H, Birkmayer W, Hornykiewicz O, Jellinger K, Seitelberger F. Brain dopamine and the syndromes of Parkinson and Huntington. Clinical, morphological and neurochemical correlations. J Neurol Sci. 1973;20:415–455. doi: 10.1016/0022-510x(73)90175-5. [DOI] [PubMed] [Google Scholar]
- Berrettini W. Evidence for shared susceptibility in bipolar disorder and schizophrenia. Am J Med Genet C Semin Med Genet. 2003;123C:59–64. doi: 10.1002/ajmg.c.20014. [DOI] [PubMed] [Google Scholar]
- Berrettini WH. Susceptibility loci for bipolar disorder: overlap with inherited vulnerability to schizophrenia. Biol Psychiatry. 2000;47:245–251. doi: 10.1016/s0006-3223(99)00226-7. [DOI] [PubMed] [Google Scholar]
- Boks MP, Rietkerk T, van de Beek MH, Sommer IE, de Koning TJ, Kahn RS. Reviewing the role of the genes G72 and DAAO in glutamate neurotransmission in schizophrenia. Eur Neuropsychopharmacol. 2007;17:567–572. doi: 10.1016/j.euroneuro.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Brandon MC, Lott MT, Nguyen KC, Spolim S, Navathe SB, Baldi P, Wallace DC. MITOMAP: a human mitochondrial genome database--2004 update. Nucleic Acids Res. 2005a;33:D611–613. doi: 10.1093/nar/gki079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandon NJ, Schurov I, Camargo LM, Handford EJ, Duran-Jimeniz B, Hunt P, Millar JK, Porteous DJ, Shearman MS, Whiting PJ. Subcellular targeting of DISC1 is dependent on a domain independent from the Nudel binding site. Mol Cell Neurosci. 2005b;28:613–624. doi: 10.1016/j.mcn.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Brenner-Lavie H, Klein E, Ben-Shachar D. Mitochondrial complex I as a novel target for intraneuronal DA: modulation of respiration in intact cells. Biochem Pharmacol. 2009;78:85–95. doi: 10.1016/j.bcp.2009.03.024. [DOI] [PubMed] [Google Scholar]
- Brown AS, Derkits EJ. Prenatal infection and schizophrenia: a review of epidemiologic and translational studies. Am J Psychiatry. 2010;167:261–280. doi: 10.1176/appi.ajp.2009.09030361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JM, Yamamoto BK. Effects of amphetamines on mitochondrial function: role of free radicals and oxidative stress. Pharmacol Ther. 2003;99:45–53. doi: 10.1016/s0163-7258(03)00052-4. [DOI] [PubMed] [Google Scholar]
- Burmeister M, McInnis MG, Zollner S. Psychiatric genetics: progress amid controversy. Nat Rev Genet. 2008;9:527–540. doi: 10.1038/nrg2381. [DOI] [PubMed] [Google Scholar]
- Campos Y, Garcia A, Eiris J, Fuster M, Rubio JC, Martin MA, del Hoyo P, Pintos E, Castro-Gago M, Arenas J. Mitochondrial myopathy, cardiomyopathy and psychiatric illness in a Spanish family harbouring the mtDNA 3303C > T mutation. J Inherit Metab Dis. 2001;24:685–687. doi: 10.1023/a:1012719211505. [DOI] [PubMed] [Google Scholar]
- Carpenter WT, Koenig JI. The evolution of drug development in schizophrenia: past issues and future opportunities. Neuropsychopharmacology. 2008;33:2061–2079. doi: 10.1038/sj.npp.1301639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo AM, McPhie DL, Lange NT, Punzell S, Elmiligy S, Ye NZ, Froimowitz MP, Hassinger LC, Menesale EB, Sargent LW, Logan DJ, Carpenter AE, Cohen BM. Abnormalities in mitochondrial structure in cells from patients with bipolar disorder. Am J Pathol. 2010 doi: 10.2353/ajpath.2010.081068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavelier L, Jazin EE, Eriksson I, Prince J, Bave U, Oreland L, Gyllensten U. Decreased cytochrome-c oxidase activity and lack of age-related accumulation of mitochondrial DNA deletions in the brains of schizophrenics. Genomics. 1995;29:217–224. doi: 10.1006/geno.1995.1234. [DOI] [PubMed] [Google Scholar]
- Chen RW, Chuang DM. Long term lithium treatment suppresses p53 and Bax expression but increases Bcl-2 expression. A prominent role in neuroprotection against excitotoxicity. J Biol Chem. 1999;274:6039–6042. doi: 10.1074/jbc.274.10.6039. [DOI] [PubMed] [Google Scholar]
- Chen S, Owens GC, Edelman DB. Dopamine inhibits mitochondrial motility in hippocampal neurons. PLoS One. 2008;3:e2804. doi: 10.1371/journal.pone.0002804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WY, Shi YY, Zheng YL, Zhao XZ, Zhang GJ, Chen SQ, Yang PD, He L. Case-control study and transmission disequilibrium test provide consistent evidence for association between schizophrenia and genetic variation in the 22q11 gene ZDHHC8. Hum Mol Genet. 2004;13:2991–2995. doi: 10.1093/hmg/ddh322. [DOI] [PubMed] [Google Scholar]
- Chipuk JE, Green DR. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. 2008;18:157–164. doi: 10.1016/j.tcb.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chumakov I, Blumenfeld M, Guerassimenko O, Cavarec L, Palicio M, Abderrahim H, Bougueleret L, Barry C, Tanaka H, La Rosa P, Puech A, Tahri N, Cohen-Akenine A, Delabrosse S, Lissarrague S, Picard FP, Maurice K, Essioux L, Millasseau P, Grel P, Debailleul V, Simon AM, Caterina D, Dufaure I, Malekzadeh K, Belova M, Luan JJ, Bouillot M, Sambucy JL, Primas G, Saumier M, Boubkiri N, Martin-Saumier S, Nasroune M, Peixoto H, Delaye A, Pinchot V, Bastucci M, Guillou S, Chevillon M, Sainz-Fuertes R, Meguenni S, Aurich-Costa J, Cherif D, Gimalac A, Van Duijn C, Gauvreau D, Ouellette G, Fortier I, Raelson J, Sherbatich T, Riazanskaia N, Rogaev E, Raeymaekers P, Aerssens J, Konings F, Luyten W, Macciardi F, Sham PC, Straub RE, Weinberger DR, Cohen N, Cohen D. Genetic and physiological data implicating the new human gene G72 and the gene for D-amino acid oxidase in schizophrenia. Proc Natl Acad Sci U S A. 2002;99:13675–13680. doi: 10.1073/pnas.182412499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clay Montier LL, Deng JJ, Bai Y. Number matters: control of mammalian mitochondrial DNA copy number. J Genet Genomics. 2009;36:125–131. doi: 10.1016/S1673-8527(08)60099-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craddock N, O’Donovan MC, Owen MJ. Genes for schizophrenia and bipolar disorder? Implications for psychiatric nosology. Schizophr Bull. 2006;32:9–16. doi: 10.1093/schbul/sbj033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dager SR, Friedman SD, Parow A, Demopulos C, Stoll AL, Lyoo IK, Dunner DL, Renshaw PF. Brain metabolic alterations in medication-free patients with bipolar disorder. Arch Gen Psychiatry. 2004;61:450–458. doi: 10.1001/archpsyc.61.5.450. [DOI] [PubMed] [Google Scholar]
- Deicken RF, Calabrese G, Merrin EL, Vinogradov S, Fein G, Weiner MW. Asymmetry of temporal lobe phosphorous metabolism in schizophrenia: a 31phosphorous magnetic resonance spectroscopic imaging study. Biol Psychiatry. 1995a;38:279–286. doi: 10.1016/0006-3223(94)00372-A. [DOI] [PubMed] [Google Scholar]
- Deicken RF, Fein G, Weiner MW. Abnormal frontal lobe phosphorous metabolism in bipolar disorder. Am J Psychiatry. 1995b;152:915–918. doi: 10.1176/ajp.152.6.915. [DOI] [PubMed] [Google Scholar]
- Deicken RF, Merrin EL, Floyd TC, Weiner MW. Correlation between left frontal phospholipids and Wisconsin Card Sort Test performance in schizophrenia. Schizophr Res. 1995c;14:177–181. doi: 10.1016/0920-9964(94)00036-8. [DOI] [PubMed] [Google Scholar]
- DeLisi LE, Razi K, Stewart J, Relja M, Shields G, Smith AB, Wellman N, Larach VW, Loftus J, Vita A, Comazzi M, Crow TJ. No evidence for a parent-of-origin effect detected in the pattern of inheritance of schizophrenia. Biol Psychiatry. 2000;48:706–709. doi: 10.1016/s0006-3223(00)00939-2. [DOI] [PubMed] [Google Scholar]
- DiMauro S, Schon EA. Mitochondrial disorders in the nervous system. Annu Rev Neurosci. 2008;31:91–123. doi: 10.1146/annurev.neuro.30.051606.094302. [DOI] [PubMed] [Google Scholar]
- Dror N, Klein E, Karry R, Sheinkman A, Kirsh Z, Mazor M, Tzukerman M, Ben-Shachar D. State-dependent alterations in mitochondrial complex I activity in platelets: a potential peripheral marker for schizophrenia. Mol Psychiatry. 2002;7:995–1001. doi: 10.1038/sj.mp.4001116. [DOI] [PubMed] [Google Scholar]
- Dubovsky SL, Murphy J, Thomas M, Rademacher J. Abnormal intracellular calcium ion concentration in platelets and lymphocytes of bipolar patients. Am J Psychiatry. 1992;149:118–120. doi: 10.1176/ajp.149.1.118. [DOI] [PubMed] [Google Scholar]
- Einat H, Yuan P, Manji HK. Increased anxiety-like behaviors and mitochondrial dysfunction in mice with targeted mutation of the Bcl-2 gene: further support for the involvement of mitochondrial function in anxiety disorders. Behav Brain Res. 2005;165:172–180. doi: 10.1016/j.bbr.2005.06.012. [DOI] [PubMed] [Google Scholar]
- Ekstrand MI, Falkenberg M, Rantanen A, Park CB, Gaspari M, Hultenby K, Rustin P, Gustafsson CM, Larsson NG. Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Hum Mol Genet. 2004;13:935–944. doi: 10.1093/hmg/ddh109. [DOI] [PubMed] [Google Scholar]
- Fattal O, Budur K, Vaughan AJ, Franco K. Review of the literature on major mental disorders in adult patients with mitochondrial diseases. Psychosomatics. 2006;47:1–7. doi: 10.1176/appi.psy.47.1.1. [DOI] [PubMed] [Google Scholar]
- Frahm T, Mohamed SA, Bruse P, Gemund C, Oehmichen M, Meissner C. Lack of age-related increase of mitochondrial DNA amount in brain, skeletal muscle and human heart. Mech Ageing Dev. 2005;126:1192–1200. doi: 10.1016/j.mad.2005.06.008. [DOI] [PubMed] [Google Scholar]
- Frey BN, Stanley JA, Nery FG, Monkul ES, Nicoletti MA, Chen HH, Hatch JP, Caetano SC, Ortiz O, Kapczinski F, Soares JC. Abnormal cellular energy and phospholipid metabolism in the left dorsolateral prefrontal cortex of medication-free individuals with bipolar disorder: an in vivo 1H MRS study. Bipolar Disord. 2007;9(Suppl 1):119–127. doi: 10.1111/j.1399-5618.2007.00454.x. [DOI] [PubMed] [Google Scholar]
- Fujimoto T, Nakano T, Takano T, Hokazono Y, Asakura T, Tsuji T. Study of chronic schizophrenics using 31P magnetic resonance chemical shift imaging. Acta Psychiatr Scand. 1992;86:455–462. doi: 10.1111/j.1600-0447.1992.tb03297.x. [DOI] [PubMed] [Google Scholar]
- Fuke S, Kametani M, Kato T. Quantitative analysis of the 4977-bp common deletion of mitochondrial DNA in postmortem frontal cortex from patients with bipolar disorder and schizophrenia. Neurosci Lett. 2008;439:173–177. doi: 10.1016/j.neulet.2008.05.009. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Blomgren K, Kroemer G. Mitochondrial membrane permeabilization in neuronal injury. Nat Rev Neurosci. 2009;10:481–494. doi: 10.1038/nrn2665. [DOI] [PubMed] [Google Scholar]
- Gavin DP, Kartan S, Chase K, Jayaraman S, Sharma RP. Histone deacetylase inhibitors and candidate gene expression: An in vivo and in vitro approach to studying chromatin remodeling in a clinical population. J Psychiatr Res. 2009;43:870–876. doi: 10.1016/j.jpsychires.2008.12.006. [DOI] [PubMed] [Google Scholar]
- Gershon ES, Hamovit J, Guroff JJ, Dibble E, Leckman JF, Sceery W, Targum SD, Nurnberger JI, Jr., Goldin LR, Bunney WE., Jr. A family study of schizoaffective, bipolar I, bipolar II, unipolar, and normal control probands. Arch Gen Psychiatry. 1982;39:1157–1167. doi: 10.1001/archpsyc.1982.04290100031006. [DOI] [PubMed] [Google Scholar]
- Grover S, Padhy SK, Das CP, Vasishta RK, Sharan P, Chakrabarti S. Mania as a first presentation in mitochondrial myopathy. Psychiatry Clin Neurosci. 2006;60:774–775. doi: 10.1111/j.1440-1819.2006.01599.x. [DOI] [PubMed] [Google Scholar]
- Gulyas AI, Buzsaki G, Freund TF, Hirase H. Populations of hippocampal inhibitory neurons express different levels of cytochrome c. Eur J Neurosci. 2006;23:2581–2594. doi: 10.1111/j.1460-9568.2006.04814.x. [DOI] [PubMed] [Google Scholar]
- Hattori E, Liu C, Badner JA, Bonner TI, Christian SL, Maheshwari M, Detera-Wadleigh SD, Gibbs RA, Gershon ES. Polymorphisms at the G72/G30 gene locus, on 13q33, are associated with bipolar disorder in two independent pedigree series. Am J Hum Genet. 2003;72:1131–1140. doi: 10.1086/374822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckers S, Stone D, Walsh J, Shick J, Koul P, Benes FM. Differential hippocampal expression of glutamic acid decarboxylase 65 and 67 messenger RNA in bipolar disorder and schizophrenia. Arch Gen Psychiatry. 2002;59:521–529. doi: 10.1001/archpsyc.59.6.521. [DOI] [PubMed] [Google Scholar]
- Herberth M, Koethe D, Cheng TM, Krzyszton ND, Schoeffmann S, Guest PC, Rahmoune H, Harris LW, Kranaster L, Leweke FM, Bahn S. Impaired glycolytic response in peripheral blood mononuclear cells of first-onset antipsychotic-naive schizophrenia patients. Mol Psychiatry. 2010 doi: 10.1038/mp.2010.71. [DOI] [PubMed] [Google Scholar]
- Hercher L, Bruenner G. Living with a child at risk for psychotic illness: the experience of parents coping with 22q11 deletion syndrome: an exploratory study. Am J Med Genet A. 2008;146A:2355–2360. doi: 10.1002/ajmg.a.32466. [DOI] [PubMed] [Google Scholar]
- Hotchkiss RS, Strasser A, McDunn JE, Swanson PE. Cell death. N Engl J Med. 2009;361:1570–1583. doi: 10.1056/NEJMra0901217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iborra FJ, Kimura H, Cook PR. The functional organization of mitochondrial genomes in human cells. BMC Biol. 2004;2:9. doi: 10.1186/1741-7007-2-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inuwa IM, Peet M, Williams MA. QSAR modeling and transmission electron microscopy stereology of altered mitochondrial ultrastructure of white blood cells in patients diagnosed as schizophrenic and treated with antipsychotic drugs. Biotech Histochem. 2005;80:133–137. doi: 10.1080/10520290500303349. [DOI] [PubMed] [Google Scholar]
- Isaev NK, Stelmashook EV, Dirnagl U, Plotnikov EY, Kuvshinova EA, Zorov DB. Mitochondrial free radical production induced by glucose deprivation in cerebellar granule neurons. Biochemistry (Mosc) 2008;73:149–155. doi: 10.1134/s0006297908020053. [DOI] [PubMed] [Google Scholar]
- Ishizuka K, Paek M, Kamiya A, Sawa A. A review of Disrupted-In-Schizophrenia-1 (DISC1): neurodevelopment, cognition, and mental conditions. Biol Psychiatry. 2006;59:1189–1197. doi: 10.1016/j.biopsych.2006.03.065. [DOI] [PubMed] [Google Scholar]
- Iwamoto K, Bundo M, Kato T. Altered expression of mitochondria-related genes in postmortem brains of patients with bipolar disorder or schizophrenia, as revealed by large-scale DNA microarray analysis. Hum Mol Genet. 2005;14:241–253. doi: 10.1093/hmg/ddi022. [DOI] [PubMed] [Google Scholar]
- Jablensky A. Subtyping schizophrenia: implications for genetic research. Mol Psychiatry. 2006;11:815–836. doi: 10.1038/sj.mp.4001857. [DOI] [PubMed] [Google Scholar]
- James R, Adams RR, Christie S, Buchanan SR, Porteous DJ, Millar JK. Disrupted in Schizophrenia 1 (DISC1) is a multicompartmentalized protein that predominantly localizes to mitochondria. Mol Cell Neurosci. 2004;26:112–122. doi: 10.1016/j.mcn.2004.01.013. [DOI] [PubMed] [Google Scholar]
- Jensen JE, Miller J, Williamson PC, Neufeld RW, Menon RS, Malla A, Manchanda R, Schaefer B, Densmore M, Drost DJ. Grey and white matter differences in brain energy metabolism in first episode schizophrenia: 31P-MRS chemical shift imaging at 4 Tesla. Psychiatry Res. 2006;146:127–135. doi: 10.1016/j.pscychresns.2005.11.004. [DOI] [PubMed] [Google Scholar]
- Jou MJ, Peng TI, Wu HY, Wei YH. Enhanced generation of mitochondrial reactive oxygen species in cybrids containing 4977-bp mitochondrial DNA deletion. Ann N Y Acad Sci. 2005;1042:221–228. doi: 10.1196/annals.1338.024. [DOI] [PubMed] [Google Scholar]
- Kakiuchi C, Ishiwata M, Kametani M, Nelson C, Iwamoto K, Kato T. Quantitative analysis of mitochondrial DNA deletions in the brains of patients with bipolar disorder and schizophrenia. Int J Neuropsychopharmacol. 2005;8:515–522. doi: 10.1017/S1461145705005213. [DOI] [PubMed] [Google Scholar]
- Karayiorgou M, Simon TJ, Gogos JA. 22q11.2 microdeletions: linking DNA structural variation to brain dysfunction and schizophrenia. Nat Rev Neurosci. 2010;11:402–416. doi: 10.1038/nrn2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsgodt KH, Sun D, Jimenez AM, Lutkenhoff ES, Willhite R, van Erp TG, Cannon TD. Developmental disruptions in neural connectivity in the pathophysiology of schizophrenia. Dev Psychopathol. 2008;20:1297–1327. doi: 10.1017/S095457940800062X. [DOI] [PubMed] [Google Scholar]
- Karry R, Klein E, Ben Shachar D. Mitochondrial complex I subunits expression is altered in schizophrenia: a postmortem study. Biol Psychiatry. 2004;55:676–684. doi: 10.1016/j.biopsych.2003.12.012. [DOI] [PubMed] [Google Scholar]
- Kasahara T, Kubota M, Miyauchi T, Noda Y, Mouri A, Nabeshima T, Kato T. Mice with neuron-specific accumulation of mitochondrial DNA mutations show mood disorder-like phenotypes. Mol Psychiatry. 2006;11:577–593. 523. doi: 10.1038/sj.mp.4001824. [DOI] [PubMed] [Google Scholar]
- Kasahara T, Kubota M, Miyauchi T, Ishiwata M, Kato T. A marked effect of electroconvulsive stimulation on behavioral aberration of mice with neuron-specific mitochondrial DNA defects. PLoS One. 2008;3:e1877. doi: 10.1371/journal.pone.0001877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato T, Takahashi S, Shioiri T, Inubushi T. Alterations in brain phosphorous metabolism in bipolar disorder detected by in vivo 31P and 7Li magnetic resonance spectroscopy. J Affect Disord. 1993;27:53–59. doi: 10.1016/0165-0327(93)90097-4. [DOI] [PubMed] [Google Scholar]
- Kato T, Takahashi S, Shioiri T, Murashita J, Hamakawa H, Inubushi T. Reduction of brain phosphocreatine in bipolar II disorder detected by phosphorus-31 magnetic resonance spectroscopy. J Affect Disord. 1994;31:125–133. doi: 10.1016/0165-0327(94)90116-3. [DOI] [PubMed] [Google Scholar]
- Kato T, Shioiri T, Murashita J, Hamakawa H, Takahashi Y, Inubushi T, Takahashi S. Lateralized abnormality of high energy phosphate metabolism in the frontal lobes of patients with bipolar disorder detected by phase-encoded 31P-MRS. Psychol Med. 1995;25:557–566. doi: 10.1017/s003329170003347x. [DOI] [PubMed] [Google Scholar]
- Kato T, Winokur G, Coryell W, Keller MB, Endicott J, Rice J. Parent-of-origin effect in transmission of bipolar disorder. Am J Med Genet. 1996;67:546–550. doi: 10.1002/(SICI)1096-8628(19961122)67:6<546::AID-AJMG6>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Kato T, Stine OC, McMahon FJ, Crowe RR. Increased levels of a mitochondrial DNA deletion in the brain of patients with bipolar disorder. Biol Psychiatry. 1997;42:871–875. doi: 10.1016/S0006-3223(97)00012-7. [DOI] [PubMed] [Google Scholar]
- Kato T, Murashita J, Kamiya A, Shioiri T, Kato N, Inubushi T. Decreased brain intracellular pH measured by 31P-MRS in bipolar disorder: a confirmation in drug-free patients and correlation with white matter hyperintensity. Eur Arch Psychiatry Clin Neurosci. 1998;248:301–306. doi: 10.1007/s004060050054. [DOI] [PubMed] [Google Scholar]
- Kato T, Kato N. Mitochondrial dysfunction in bipolar disorder. Bipolar Disord. 2000;2:180–190. doi: 10.1034/j.1399-5618.2000.020305.x. [DOI] [PubMed] [Google Scholar]
- Kato T, Kunugi H, Nanko S, Kato N. Association of bipolar disorder with the 5178 polymorphism in mitochondrial DNA. Am J Med Genet. 2000;96:182–186. [PubMed] [Google Scholar]
- Kato T. DNA polymorphisms and bipolar disorder. Am J Psychiatry. 2001;158:1169–1170. doi: 10.1176/appi.ajp.158.7.1169-b. [DOI] [PubMed] [Google Scholar]
- Kato T, Ishiwata M, Mori K, Washizuka S, Tajima O, Akiyama T, Kato N. Mechanisms of altered Ca2+ signalling in transformed lymphoblastoid cells from patients with bipolar disorder. Int J Neuropsychopharmacol. 2003;6:379–389. doi: 10.1017/S1461145703003717. [DOI] [PubMed] [Google Scholar]
- Kazuno AA, Munakata K, Nagai T, Shimozono S, Tanaka M, Yoneda M, Kato N, Miyawaki A, Kato T. Identification of mitochondrial DNA polymorphisms that alter mitochondrial matrix pH and intracellular calcium dynamics. PLoS Genet. 2006;2:e128. doi: 10.1371/journal.pgen.0020128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazuno AA, Munakata K, Kato N, Kato T. Mitochondrial DNA-dependent effects of valproate on mitochondrial calcium levels in transmitochondrial cybrids. Int J Neuropsychopharmacol. 2008;11:71–78. doi: 10.1017/S1461145707007614. [DOI] [PubMed] [Google Scholar]
- Kazuno AA, Munakata K, Mori K, Nanko S, Kunugi H, Nakamura K, Mori N, Yamada K, Yoshikawa T, Kato N, Kato T. Mitochondrial DNA haplogroup analysis in patients with bipolar disorder. Am J Med Genet B Neuropsychiatr Genet. 2009;150B:243–247. doi: 10.1002/ajmg.b.30804. [DOI] [PubMed] [Google Scholar]
- King TD, Bijur GN, Jope RS. Caspase-3 activation induced by inhibition of mitochondrial complex I is facilitated by glycogen synthase kinase-3beta and attenuated by lithium. Brain Res. 2001;919:106–114. doi: 10.1016/s0006-8993(01)03005-0. [DOI] [PubMed] [Google Scholar]
- Kirk R, Furlong RA, Amos W, Cooper G, Rubinsztein JS, Walsh C, Paykel ES, Rubinsztein DC. Mitochondrial genetic analyses suggest selection against maternal lineages in bipolar affective disorder. Am J Hum Genet. 1999;65:508–518. doi: 10.1086/302507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemm S, Rzanny R, Riehemann S, Volz HP, Schmidt B, Gerhard UJ, Filz C, Schonberg A, Mentzel HJ, Kaiser WA, Blanz B. Cerebral phosphate metabolism in first-degree relatives of patients with schizophrenia. Am J Psychiatry. 2001;158:958–960. doi: 10.1176/appi.ajp.158.6.958. [DOI] [PubMed] [Google Scholar]
- Klushnik TP, Spunde A, Yakovlev AG, Khuchua ZA, Saks VA, Vartanyan ME. Intracellular alterations of the creatine kinase isoforms in brains of schizophrenic patients. Mol Chem Neuropathol. 1991;15:271–280. doi: 10.1007/BF03161065. [DOI] [PubMed] [Google Scholar]
- Konradi C, Eaton M, MacDonald ML, Walsh J, Benes FM, Heckers S. Molecular evidence for mitochondrial dysfunction in bipolar disorder. Arch Gen Psychiatry. 2004;61:300–308. doi: 10.1001/archpsyc.61.3.300. [DOI] [PubMed] [Google Scholar]
- Kroll JL. New directions in the conceptualization of psychotic disorders. Curr Opin Psychiatry. 2007;20:573–577. doi: 10.1097/YCO.0b013e3282f08759. [DOI] [PubMed] [Google Scholar]
- Kung L, Roberts RC. Mitochondrial pathology in human schizophrenic striatum: a postmortem ultrastructural study. Synapse. 1999;31:67–75. doi: 10.1002/(SICI)1098-2396(199901)31:1<67::AID-SYN9>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Kusumi I, Koyama T, Yamashita I. Thrombin-induced platelet calcium mobilization is enhanced in bipolar disorders. Biol Psychiatry. 1992;32:731–734. doi: 10.1016/0006-3223(92)90305-j. [DOI] [PubMed] [Google Scholar]
- Kvajo M, Dhilla A, Swor DE, Karayiorgou M, Gogos JA. Evidence implicating the candidate schizophrenia/bipolar disorder susceptibility gene G72 in mitochondrial function. Mol Psychiatry. 2008;13:685–696. doi: 10.1038/sj.mp.4002052. [DOI] [PubMed] [Google Scholar]
- Kyle UG, Pichard C. The Dutch Famine of 1944-1945: a pathophysiological model of long-term consequences of wasting disease. Curr Opin Clin Nutr Metab Care. 2006;9:388–394. doi: 10.1097/01.mco.0000232898.74415.42. [DOI] [PubMed] [Google Scholar]
- Lan TH, Beaty TH, DePaulo JR, McInnis MG. Parent-of-origin effect in the segregation analysis of bipolar affective disorder families. Psychiatr Genet. 2007;17:93–101. doi: 10.1097/YPG.0b013e328013e604. [DOI] [PubMed] [Google Scholar]
- Lang UE, Puls I, Muller DJ, Strutz-Seebohm N, Gallinat J. Molecular mechanisms of schizophrenia. Cell Physiol Biochem. 2007;20:687–702. doi: 10.1159/000110430. [DOI] [PubMed] [Google Scholar]
- Lee HC, Wei YH. Mitochondrial biogenesis and mitochondrial DNA maintenance of mammalian cells under oxidative stress. Int J Biochem Cell Biol. 2005;37:822–834. doi: 10.1016/j.biocel.2004.09.010. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Levitt P. Schizophrenia as a disorder of neurodevelopment. Annu Rev Neurosci. 2002;25:409–432. doi: 10.1146/annurev.neuro.25.112701.142754. [DOI] [PubMed] [Google Scholar]
- Li J, Norwood DL, Mao LF, Schulz H. Mitochondrial metabolism of valproic acid. Biochemistry. 1991;30:388–394. doi: 10.1021/bi00216a012. [DOI] [PubMed] [Google Scholar]
- Li T, Ma X, Sham PC, Sun X, Hu X, Wang Q, Meng H, Deng W, Liu X, Murray RM, Collier DA. Evidence for association between novel polymorphisms in the PRODH gene and schizophrenia in a Chinese population. Am J Med Genet B Neuropsychiatr Genet. 2004;129B:13–15. doi: 10.1002/ajmg.b.30049. [DOI] [PubMed] [Google Scholar]
- Lin PI, Mitchell BD. Approaches for unraveling the joint genetic determinants of schizophrenia and bipolar disorder. Schizophr Bull. 2008;34:791–797. doi: 10.1093/schbul/sbn050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CY, Lee CF, Wei YH. Quantitative effect of 4977 bp deletion of mitochondrial DNA on the susceptibility of human cells to UV-induced apoptosis. Mitochondrion. 2007;7:89–95. doi: 10.1016/j.mito.2006.11.020. [DOI] [PubMed] [Google Scholar]
- Liu H, Heath SC, Sobin C, Roos JL, Galke BL, Blundell ML, Lenane M, Robertson B, Wijsman EM, Rapoport JL, Gogos JA, Karayiorgou M. Genetic variation at the 22q11 PRODH2/DGCR6 locus presents an unusual pattern and increases susceptibility to schizophrenia. Proc Natl Acad Sci U S A. 2002;99:3717–3722. doi: 10.1073/pnas.042700699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald ML, Naydenov A, Chu M, Matzilevich D, Konradi C. Decrease in creatine kinase messenger RNA expression in the hippocampus and dorsolateral prefrontal cortex in bipolar disorder. Bipolar Disord. 2006;8:255–264. doi: 10.1111/j.1399-5618.2006.00302.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado-Vieira R, Manji HK, Zarate CA., Jr. The role of lithium in the treatment of bipolar disorder: convergent evidence for neurotrophic effects as a unifying hypothesis. Bipolar Disord. 2009;11(Suppl 2):92–109. doi: 10.1111/j.1399-5618.2009.00714.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier W, Zobel A, Wagner M. Schizophrenia and bipolar disorder: differences and overlaps. Curr Opin Psychiatry. 2006;19:165–170. doi: 10.1097/01.yco.0000214342.52249.82. [DOI] [PubMed] [Google Scholar]
- Mancuso M, Ricci G, Choub A, Filosto M, DiMauro S, Davidzon G, Tessa A, Santorelli FM, Murri L, Siciliano G. Autosomal dominant psychiatric disorders and mitochondrial DNA multiple deletions: report of a family. J Affect Disord. 2008;106:173–177. doi: 10.1016/j.jad.2007.05.016. [DOI] [PubMed] [Google Scholar]
- Marchbanks RM, Ryan M, Day IN, Owen M, McGuffin P, Whatley SA. A mitochondrial DNA sequence variant associated with schizophrenia and oxidative stress. Schizophr Res. 2003;65:33–38. doi: 10.1016/s0920-9964(03)00011-2. [DOI] [PubMed] [Google Scholar]
- Maurer I, Moller HJ. Inhibition of complex I by neuroleptics in normal human brain cortex parallels the extrapyramidal toxicity of neuroleptics. Mol Biochem. 1997;174:255–259. [PubMed] [Google Scholar]
- Maurer I, Zierz S, Moller H. Evidence for a mitochondrial oxidative phosphorylation defect in brains from patients with schizophrenia. Schizophr Res. 2001;48:125–136. doi: 10.1016/s0920-9964(00)00075-x. [DOI] [PubMed] [Google Scholar]
- Maurer IC, Schippel P, Volz HP. Lithium-induced enhancement mitochondrial oxidative phosphorylation in human brain tissue. Bipolar Disord. 2009;11:515–522. doi: 10.1111/j.1399-5618.2009.00729.x. [DOI] [PubMed] [Google Scholar]
- Maynard TM, Meechan DW, Dudevoir ML, Gopalakrishna D, Peters AZ, Heindel CC, Sugimoto TJ, Wu Y, Lieberman JA, Lamantia AS. Mitochondrial localization and function of a subset of 22q11 deletion syndrome candidate genes. Mol Cell Neurosci. 2008;39:439–451. doi: 10.1016/j.mcn.2008.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInerny SC, Brown AL, Smith DW. Region-specific changes mitochondrial D-loop in aged rat CNS. Mech Ageing Dev. 2009;130:343–349. doi: 10.1016/j.mad.2009.01.008. [DOI] [PubMed] [Google Scholar]
- McMahon FJ, Stine OC, Meyers DA, Simpson SG, DePaulo JR. Patterns of maternal transmission in bipolar affective disorder. Am J Hum Genet. 1995;56:1277–1286. [PMC free article] [PubMed] [Google Scholar]
- McMahon FJ, Chen YS, Patel S, Kokoszka J, Brown MD, Torroni A, DePaulo JR, Wallace DC. Mitochondrial DNA sequence diversity in bipolar affective disorder. Am J Psychiatry. 2000;157:1058–1064. doi: 10.1176/appi.ajp.157.7.1058. [DOI] [PubMed] [Google Scholar]
- Michaelis M, Suhan T, Michaelis UR, Beek K, Rothweiler F, Tausch L, Werz O, Eikel D, Zornig M, Nau H, Fleming I, Doerr HW, Cinatl J., Jr. Valproic acid induces extracellular signal-regulated kinase 1/2 activation and inhibits apoptosis in endothelial cells. Cell Death Differ. 2006;13:446–453. doi: 10.1038/sj.cdd.4401759. [DOI] [PubMed] [Google Scholar]
- Middleton FA, Mirnics K, Pierri JN, Lewis DA, Levitt P. Gene expression profiling reveals alterations of specific metabolic pathways in schizophrenia. J Neurosci. 2002;22:2718–2729. doi: 10.1523/JNEUROSCI.22-07-02718.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mill J, Tang T, Kaminsky Z, Khare T, Yazdanpanah S, Bouchard L, Jia P, Assadzadeh A, Flanagan J, Schumacher A, Wang SC, Petronis A. Epigenomic profiling reveals DNA-methylation changes associated with major psychosis. Am J Hum Genet. 2008;82:696–711. doi: 10.1016/j.ajhg.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millar JK, James R, Christie S, Porteous DJ. Disrupted in schizophrenia 1 (DISC1): subcellular targeting and induction of ring mitochondria. Mol Cell Neurosci. 2005;30:477–484. doi: 10.1016/j.mcn.2005.08.021. [DOI] [PubMed] [Google Scholar]
- Modica-Napolitano JS, Lagace CJ, Brennan WA, Aprille JR. Differential effects of typical and atypical neuroleptics on mitochondrial function in vitro. Arch Pharm Res. 2003;26:951–959. doi: 10.1007/BF02980205. [DOI] [PubMed] [Google Scholar]
- Munakata K, Tanaka M, Mori K, Washizuka S, Yoneda M, Tajima O, Akiyama T, Nanko S, Kunugi H, Tadokoro K, Ozaki N, Inada T, Sakamoto K, Fukunaga T, Iijima Y, Iwata N, Tatsumi M, Yamada K, Yoshikawa T, Kato T. Mitochondrial DNA 3644T-->C mutation associated with bipolar disorder. Genomics. 2004;84:1041–1050. doi: 10.1016/j.ygeno.2004.08.015. [DOI] [PubMed] [Google Scholar]
- Munakata K, Iwamoto K, Bundo M, Kato T. Mitochondrial DNA 3243A>G mutation and increased expression of LARS2 gene in the brains of patients with bipolar disorder and schizophrenia. Biol Psychiatry. 2005;57:525–532. doi: 10.1016/j.biopsych.2004.11.041. [DOI] [PubMed] [Google Scholar]
- Murphy AN, Bredesen DE, Cortopassi G, Wang E, Fiskum G. Bcl-2 potentiates the maximal calcium uptake capacity of neural cell mitochondria. Proc Natl Acad Sci U S A. 1996;93:9893–9898. doi: 10.1073/pnas.93.18.9893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naydenov AV, MacDonald ML, Ongur D, Konradi C. Differences in lymphocyte electron transport gene expression levels between subjects with bipolar disorder and normal controls in response to glucose deprivation stress. Arch Gen Psychiatry. 2007;64:555–564. doi: 10.1001/archpsyc.64.5.555. [DOI] [PubMed] [Google Scholar]
- Newcomer JW. Abnormalities of glucose metabolism associated with atypical antipsychotic drugs. J Clin Psychiatry. 2004;65(Suppl 18):36–46. [PubMed] [Google Scholar]
- Ongur D, Prescot AP, Jensen JE, Cohen BM, Renshaw PF. Creatine abnormalities in schizophrenia and bipolar disorder. Psychiatry Res. 2009;172:44–48. doi: 10.1016/j.pscychresns.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol. 2003;4:552–565. doi: 10.1038/nrm1150. [DOI] [PubMed] [Google Scholar]
- Otte DM, Bilkei-Gorzo A, Filiou MD, Turck CW, Yilmaz O, Holst MI, Schilling K, Abou-Jamra R, Schumacher J, Benzel I, Kunz WS, Beck H, Zimmer A. Behavioral changes in G72/G30 transgenic mice. Eur Neuropsychopharmacol. 2009;19:339–348. doi: 10.1016/j.euroneuro.2008.12.009. [DOI] [PubMed] [Google Scholar]
- Ousley O, Rockers K, Dell ML, Coleman K, Cubells JF. A review of neurocognitive and behavioral profiles associated with 22q11 deletion syndrome: implications for clinical evaluation and treatment. Curr Psychiatry Rep. 2007;9:148–158. doi: 10.1007/s11920-007-0085-8. [DOI] [PubMed] [Google Scholar]
- Ouyang YB, Giffard RG. Cellular neuroprotective mechanisms in cerebral ischemia: Bcl-2 family proteins and protection of mitochondrial function. Cell Calcium. 2004;36:303–311. doi: 10.1016/j.ceca.2004.02.015. [DOI] [PubMed] [Google Scholar]
- Pennington K, Beasley CL, Dicker P, Fagan A, English J, Pariante CM, Wait R, Dunn MJ, Cotter DR. Prominent synaptic and metabolic abnormalities revealed by proteomic analysis of the dorsolateral prefrontal cortex in schizophrenia and bipolar disorder. Mol Psychiatry. 2008;13:1102–1117. doi: 10.1038/sj.mp.4002098. [DOI] [PubMed] [Google Scholar]
- Pereira RS, Bertocchi AP, Vercesi AE. Protective effect of trifluoperazine on the mitochondrial damage induced by Ca2+ plus prooxidants. Biochem Pharmacol. 1992;44:1795–1801. doi: 10.1016/0006-2952(92)90074-s. [DOI] [PubMed] [Google Scholar]
- Pettegrew JW, Keshavan MS, Panchalingam K, Strychor S, Kaplan DB, Tretta MG, Allen M. Alterations in brain high-energy phosphate and membrane phospholipid metabolism in first-episode, drug-naive schizophrenics. A pilot study of the dorsal prefrontal cortex by in vivo phosphorus 31 nuclear magnetic resonance spectroscopy. Arch Gen Psychiatry. 1991;48:563–568. doi: 10.1001/archpsyc.1991.01810300075011. [DOI] [PubMed] [Google Scholar]
- Plant N, Barber P, Horner E, Cockburn CL, Gibson G, Bugelski P, Lord P. Differential gene expression in rats following subacute exposure to the anticonvulsant sodium valproate. Toxicol Appl Pharmacol. 2002;183:127–134. [PubMed] [Google Scholar]
- Post RM. Comparative pharmacology of bipolar disorder and schizophrenia. Schizophr Res. 1999;39:153–158. doi: 10.1016/s0920-9964(99)00115-2. discussion 163. [DOI] [PubMed] [Google Scholar]
- Prata D, Breen G, Osborne S, Munro J, St Clair D, Collier D. Association of DAO and G72(DAOA)/G30 genes with bipolar affective disorder. Am J Med Genet B Neuropsychiatr Genet. 2008;147B:914–917. doi: 10.1002/ajmg.b.30682. [DOI] [PubMed] [Google Scholar]
- Prayson RA, Wang N. Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes (MELAS) syndrome: an autopsy report. Arch Pathol Lab Med. 1998;122:978–981. [PubMed] [Google Scholar]
- Prince JA, Yassin MS, Oreland L. Neuroleptic-induced mitochondrial enzyme alterations in the rat brain. J Pharmacol Exp Ther. 1997a;280:261–267. [PubMed] [Google Scholar]
- Prince JA, Yassin MS, Oreland L. Normalization of cytochrome-c oxidase activity in the rat brain by neuroleptics after chronic treatment with PCP or methamphetamine. Neuropharmacology. 1997b;36:1665–1678. doi: 10.1016/s0028-3908(97)00152-4. [DOI] [PubMed] [Google Scholar]
- Pysh JJ, Khan T. Variations in mitochondrial structure and content of neurons and neuroglia in rat brain: an electron microscopic study. Brain Res. 1972;36:1–18. doi: 10.1016/0006-8993(72)90762-7. [DOI] [PubMed] [Google Scholar]
- Quiroz JA, Gray NA, Kato T, Manji HK. Mitochondrially mediated plasticity in the pathophysiology and treatment of bipolar disorder. Neuropsychopharmacology. 2008;33:2551–2565. doi: 10.1038/sj.npp.1301671. [DOI] [PubMed] [Google Scholar]
- Quiroz JA, Machado-Vieira R, Zarate CA, Jr., Manji HK. Novel insights into lithium’s mechanism of action: neurotrophic and neuroprotective effects. Neuropsychobiology. 2010;62:50–60. doi: 10.1159/000314310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regenold WT, Thapar RK, Marano C, Gavirneni S, Kondapavuluru PV. Increased prevalence of type 2 diabetes mellitus among psychiatric inpatients with bipolar I affective and schizoaffective disorders independent of psychotropic drug use. J Affect Disord. 2002;70:19–26. doi: 10.1016/s0165-0327(01)00456-6. [DOI] [PubMed] [Google Scholar]
- Regenold WT, Phatak P, Marano CM, Sassan A, Conley RR, Kling MA. Elevated cerebrospinal fluid lactate concentrations in patients with bipolar disorder and schizophrenia: implications for the mitochondrial dysfunction hypothesis. Biol Psychiatry. 2009;65:489–494. doi: 10.1016/j.biopsych.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripova D, Strunecka A, Nemcova V, Farska I. Phospholipids and calcium alterations in platelets of schizophrenic patients. Physiol Res. 1997;46:59–68. [PubMed] [Google Scholar]
- Rizzuto R, Pozzan T. Microdomains of intracellular Ca2+: molecular determinants and functional consequences. Physiol Rev. 2006;86:369–408. doi: 10.1152/physrev.00004.2005. [DOI] [PubMed] [Google Scholar]
- Rollins B, Martin MV, Sequeira PA, Moon EA, Morgan LZ, Watson SJ, Schatzberg A, Akil H, Myers RM, Jones EG, Wallace DC, Bunney WE, Vawter MP. Mitochondrial variants in schizophrenia, bipolar disorder, and major depressive disorder. PLoS One. 2009;4:e4913. doi: 10.1371/journal.pone.0004913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell JW, Golovoy D, Vincent AM, Mahendru P, Olzmann JA, Mentzer A, Feldman EL. High glucose-induced oxidative stress and mitochondrial dysfunction in neurons. FASEB J. 2002;16:1738–1748. doi: 10.1096/fj.01-1027com. [DOI] [PubMed] [Google Scholar]
- Sagara Y. Induction of reactive oxygen species in neurons by haloperidol. J Neurochem. 1998;71:1002–1012. doi: 10.1046/j.1471-4159.1998.71031002.x. [DOI] [PubMed] [Google Scholar]
- Seeman P. Dopamine receptors and the dopamine hypothesis of schizophrenia. Synapse. 1987;1:133–152. doi: 10.1002/syn.890010203. [DOI] [PubMed] [Google Scholar]
- Shalbuyeva N, Brustovetsky T, Brustovetsky N. Lithium desensitizes brain mitochondria to calcium, antagonizes permeability transition, and diminishes cytochrome C release. J Biol Chem. 2007;282:18057–18068. doi: 10.1074/jbc.M702134200. [DOI] [PubMed] [Google Scholar]
- Shao L, Martin MV, Watson SJ, Schatzberg A, Akil H, Myers RM, Jones EG, Bunney WE, Vawter MP. Mitochondrial involvement in psychiatric disorders. Ann Med. 2008;40:281–295. doi: 10.1080/07853890801923753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma RP, Rosen C, Kartan S, Guidotti A, Costa E, Grayson DR, Chase K. Valproic acid and chromatin remodeling in schizophrenia and bipolar disorder: preliminary results from a clinical population. Schizophr Res. 2006;88:227–231. doi: 10.1016/j.schres.2006.07.015. [DOI] [PubMed] [Google Scholar]
- Shmookler Reis RJ, Goldstein S. Mitochondrial DNA in mortal and immortal human cells. Genome number, integrity, and methylation. J Biol Chem. 1983;258:9078–9085. [PubMed] [Google Scholar]
- Silva JP, Kohler M, Graff C, Oldfors A, Magnuson MA, Berggren PO, Larsson NG. Impaired insulin secretion and beta-cell loss in tissue-specific knockout mice with mitochondrial diabetes. Nat Genet. 2000;26:336–340. doi: 10.1038/81649. [DOI] [PubMed] [Google Scholar]
- Stanley JA, Williamson PC, Drost DJ, Carr TJ, Rylett RJ, Morrison-Stewart S, Thompson RT. Membrane phospholipid metabolism and schizophrenia: an in vivo 31P-MR spectroscopy study. Schizophr Res. 1994;13:209–215. doi: 10.1016/0920-9964(94)90044-2. [DOI] [PubMed] [Google Scholar]
- Stelmashook EV, Isaev NK, Plotnikov EY, Uzbekov RE, Alieva IB, Arbeille B, Zorov DB. Effect of transitory glucose deprivation on mitochondrial structure and functions in cultured cerebellar granule neurons. Neurosci Lett. 2009;461:140–144. doi: 10.1016/j.neulet.2009.05.073. [DOI] [PubMed] [Google Scholar]
- Stoffel M, Karayiorgou M, Espinosa R, 3rd, Beau MM. The human mitochondrial citrate transporter gene (SLC20A3) maps to chromosome band 22q11 within a region implicated in DiGeorge syndrome, velo-cardio-facial syndrome and schizophrenia. Hum Genet. 1996;98:113–115. doi: 10.1007/s004390050169. [DOI] [PubMed] [Google Scholar]
- Stork C, Renshaw PF. Mitochondrial dysfunction in bipolar disorder: evidence from magnetic resonance spectroscopy research. Mol Psychiatry. 2005;10:900–919. doi: 10.1038/sj.mp.4001711. [DOI] [PubMed] [Google Scholar]
- Struewing IT, Barnett CD, Tang T, Mao CD. Lithium increases PGC-1alpha expression and mitochondrial biogenesis in primary bovine aortic endothelial cells. FEBS J. 2007;274:2749–2765. doi: 10.1111/j.1742-4658.2007.05809.x. [DOI] [PubMed] [Google Scholar]
- Thaker GK. Neurophysiological endophenotypes across bipolar and schizophrenia psychosis. Schizophr Bull. 2008;34:760–773. doi: 10.1093/schbul/sbn049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno H, Nishigaki Y, Kong QP, Fuku N, Kojima S, Iwata N, Ozaki N, Tanaka M. Analysis of mitochondrial DNA variants in Japanese patients with schizophrenia. Mitochondrion. 2009;9:385–393. doi: 10.1016/j.mito.2009.06.003. [DOI] [PubMed] [Google Scholar]
- Uranova N, Orlovskaya D, Vikhreva O, Zimina I, Kolomeets N, Vostrikov V, Rachmanova V. Electron microscopy of oligodendroglia in severe mental illness. Brain Res Bull. 2001;55:597–610. doi: 10.1016/s0361-9230(01)00528-7. [DOI] [PubMed] [Google Scholar]
- Uranova N, Bonartsev P, Brusov O, Morozova M, Rachmanova V, Orlovskaya D. The ultrastructure of lymphocytes in schizophrenia. World J Biol Psychiatry. 2007;8:30–37. doi: 10.1080/15622970600960207. [DOI] [PubMed] [Google Scholar]
- Valvassori SS, Rezin GT, Ferreira CL, Moretti M, Goncalves CL, Cardoso MR, Streck EL, Kapczinski F, Quevedo J. Effects of mood stabilizers on mitochondrial respiratory chain activity in brain of rats treated with d-amphetamine. J Psychiatr Res. 2010 doi: 10.1016/j.jpsychires.2010.02.009. [DOI] [PubMed] [Google Scholar]
- Vawter MP, Tomita H, Meng F, Bolstad B, Li J, Evans S, Choudary P, Atz M, Shao L, Neal C, Walsh DM, Burmeister M, Speed T, Myers R, Jones EG, Watson SJ, Akil H, Bunney WE. Mitochondrial-related gene expression changes are sensitive to agonal-pH state: implications for brain disorders. Mol Psychiatry. 2006;11:615, 663–679. doi: 10.1038/sj.mp.4001830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC. Mitochondrial diseases in man and mouse. Science. 1999;283:1482–1488. doi: 10.1126/science.283.5407.1482. [DOI] [PubMed] [Google Scholar]
- Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Lu YY. Mitochondrial DNA 4977-bp deletion correlated with reactive oxygen species production and manganese superoxidedismutase expression in gastric tumor cells. Chin Med J (Engl) 2009;122:431–436. [PubMed] [Google Scholar]
- Wang JF, Shao L, Sun X, Young LT. Glutathione S-transferase is a novel target for mood stabilizing drugs in primary cultured neurons. J Neurochem. 2004;88:1477–1484. doi: 10.1046/j.1471-4159.2003.02276.x. [DOI] [PubMed] [Google Scholar]
- Washizuka S, Ikeda A, Kato N, Kato T. Possible relationship between mitochondrial DNA polymorphisms and lithium response in bipolar disorder. Int J Neuropsychopharmacol. 2003a;6:421–424. doi: 10.1017/S1461145703003778. [DOI] [PubMed] [Google Scholar]
- Washizuka S, Kakiuchi C, Mori K, Kunugi H, Tajima O, Akiyama T, Nanko S, Kato T. Association of mitochondrial complex I subunit gene NDUFV2 at 18p11 with bipolar disorder. Am J Med Genet B Neuropsychiatr Genet. 2003b;120B:72–78. doi: 10.1002/ajmg.b.20041. [DOI] [PubMed] [Google Scholar]
- Washizuka S, Kakiuchi C, Mori K, Tajima O, Akiyama T, Kato T. Expression of mitochondria-related genes in lymphoblastoid cells from patients with bipolar disorder. Bipolar Disord. 2005;7:146–152. doi: 10.1111/j.1399-5618.2005.00184.x. [DOI] [PubMed] [Google Scholar]
- Washizuka S, Kametani M, Sasaki T, Tochigi M, Umekage T, Kohda K, Kato T. Association of mitochondrial complex I subunit gene NDUFV2 at 18p11 with schizophrenia in the Japanese population. Am J Med Genet B Neuropsychiatr Genet. 2006;141B:301–304. doi: 10.1002/ajmg.b.30285. [DOI] [PubMed] [Google Scholar]
- Washizuka S, Iwamoto K, Kakiuchi C, Bundo M, Kato T. Expression of mitochondrial complex I subunit gene NDUFV2 in the lymphoblastoid cells derived from patients with bipolar disorder and schizophrenia. Neurosci Res. 2009;63:199–204. doi: 10.1016/j.neures.2008.12.004. [DOI] [PubMed] [Google Scholar]
- Wasserman MJ, Corson TW, Sibony D, Cooke RG, Parikh SV, Pennefather PS, Li PP, Warsh JJ. Chronic lithium treatment attenuates intracellular calcium mobilization. Neuropsychopharmacology. 2004;29:759–769. doi: 10.1038/sj.npp.1300400. [DOI] [PubMed] [Google Scholar]
- Williams NM, Green EK, Macgregor S, Dwyer S, Norton N, Williams H, Raybould R, Grozeva D, Hamshere M, Zammit S, Jones L, Cardno A, Kirov G, Jones I, O’Donovan MC, Owen MJ, Craddock N. Variation at the DAOA/G30 locus influences susceptibility to major mood episodes but not psychosis in schizophrenia and bipolar disorder. Arch Gen Psychiatry. 2006;63:366–373. doi: 10.1001/archpsyc.63.4.366. [DOI] [PubMed] [Google Scholar]
- Wredenberg A, Wibom R, Wilhelmsson H, Graff C, Wiener HH, Burden SJ, Oldfors A, Westerblad H, Larsson NG. Increased mitochondrial mass in mitochondrial myopathy mice. Proc Natl Acad Sci U S A. 2002;99:15066–15071. doi: 10.1073/pnas.232591499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Li PP, Kennedy JL, Green M, Hughes B, Cooke RG, Parikh SV, Warsh JJ. Further support for association of the mitochondrial complex I subunit gene NDUFV2 with bipolar disorder. Bipolar Disord. 2008;10:105–110. doi: 10.1111/j.1399-5618.2008.00535.x. [DOI] [PubMed] [Google Scholar]
- Youssoufian H, Pyeritz RE. Mechanisms and consequences of somatic mosaicism in humans. Nat Rev Genet. 2002;3:748–758. doi: 10.1038/nrg906. [DOI] [PubMed] [Google Scholar]
- Yurgelun-Todd DA, Renshaw PF, Gruber SA, Ed M, Waternaux C, Cohen BM. Proton magnetic resonance spectroscopy of the temporal lobes in schizophrenics and normal controls. Schizophr Res. 1996;19:55–59. doi: 10.1016/0920-9964(95)00071-2. [DOI] [PubMed] [Google Scholar]
- Zhang J, Li X, Wang Y, Ji J, Yang F, Feng G, Wan P, Lindpaintner K, He L, He G. Association study on the mitochondrial gene NDUFV2 and bipolar disorder in the Chinese Han population. J Neural Transm. 2009;116:357–361. doi: 10.1007/s00702-009-0185-1. [DOI] [PubMed] [Google Scholar]