

Abstract
Quinoline antimalarial drugs bind both monomeric and dimeric forms of free heme, with distinct preferences depending on the chemical environment. Under biological conditions, chloroquine (CQ) appears to prefer to bind to μ-oxo dimeric heme, while quinine (QN) preferentially binds monomer. To further explore this important distinction, we study three newly synthesized and several commercially available QN analogues lacking various functional groups. We find that removal of the QN hydroxyl lowers heme affinity, hemozoin (Hz) inhibition efficiency, and antiplasmodial activity. Elimination of the rigid quinuclidyl ring has similar effects, but elimination of either the vinyl or methoxy group does not. Replacing the quinuclidyl N with a less rigid tertiary aliphatic N only partially restores activity. To further study these trends, we probe drug-heme interactions via NMR studies with both Fe and Zn protoporphyrin IX (FPIX, ZnPIX) for QN, dehydroxyQN (DHQN), dequinuclidylQN (DQQN), and deamino-dequinuclidylQN (DADQQN). Magnetic susceptibility measurements in the presence of FPIX demonstrate that these compounds differentially perturb FPIX monomer-dimer equilibrium. We also isolate the QN-FPIX complex formed under mild aqueous conditions and analyze it by mass spectrometry, as well as fluorescence, vibrational, and solid state NMR spectroscopies. The data elucidate key features of QN pharmacology and allow us to propose a refined model for the preferred binding of QN to monomeric FPIX under biologically relevant conditions. With this model in hand, we also propose how QN, CQ, and amodiaquine (AQ) differ in their ability to inhibit Hz formation.
Introduction
Malaria remains one of the world’s most tragic infectious diseases, afflicting hundreds of millions and killing millions worldwide annually [1]. The lack of an effective vaccine coupled with the continued spread and evolution of resistance to antimalarial drugs [1–3], makes development of additional inexpensive, novel, and efficacious antimalarial drugs particularly urgent. To achieve this goal, a better understanding of the molecular mechanism of action of effective antimalarial drugs as well as the mechanism(s) of parasite antimalarial drug resistance is needed.
Quinolines are an extremely important class of antimalarial drugs. The cinchona alkaloid quinine (QN)1 was effectively used as early as the 17th century [4] and remained a first line drug therapy vs malaria until the discovery and widespread introduction of chloroquine (CQ) [5]. Today, the World Health Organization (WHO) currently recommends the use of QN in the absence of artemisin combination therapies (ARTs) to treat CQ resistant (CQR) P. falciparum malaria. In the laboratory, it is known that QN remains active vs some CQR strains of P. falciparum, but the molecular explanation for this is unknown. This is in part due to the fact that, surprisingly, despite extensive use of the drug, the molecular mechanism of action of QN has not been fully elucidated. Most current research on quinoline-based antimalarials has focused on 4-amino and 8-amino quinolines, even though it is clear a QN pharmacophore has specific advantages.
Quinoline antimalarials are believed to target heme detoxification within the digestive vacuole (DV) of the malaria parasite [6–9]. Studies directed at understanding this include genetic, cell biological, and molecular pharmacological approaches. Genetically, QN resistance (QNR) has been linked to key mutations and/or over-expression of the P. falciparum chloroquine resistance transporter (PfCRT) [10,11], the P. falciparum multidrug resistance protein (PfMDR1) [12–15], and either mutation or increased activity of the P. falciparum Na+/H+ exchanger (PfNHE) [16,17]. The existence of various combinations of these multiple genetic events in various QNR strains, leading to combinations of altered function in three membrane proteins, presumably explains why levels of QNR are distributed across a wide range (e.g. 2, 3, 4, 5,… 9-fold, etc.). In contrast, across all strains that have been examined, P. falciparum CQR conferred predominantly by mutations in a single gene (pfcrt) [11] is typically characterized as a ≥ 10-fold increase in growth inhibition IC50, with no increases seen between 1- and 10-fold. In the presence of PfCRT mutations conferring this 10-fold jump in IC50, it is believed that mutation and/or increased expression of PfMDR1 can then further modulate quinoline drug resistance patterns.
In terms of molecular pharmacology, similar to CQ, QN is known to inhibit the crystallization of toxic ferriprotoporphyrin IX (FPIX) heme to non toxic hemozoin (Hz) [6–9]. Additional data defining non-covalent binding of QN to free μ-oxo dimeric heme [18] suggests inhibition of Hz formation could be by non-covalent binding to pre-crystalline dimeric forms of FPIX. However, several forms of FPIX exist in biological environments, including reduced and oxidized monomer, μ-oxo dimer, and tethered head-to-tail dimer [19]. QN binding to the different forms is not fully defined, and it is not known which form(s) are most relevant for QN-mediated inhibition of Hz formation. Recent studies [20,21] suggest CQ prefers binding to dimeric forms of FPIX while QN prefers binding to monomer, and that equilibria between the various complexes are significantly influenced by partitioning between aqueous and hydrophobic (i.e. membrane lipid) phases.
Earlier evidence based on spectroscopic comparisons of QN adducts to those of N-coordinated ligand complexes of tetraphenyl-porphyrinato iron(III) and chloroiron(III) protoporphyrin IX dimethyl ester appeared to eliminate possible Fe-quinolynyl N covalent coordination for a QN-monomeric FPIX complex [22]. Based on 13C NMR results, these authors hypothesized alkoxide coordination between the hydroxyl group of QN and iron center of FPIX, but the data did not allow for determination of QN-FPIX stoichiometry. Constantinidis et al studied drug-heme interaction between QN and urohemin I/uroporphyrin I [23], and their results were also consistent with the QN hydroxyl acting as a covalent axial ligand to heme iron. These data further suggested a 2:1 (urohemin:QN) binding stoichiometry, consistent with QN as an axial ligand to the μ-oxo dimer form of FPIX. Other studies have shown that stereochemical configuration is crucial to the activity of QN-related alkaloids. For example, threo-epi cinchona alkaloids are known to be significantly less active than the erythro-alkaloids [24–26].
Using magnetic susceptibility measurements, we have recently shown that unlike CQ, QN promotes formation of monomeric rather than dimeric FPIX [21]. This suggests that structural differences among quinoline antimalarials may result in different mechanisms of Hz inhibition. If so, this might then form the basis for understanding lack of strict correspondence between levels of CQR and QNR [16,17]. Egan and colleagues have recently obtained a very informative crystal structure of a halofantrine (HF)-FPIX complex that shows alkoxide coordination to FPIX, but were unsuccessful in obtaining diffraction quality QN-FPIX crystals [27]. Nonetheless, based on the HF-FPIX structure and energy profile similarities between QN, QD and HF, these workers propose that QN-FPIX interaction involves covalent coordination between the hydroxyl moiety of QN and the Fe3+ center of FPIX. This model is consistent with previous studies mentioned above [22]. Thus, the majority of molecular studies have implicated that the hydroxyl group of QN interacts with FPIX Fe3+ in some fashion [22–27] but there are conflicting data and interpretation regarding the covalent vs. non-covalent nature of that interaction, QN:FPIX stoichiometry, and QN structural features that influence binding to FPIX.
In this study, we synthesize three novel QN analogs, and use these together with commercially available analogs to define QN structural features necessary for interaction with FPIX and for biological activity. We assess antiplasmodial and in vitro Hz inhibitory activities as well as FPIX binding affinities for these compounds. We measure FPIX magnetic moment in the presence of several analogs and compare these results to earlier results for QN. We perform solution NMR studies in the presence of ZnPIX to quantify effects of heme binding on proton chemical shifts, and use these shifts to estimate distance between drug and heme aromatic rings. We also report the first isolation of a QN-FPIX complex from aqueous solution. Detailed spectroscopic analyses of this complex shows that formation of a 1:1, non-covalent complex between QN free base and monomeric FPIX occurs in aqueous solution, and that this complex likely forms an insoluble, non-crystalline aggregate in the DV that then stabilizes weak QN-FPIX interaction. Collectively, our findings allow us to propose a novel model for QN binding to monomeric FPIX, as well as biologically relevant models that distinguish QN vs. CQ inhibition of Hz formation.
Materials and Methods
Materials
All reagents and solvents were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise noted. Iodoethane was purchased from Acros Organics (Geel, Belgium). Propionic acid, sodium propionate, anhydrous sodium sulfate, clear polystyrene and sterile tissue culture flat bottom 96-well plates were purchased from Fischer Scientific (Newark, DE). Sodium dodecyl sulfate was purchased from Bio-Rad laboratories (Hercules, CA). Hemin was purchased from Fluka (Buchs, Switzerland). Zinc protoporphyrin IX was purchased from Frontier Scientific (Logan, Utah). D2O and DMSO-d6 were purchased from Cambridge Isotope Laboratories Inc. (Andover, MA).
Culturing
P. falciparum strains Dd2, GCO3, 7G8, and HB3 were obtained from the Malaria Research and Reference Reagent Resource Center (Manassas, VA). Off-the-clot, heat-inactivated pooled O+ human serum and O+ human whole blood were purchased from Biochemed Services (Winchester, VA). Sybr Green I nucleic acid dye was purchased from Invitrogen (Eugene, OR). Custom 5% O2/5% CO2/90% N2 culturing gas blend was purchased from Robert’s Oxygen (Rockville, MD). RPMI 1640, hypoxanthine, HEPES, and Giemsa stain were from Sigma-Aldrich (St. Louis, MO).
Test Compounds
Quinine dihydrochloride was purchased from VWR International (Westchester, PA). Cinchonine (CN) was purchased from Acros Organics (Geel, Belgium), quinidine (QD) hydrochloride monohydrate, halofantrine hydrochloride, and hydroquinine (HQN) were purchased from Sigma-Aldrich (St. Louis, MO). 2-((6-methoxyquinolin-4-yl)methyl)-8-vinylquinuclidine (DHQN, 2), 1-(6-methoxyquinolin-4-yl)propan-1-ol (DADQQN, 5), and 2-(diethylamino)-1-(6-methoxyquinolin-4-yl)ethanol (DQQN, 8) were synthesized as described in the Methods section.
General Methods
All commercially available reagents and solvents were used without further purification. Flash chromatography was performed on Kieselgel 60, particle size 0.032–0.063 mm. Thin layer chromatography analyses were performed on Selecto Scientific flexible TLC plates (silica gel 60 - F 254, 200 micron). NMR spectra were obtained on a 400 MHz (1H NMR) and 100 MHz (13C NMR) Varian FT-NMR spectrometer using CDCl3 as solvent unless otherwise indicated, and using tetramethylsilane (TMS) as the external standard. Mass spectroscopic measurements were performed in MeOH or ACN on a Varian500 ESI Mass spectrometer.
Synthesis of 2-((6-methoxyquinolin-4-yl)methyl)-8-vinylquinuclidine (2, Scheme 1)
Scheme 1.

Synthesis of 2-((6-methoxyquinolin-4-yl)methyl)-8-vinylquinuclidine (2, DHQN): i). TsCl, Et3N, CH2Cl2, 9 h, 0°C to rt, 34 % yield; ii). LiAlH4, THF, 12 h, rt, 27 % yield.
QN was reacted with p-toluenesulfonyl chloride in the presence of a mild base to give the tosyl-protected QN derivative 1 in 34% yield. Treatment of 1 with lithium aluminum hydride (LiAlH4) in tetrahydrofuran (THF) at room temperature resulted in the reduction of the hydroxyl functionality to give 2 (DHQN) as a yellowish oil in 27 % yield (see Supporting Information for details).
Synthesis of 1-(6-methoxyquinolin-4-yl)propan-1-ol (5, Scheme 2)
Scheme 2.

Synthesis of 1-(6-methoxyquinolin-4-yl)propan-1-ol (5, DADQQN) and 2-(diethylamino)-1-(6-methoxyquinolin-4-yl)ethanol (8, DQQN): i) SeO2, 4:1 dioxane/H2O, 90°C, 15 h, 70%; ii) EtMgBr, THF, 0°C, 6 h, 57% yield; iii) MeNO2, Et3N, MeOH, −10°C, 5 h, 66%; iv) LiAlH4, THF, 0°C to rt, 1 h, 35%; (v) EtI, Et3N, DMF, rt, 24 h, 18%.
was prepared through a Grignard reaction involving the addition of ethylmagnesium bromide to a solution of 6-methoxyquinoline-4-carbaldehyde (4) at 0 °C with continuous stirring to give the pure product 5 (DADQQN) in 57 % yield.
Synthesis of 2-(diethylamino)-1-(6-methoxyquinolin-4-yl)ethanol (8, Scheme 2)
6-Methoxy-4-methylquinoline (3) was oxidized with selenium dioxide in a mixture of dioxane and water to give 6-methoxyquinoline-4-carbaldehyde (4) in 70% yield. A Henry reaction with nitromethane in the presence of triethylamine afforded nitroaldol 6 in 66% yield, which was subsequently reduced to amino alcohol 7 in 35% yield using LiAlH4. In the final step, 7 was reacted with two equivalents of iodoethane in the presence of triethylamine to give QN analogue 8 (DQQN) in 18% yield (see Supporting Information for details).
Antiplasmodial Activity Measurements
All P. falciparum strains were maintained using the method of Trager and Jensen [28] with minor modifications. Briefly, cultures were maintained under an atmosphere containing 5% CO2, 5% O2 & 90% N2 gaseous mix at 2% hematocrit and 1–2% parasitemia in RPMI 1640 supplemented with 10% O+ human serum, 25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES; pH 7.4), 24 mM NaHCO3, 11 mM glucose, 0.75 mM hypoxanthine, and 20 μg/L gentamycin with regular media changes every 48 h. The antiplasmodial activity was assessed essentially as previously described [29] with minor modifications. Test compounds were diluted to make 2X stocks using complete media under sterile conditions. 100 μL of these were then transferred into 96-well plates followed by incubation at 37°C. Sorbitol synchronized cultures were utilized for the assays with > 95% of the parasites in the ring stage [30]. Cultures were diluted to give a working stock of 0.5% parasitemia and 2% hematocrit and 100 μL was transferred to each drug-preloaded well (final 1% hematocrit and 0.5% parasitemia). The plates were transferred to an airtight chamber which was then gassed and incubated at 37 °C. After 72 h, 50 μL of 10X SYBR Green I dye (diluted using complete media from a 10,000X DMSO stock) was added, the plates incubated for an additional 1 h at 37 °C to allow DNA intercalation, and fluorescence measured at 530 nm (490 nm excitation) using a Spectra GeminiEM plate reader (Molecular Devices). Data analysis was performed using Sigma Plot 10.0 software after downloading data in Excel format. For each assay, each drug dilution was analyzed in triplicate, and the results from three separate assays averaged (±SD). All drugs were tested against two quinine sensitive (QNS), and two quinine resistant (QNR) strains of P. falciparum (GCO3 and HB3, 7G8 and Dd2, respectively).
β-hematin Growth Inhibition Studies
β-hematin growth inhibition was assessed using a recently perfected 96-well plate high throughput assay [31]. Briefly, the assay uses physiologic temperature and lipid catalyst, and relies on the differential solubility properties of crystalline and non-crystalline forms of FPIX in 2.5% SDS (86.7 mM) and alkaline bicarbonate buffer (0.1 M, pH 9.1). Hemin was dissolved in 0.1 M NaOH to make a 2 mM stock. 10 μL was then transferred to 96-well plate wells, followed by propionate buffer (180 μL at desired pH) and 10 μL of sonicated phosphatidyl choline suspension (10 μL) to a final heme concentration of 100 μM. Drugs were added at different concentrations, plates were mixed, wrapped in plastic wrap, and incubated at 37 °C for 16 h. The assay was terminated by adding 100 μL of a solution of SDS dissolved in 0.1 M bicarbonate buffer (pH 9.1) {final concentration/well of SDS 2.5% (w/v), 86.7 mM}. The well contents were gently mixed and the plate incubated at room temperature for 10 minutes to allow un-dissolved Hz crystals to settle. A 50 μL aliquot from each well was then transferred to a second plate preloaded with 200 μL of SDS solution (2.5% w/v, 86.7 mM) in 0.1 M bicarbonate buffer. Absorbance of non-crystallized heme was recorded at 405 nm with a 96-well plate-adapted ELx800 BioTek absorbance microplate reader. Percent Hz formed was analyzed using Microsoft Excel (2007 Edition) and Sigma Plot 10.0 software [31].
Heme Affinity Measurements
Drug-heme affinity measurements were performed by monitoring the changes in the absorbance of heme in the presence of increasing concentrations of drug. Hemin was dissolved in DMSO to 5 mM, which was then serially diluted to 5 μM in 40% DMSO/0.2 M acetate buffer, pH 5.0. Drug solutions were prepared by dissolving the compound in DMSO and diluting to 1.5 mM in 40% DMSO/0.02 M HEPES, pH 7.4. A cuvette containing 1 mL of freshly prepared heme (5 μM) was titrated with increasing drug (0–210 μM), the sample mixed following each addition, and absorbance of heme at 402 nm recorded using an Agilent UV-VIS spectrophotometer (final volume dilution 6.54%). Solvent dilution controls were performed similarly and spectral and data analyses performed using KaleidaGraph and Sigma Plot 10.0 software. Nonlinear least squares curve fitting of the raw data was done using the Levenberg-Marquardt algorithm (initial Ka input = 0.01 μM−1) and affinity coefficients (Ka) were computed (see caption to Figure 2).
Figure 2.

Drug-heme binding curves. Affinity was measured by titrating increasing concentrations of QN (▲), QD (□), HF (△), CN (●), HQN (○), DHQN (▼), DQQN (◇), or DADQQN (■) dissolved in 40 % DMSO/acetate buffer (0.2 M, apparent pH 5.0) into a solution of heme (10 μM) followed by non-linear least squares curve fitting to determine Ka. The affinity coefficients were determined to be 3.92 × 104 (R2 = 0.99), 2.45 × 104 (R2 = 0.99), 1.87 × 104 (R2 = 1.00), 1.96 × 104 (R2 = 1.00), and 2.18 × 104 M−1 (R2 = 0.99) for HF, QN, QD, CN, and HQN, respectively. No statistically reliable coefficients could be determined for DHQN, DQQN, and DADQQN.
Magnetic Susceptibility Measurements
Magnetic susceptibility measurements were performed as previously described [21] in 40% (v/v) DMSO/100 mM phosphate buffer (pH 7.0). Both hemin (Fluka) and drug stock solutions (20 mM) were prepared in 100% DMSO-d6. Test samples (40% v/v DMSO/100 mM phosphate buffer at pH 7.0) were prepared by adding 200 μL of hemin and the corresponding test compound into a 1.5 mL microcentrifuge tube followed by the addition of 600 μL of 100 mM phosphate buffer to give a 1:1 solution of both components at 4 mM. The pH of the resulting samples was taken to be the pH of the buffered aqueous medium measured at 25°C using an Accumet Basic AB15 pH meter. The samples were transferred into 5 mm NMR tubes fitted with coaxial inserts containing the test compound in a similar solvent system. Measurements were made with a Varian Unity INOVA 500 MHz NMR spectrometer with a proton frequency of 499.789 MHz and data analyzed using the Varian VNMR version 5.1 software. Magnetic susceptibility was determined using the Evans method [32] employing the equation (Equation 1) appropriate for a superconducting magnet at 298 K:
| (1) |
where xm is the molar susceptibility of the paramagnetic substance in cm3/mol, Δν is the chemical shift difference (in ppm) between a reference proton in the sample and that in a solution lacking the paramagnetic compound, c is the concentration of FPIX in mol/mL, and xD is the diamagnetic susceptibility of heme (6.9 × 10−4 cgs units). Solvent susceptibility corrections and the solution-solvent density differences are ignored. The molar susceptibility was converted to magnetic moment (μ) using Equation 2 below, where T is the temperature (K): μ = 2.8√(xmT)
| (2) |
Solution NMR Experiments Using Zinc (II) Protoporphyrin IX
A 10 mM stock solution of ZnPIX was prepared in 0.05 M NaOH in D2O, and 10 mM drug stock solutions were prepared by dissolving the corresponding HCl salts in D2O. Equimolar drug-heme solutions (1.667 mM each) were then prepared and titrated to pH 7.0. 600 μL of this solution was transferred to a microcentrifuge tube (1.5 mL) followed by the addition of 400 μL of DMSO-d6, that is test samples contained a 1:1 drug to ZnPIX ratio at 500 μM. These solutions were then transferred to a 5 mm NMR tube for analyses. 1D & 2D proton NMR spectra were recorded and analyzed as stated above on the 500 MHz NMR spectrometer using tetramethylsilane as the external reference.
Formation and Analyses of Aqueous QN-Heme Complex
A QN-heme complex formed under aqueous conditions was prepared by first dissolving hemin in 0.1 M NaOH to give a 2 mM stock solution. This solution was slowly titrated with increasing volumes from a QN stock (100 mM in deionized water) while monitoring the pH (see Results). At a molar ratio of 1:9 (heme to QN) copious heme was observed to precipitate, leaving a clear solution of QN (see Results). The resultant precipitate was isolated via vacuum filtration and freeze-dried.
Mass spectrometry analyses using a Varian 500 MS were performed by dissolving the precipitate in ACN. IR analyses of heme, QN, and the adduct were performed in KBr pellets on a Nicolet 380 FTIR spectrometer (Thermo-Electron Corporation). Briefly, dried solid samples of either QN, heme, or the adduct were added to powdered KBr (1:3 ratio sample to KBr), ground, and pressed. Fluorescence spectra were recorded from 5 μM samples dissolved in 1:1 methanol/0.2 M HEPES (pH 7.2) using a QM 2001–4 Quantum Master Fluorometer (shown in Fig. S1 of “Supplemental Data”). Solid-state 13C CP-MAS NMR spectra were acquired on a Bruker Avance DRX-500 spectrometer operating at a 13C frequency of 125.76 MHz and using XWIN-NMR version 2.1 software. Samples were packed into a 4-mm rotor and spun at 9 kHz MAS at room temperature. For QN base, QN monohydrochloride dehydrate, and QN dihydrochloride samples, 2048 scans were collected; for the QN-FPIX complex, 15360 scans were collected. The recycle delay was 3s and the CP contact time was 1 ms. 45 kHz CP and 63 kHz TPPM decoupling were used. The spectral width was 83,333 Hz. Exponential line broadening of 100 Hz was applied to all spectra. Chemical shifts were externally referenced to TMS using the CO signal of glycine at 176.4 ppm [33].
Results
We first designed and synthesized a series of QN analogs containing the QN pharmacophore but lacking specific functional groups (DHQN, DQQN, and DADQQN; Schemes 1 & 2, Figure 1), and also obtained two additional commercially available QN analogues (CN, HQN) lacking additional groups. We calculated simple physicochemical characteristics for these (Table 1) which indicate that, in most cases, the structural alterations do not significantly perturb these relative to QN. One exception is DADQQN, which is predicted to have lower DV accumulation (lower VAR, cf. Table 1) due to the loss of the basic terminal N. For the remaining analogs, both quinolyl N and aliphatic N pKas were comparable to those for QN, making them effectively monobasic under biological conditions. Correspondingly, calculated vacuolar accumulation ratios (VAR) [34,35] for these compounds were comparable.
Figure 1.

Structures of compounds used in our analyses. CQ – chloroquine; AQ – amodiaquine; HF – halofantrine; QD – quinidine; QN – quinine; DHQN – dehydroxyquinine; CN – cinchonine; HQN – hydroquinine; DQQN – dequinuclidylquinine; DADQQN – deamino-dequinuclidylquinine.
Table 1.
Calculated physicochemical properties for QN, QD, and QN analogs under investigation.
| CPD ID | MWa | LogPb | pKac | Cyt pH7.2VARd | ||
|---|---|---|---|---|---|---|
| (Oct/H2O) | pKa1 | pKa2 | DVpH5.2 | DVpH5.6 | ||
| QN | 324.42 | 2.66 | 5.69 | 9.16 | 393 | 85.2 |
| QD | 324.81 | 3.06 | 5.49 | 9.09 | 392 | 85.1 |
| DHQN | 308.42 | 3.65 | 5.69 | 9.17 | 393 | 85.3 |
| CN | 294.39 | 2.63 | 5.10 | 8.75 | 374 | 81.9 |
| DQQN | 274.36 | 2.45 | 5.75 | 9.55 | 437 | 92.4 |
| DADQQN | 217.26 | 2.34 | 5.78 | - | 4.63 | 2.43 |
| HQN | 326.43 | 2.90 | 5.72 | 9.39 | 415 | 88.8 |
MW – Molecular weight; VAR – Vacuolar accumulation ratio;
Determined using ChemOffice Ultra 10.0,
LogP online calculator for calculation of molecular properties and drug-likeness (http://www.molinspiration.com/cgi-bin/properties),
SPARC online calculator (http://ibmlc2.chem.uga.edu/sparc) pKa1 and pKa2 represent the pKas of the quinolyl and aliphatic nitrogen respectively;
Calculated using the Henderson Hasselbalch equation (DV & Cytosolic pH as defined above; see also Natarajan JK, et al. J Med Chem. 2008, 51, 3466).
Antiplasmodial activities for these compounds were measured vs. two QNS (HB3, GCO3) and two QNR (Dd2, 7G8) strains of P. falciparum using the SYBR Green I assay ([29], Table 2). Results show that QD and CN are approximately 4–5 fold more active than QN, and that QN and HQN had quite similar activity. DQQN, DADQQN, and DHQN were the least active, exhibiting IC50s ~ 7–100 fold higher than QN, respectively, depending on the strain (Table 2). These results demonstrate that elimination of the methoxy group improves the activity of QN such that it is then similar to QD and that loss of the vinyl group has no effect on QN antiplasmodial activity. In contrast, IC50s for DHQN and DADQQN were, in general, > 10 μM for both QNS & QNR strains, indicating that loss of the hydroxyl moiety or the rigid quinuclidine ring essentially destroys activity. Interestingly, replacement of a tertiary aliphatic N at the same position as that found in the QN quinuclidine ring (DQQN) partially restored activity.
Table 2.
Antiplasmodial activities against two QNS strains (GCO3, HB3) and two QNR strains (Dd2, 7G8)
| CPD ID | Experimental IC50a (nM) |
|||||
|---|---|---|---|---|---|---|
| HB3b | Dd2c | SId | GCO3b | 7G8c | SIe | |
| QN | 81±8.4 | 320±50 | 4.0 | 99 ± 13 | 350±39 | 3.6 |
| QD | 18 ± 2.2 | 90 ± 8.4 | 5.0 | 28 ± 7.6 | 110±2.3 | 3.7 |
| HF | 1.6 ±0.7 | 3.2 ±0.4 | 2.0 | 1.4 ±0.3 | 1.8 ±0.3 | 1.3 |
| DHQN | >10,000 | >10,000 | - | >10,000 | >10,000 | - |
| CN | 22 ±6.8 | 82 ± 8.1 | 3.8 | 19 ± 3.4 | 95 ± 18 | 5.1 |
| DQQN | 2600 ± 58 | 2100 ± 410 | 0.83 | 2700 ± 280 | 1600 ± 150 | 0.6 |
| DADQQN | 9600 ± 290 | >10,000 | - | >10,000 | >10,000 | - |
| HQN | 100 ±19 | 410 ±57 | 4.0 | 110 ±14 | 490 ±53 | 4.4 |
The experimental IC50s are an average of three separate determinations (± SD); Strain phenotype:
QNS,
QNR;
SId – (IC50 Dd2/IC50 HB3),
SIe – (IC50 7G8/IC50 GCO3).
As mentioned, inhibition of parasite Hz formation is hypothesized to be the principle basis of antiplasmodial activity for the cinchona alkaloids [6–9]. Thus, we investigated the ability of these compounds to inhibit β-hematin (Hz) crystal growth in vitro (BHIA activity) in order define the significance of QN functional groups in this process. A similar trend was observed at both pH 5.2 and pH 5.6 [36] (Table 3). We observed ≥ 5-fold difference between BHIA at pH 5.6 vs. pH 5.2 for all compounds with the exception of CN, which was only 2-fold different at the two pH, and DADQQN which had quite poor activity at both. At pH 5.6, the BHIA IC50s of QD, QN, CN, and HQN were conspicuously lower than DHQN, DADQQN, and DQQN IC50, and these three analogs had even poorer IC50s (> 2 mM) at pH 5.2. These results demonstrate the importance of the hydroxyl and rigid quinuclidyl moeity for β-hematin crystal growth inhibition by QN. However, we do not find a strong correlation when plotting antiplasmodial activity vs BHIA for these QN analogues (data not shown).
Table 3.
In vitro β-hematin growth inhibitory activities (BHIA)
| CPD | BHIA IC50a (μM) |
|
|---|---|---|
| pH 5.2 | pH 5.6 | |
| QN | 260 ± 15 | 49 ± 0.4 |
| QD | 130 ± 2.5 | 23 ± 2.3 |
| HF | 100 ± 3.1 | 24 ± 4.7 |
| DHQN | > 2000 | 230 ± 24 |
| CN | 370 ± 11 | 170 ± 36 |
| DQQN | > 2000 | 210 ± 12 |
| DADQQN | > 2000 | 1900 ± 53 |
| HQN | 280 ± 9.0 | 44 ± 1.6 |
The experimental IC50s are averages of two separate determinations each performed in triplicate (± SE).
We next tested if BHIA is related to affinity for monomeric heme since we have recently concluded that QN, unlike CQ, promotes formation of monomeric heme [21]. Although it would be ideal to perform these experiments under physiologically relevant aqueous conditions, drug-FPIX titrations were performed in 40% aqueous DMSO since monomeric FPIX is unstable in aqueous solution [37]. The results indicate significant differences in heme binding for the compounds (Figure 2), with HF, QD, and CN showing similar binding that is also higher affinity than that for QN and HQN. DHQN, DADQQN, and DQQN showed little to no heme binding under these conditions, in general consistent with the above data showing reduced efficiency of these three molecules in inhibiting β-hematin crystal growth.
We next measured the magnetic susceptibility of FPIX (4 mM) in the presence of test compounds (4 mM) in 40% DMSO/phosphate buffer (100 mM), pH 7.0. These measurements easily distinguish presence of the μ-oxo bridged vs. monomeric FPIX species and have been used previously to determine preference of a particular FPIX species for different quinoline drugs [21]. Our results (Table 4, far right column) clearly show significantly different perturbations in monomer-dimer equilibria upon addition of QN vs. DHQN, DQQN, or DADQQN, as determined by comparing heme μ measured in the presence of these compounds. For QN, QD, and HF, a magnetic moment of 5.45 μB was observed, which is close to the upper limiting value of 5.6 μB corresponding to the high spin monomeric species [21], indicating, as expected, that these drugs strongly stabilize FPIX monomer in solution. In contrast, DHQN, DQQN, and DADQQN exhibited moments of 2.52, 2.89 and 2.83μB, respectively. These are similar, but not identical to, μB previously measured in the presence of CQ [19] and are consistent with the presence of low spin μ-oxo dimeric FPIX.
Table 4.
Changes in chemical shifts and magnetic moments (μ).
| CPD ID | Proton #a/Change in Chemical Shiftb (ppm) |
μc | |||||
|---|---|---|---|---|---|---|---|
| 2 | 3 | 5 | 6 | 7 | 8 | ||
| CQ | −0.20 | −0.09 | −0.20 | −0.18 | - | −0.14 | 2.14 |
| AQ | −0.11 | −0.06 | −0.19 | −0.06 | - | −0.15 | 4.48 |
| DHQN | −0.23 | −0.44 | −0.40 | - | −0.13 | −0.17 | 2.52 |
| DQQN | −0.03 | −0.04 | −0.07 | - | −0.04 | −0.03 | 2.89 |
| DADQQN | 0.00 | 0.00 | 0.00 | - | −0.01 | 0.00 | 2.83 |
| QN | −0.03 | −0.04 | −0.03 | - | −0.03 | −0.03 | 5.45 |
| QD | −0.03 | −0.04 | −0.04 | - | −0.03 | −0.03 | 6.01 |
| HF | - | - | - | - | - | - | 5.87 |
Quinoline protons are numbered according to the IUPAC numbering system of the corresponding carbon atoms (see Fig. 6).
Chemical shift difference between the corresponding proton in the presence and absence of heme (ZnPIX in 40% DMSO-d6/D2O pH 7.0). The negative sign indicates an up-field shift relative to the pure drug.
Measured magnetic moments for FPIX in 40% DMSO-d6/Phosphate buffer (0.1 M) pH 7.0 in the presence of the corresponding drug.
Additional NMR studies using non-paramagnetic ZnPIX were used to further probe these drug-heme interactions. We measured perturbations in chemical shift for the quinoline protons in the presence vs. absence of ZnPIX, knowing that different values for these shifts illustrate different distances between the quinoline and porphyrin ring systems [18]. As expected [18], relatively large changes in chemical shift for CQ aromatic ring protons 2, 3, 5, and 6 indicate a short (approximately 3–4 Å, see [18]) CQ-FPIX interplanar distance, whereas for QN the very small change for equivalent proton shifts indicates a much wider (estimated 6 Å [18]) distance between the rings (Table 4).
To further define the interaction between QN and FPIX, we attempted to isolate the QN-FPIX complex formed under aqueous conditions. One method we perfected, based on simple titration as described in Methods, leads to precipitation of a greenish, highly fluorescent (see Supplemental Fig. 1) precipitate from aqueous solution. This precipitate was initially isolated at pH ~8.5 but can be obtained in lower yield at pH 6.0–7.0. We analyzed the presumed QN-FPIX adduct by electrospray ionization - mass spectrometry (ESI -MS) in positive ion mode and obtained minor peaks at m/z 325.2 and 616.2, corresponding to free QNH+ and free FPIX monomer, respectively, and clear major peaks at m/z 938.5, 940.4, and 941.3 (Figure 3). The major peaks coincide with the calculated m/z values (M – 2H)+ = 938.58, (M)+ = 940.35, and (M + H)+ = 941.36 for C54H55FeN6O6, corresponding to 1:1 QN-FPIX complexes. One simple interpretation of the data is that the three peaks correspond to complexes between QN free base and FPIX with deprotonated propionate side chains, QN free base and protonated FPIX, and QN free base and protonated FPIX but including 13C natural abundance for the 54 carbons, respectively.
Figure 3.

Mass spectrum of the QN-FPIX adduct in ACN. The spectrum shows m/z peaks at 325.2 corresponding to (M + H)+ for QN free base, m/z 616.2 corresponding to (M)+ for FPIX, and 938.5, 940.4, 941.3 which match the calculated m/z values (M − 2H)+ = 938.58, (M)+ = 940.35, and (M + H)+ = 941.36 for C54H55FeN6O6 that correspond to a 1:1 QN-heme complex. The peaks at m/z 498.2, 771.6, and 859.8 are possible FPIX fragments.
IR spectral analyses of the adduct (Figure 4A,B, top spectrum “W”) revealed a sum of QN (“X”) and heme (“Y”) vibrational peaks, but the conspicuous absence of peaks indicative of QNH+ (Figure 4 “Z”), including the strong N-H stretch centered near 2575 cm−1 (Figure 4A), as well as the prominent 1702 cm−1 heme propionate carbonyl stretch (Figure 4B). Thus, surprisingly, the isolated complex is most likely QN free base complexed with fully ionized monomeric FPIX.
Figure 4.

IR spectra for the QN-heme adduct (W, dotted line), QN free base (X, dashed line), hematin (Y, continuous line), and QN hydrochloride salt (Z, dash-dot line). A) The broad peak at 2687-2400cm−1 in the QN hydrochloride spectrum that corresponds to the N-H stretch of the protonated quinuclidyl nitrogen is absent in the QN free base, heme, and the QN-FPIX adduct spectra; B) the strong peak at 1702 cm−1 in the heme spectrum that corresponds to the carbonyl stretch of the heme carboxylic acids is absent in the QN free base, QN hydrochloride, and QN-FPIX adduct spectra.
Solid state 13C NMR analyses of the precipitated adduct (Figure 5B) shows clear aromatic and aliphatic QN peaks and an obvious lack of contact shifts previously observed for similar CQ-FPIX precipitates [38]. In particular, there is only a 3 ppm shift in the peak at ~ 70 ppm, which corresponds to the carbon bearing the QN hydroxyl group. These data clearly indicate a non-covalent interaction between QN and FPIX. Consistent with IR analysis, the pattern of adduct peaks is most similar to the spectrum for QN free base (Figure 5A), but differs significantly relative to the spectra for the mono- and di-protonated salts (Figure 5C,D, respectively).
Figure 5.

Solid state 13C CP-MAS NMR spectra for QN free base (A), QN-FPIX adduct (B), QN hydrochloride salt (C), and QN dihydrochloride salt (D). The corresponding chemical shifts for QN free base: δ 18.8, 28.8, 43.0, 53.5, 59.0, 70.8, 100.1, 113.8, 125.7, 131.4, 143.7,153.1,158.4; QN-FPIX adduct: 19.6, 28.5, 42.8, 58.9, 71.0, 101.1, 118.6, 131.5, 147.2, 158.4; QN hydrochloride salt: 20.0, 27.9, 39.0, 45.1, 57.3, 62.1, 66.7, 86.4, 100.0, 118.5, 131.5, 144.3, 159.3; QN dihydrochloride salt: 19.6, 24.6, 31.2, 40.1, 45.5, 58.4, 65.2, 76.3, 104.5, 108.3, 117.2, 124.1, 143.2, 152.7, 159.1.
Thus, in aqueous solution, QN appears to preferentially bind to ionized monomeric FPIX in the free base form. As shown in Figure 6, this complex must be non-covalent, with ring systems approximately 6 Å apart. To further stabilize such a complex, and consistent with the critical nature of cinchona alkaloid stereochemistry as well as our results with QN analogues in this paper, we propose that a strong hydrogen bond between the QN hydroxyl H and the quinuclidinyl N is essential. Presumably due to interaction of the hydroxyl O with heme Fe, the more polarized O-H bond of QN enhances this hydrogen bonding.
Figure 6.

Proposed QN-FPIX adduct structure. The model involves coordination between the –OH group of QN and Fe of FPIX, aided by the formation of a strong hydrogen bond between the –OH proton and the quinuclidyl nitrogen, resulting in the formation of a five-membered ring. Note also that in this model the quinoline ring is tilted away from porphyrin ring system resulting in minimal π- π interactions. Further, the methoxy group of QN is oriented in such a way as to be spatially close to the vinyl group as observed in NOESY experiments with ZnPIX (data not shown).
Discussion
The data in this paper strongly support a model for QN-FPIX binding depicted in Figure 6. This model is consistent with some previously published data, but includes important new information. The complex we isolate does involve FPIX monomer, not dimer, has a 1:1 stoichiometry similar to the conclusions of [22,27], and does involve interaction between the QN hydroxyl O and the Fe center of FPIX, which is also consistent with [22–27]. However, the data in this paper clearly shows that in aqueous environments the interaction is non-covalent, eliminating models wherein QN-FPIX interaction is stabilized by Fe–O or Fe–N covalent (dative covalent) bonds. Also, the data in this paper show (surprisingly) that QN prefers to associate with ionized FPIX in the free base form. This preference is likely a consequence of the stabilizing hydroxyl H/quinuclidine N hydrogen bond geometry that we propose (Figure 6).
Both CQ and QN are potent antiplasmodial drugs, both bind to heme and inhibit Hz formation, but clearly their heme binding preferences differ in interesting and important ways. In aqueous solution, CQ clearly prefers binding to μ-oxo dimeric FPIX, whereas QN prefers binding to monomeric FPIX. The QN:FPIX monomer species forms under aqueous conditions, but is clearly more stable in either aggregate form (e.g. data in this paper) or as a complex partitioned into the hydrophobic phase [20]. Two points from the recent literature further illuminate the pharmacologic significance of these conclusions. First, lower pH that promotes aggregation of μ-oxo dimeric heme also appears to accelerate Hz formation [21,31,39], as does the presence of lipid [31,39]. Lipid nanospheres are found in the P. falciparum DV and nascent Hz crystals appear to be associated with these. Thus, it is reasonable to propose that, biologically, QN is most potent in preventing Hz formation as a free base QN-monomeric FPIX complex either imbedded in lipid or existing as an amorphous solid state aggregate stabilized by low DV pH. In constrast, even though CQ is capable of interacting with monomeric FPIX via a dative Fe quinolinal N bond under specific conditions [38], it much more strongly prefers to bind to μ-oxo dimer in aqueous solution. Interestingly, data in Table 4, along with previous results [40], suggests that AQ, which is slightly more potent than either QN or CQ, may inhibit Hz formation by a combination of these two effects.
These features can be incorporated into two simple models for how the formation of Hz from free monomeric FPIX is inhibited by different quinoline antimalarials (Figure 7). The principle difference is that in the first “cyclic” model (Fig. 7A) Hz is envisioned to be formed from either monomeric FPIX or μ-oxo dimeric FPIX. It is well known that acid aggregation of μ-oxo dimer accelerates formation of Hz [6–9, 39] but it is not known if μ-oxo dimer directly converts to head-to-tail dimer (“HT dimer”, the unit cell of crystalline Hz) within the aggregated state or, alternatively, proceeds to HT dimer via dilute monomeric FPIX. It is also possible that a non covalent π – π stabilized heme dimer is a third intermediate in conversion to HT dimer, as has been proposed by two other groups [41, 42].
Figure 7.
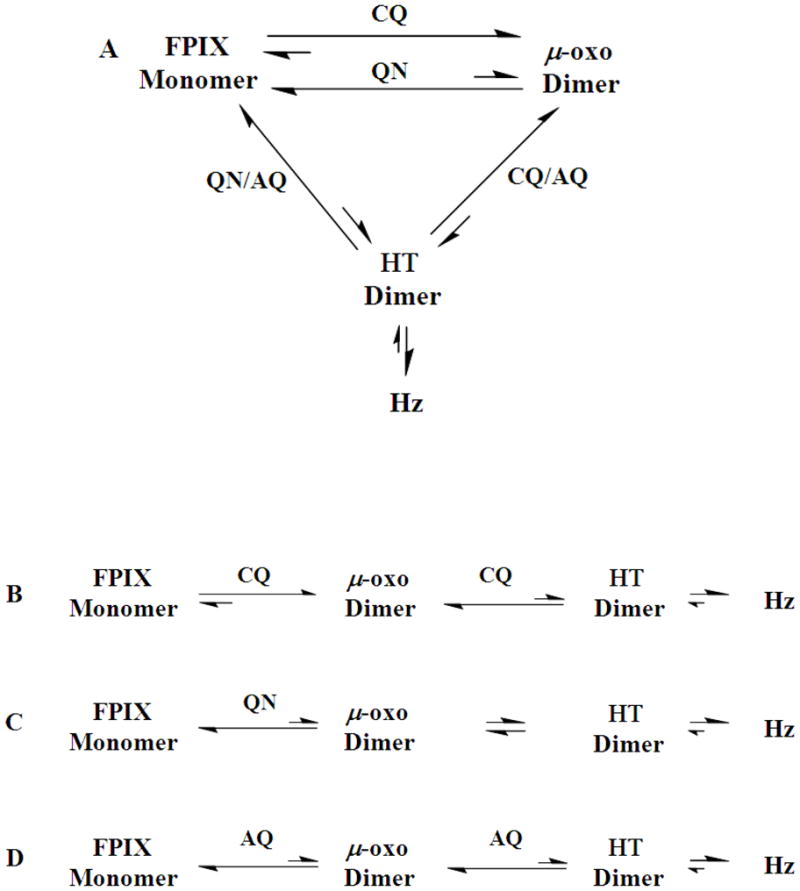
The effect of CQ, AQ, & QN on the hemozoin (Hz) crystallization process. A) Possible cyclic pathway for Hz crystallization. Both the monomer and μ-oxo dimer are envisioned to be possible direct precursors to the head-to-tail (HT) dimer which then crystallizes to Hz. Introduction of CQ shifts equilibrium away from both the HT dimer and the monomer by promoting μ-oxo dimer. QN shifts equilibrium away from the HT and μ-oxo dimers by promoting formation of the monomer. AQ interacts with both monomer and μ-oxo dimer providing a possible rationale for this drugs’ higher potency in Hz inhibition relative to CQ/QN. B–D) Alternate linear pathway for Hz crystallization envisioning that μ-oxo dimer is the direct precursor to HT dimer. B) Introduction of CQ to the linear pathway shifts equilibria towards the μ-oxo dimer. C) QN shifts equilibrium towards the monomer limiting conversion to μ-oxo dimer. D) AQ stabilizes both monomer and dimer, which synergistically shifts equilibrium away from HT dimer.
As diagramed in Fig. 7B–D, an alternate “linear” model is that μ-oxo dimer FPIX is an obligate precursor to HT dimer. We suggest direct conversion from μ-oxo to HT dimers within an acid and lipid stabilized heme aggregate is the physiologically preferred route, as is consistent with the known pH of the parasite DV [36], the presence of DV lipid nanospheres [39], and the pH-dependent aqueous vs. lipid partitioning behavior of FPIX [20,21]. Via the cyclic model (Fig. 7A) CQ and QN would have opposing effects on stabilizing μ-oxo dimer vs. monomer, respectively, but either of these effects could also drive equilibrium away from HT dimer and hence inhibit conversion to Hz, if HT dimer forms equally well from either monomer or μ-oxo dimer. However, we argue that this predicts QN-CQ synergy that to the best of our knowedge has not ever been observed. Via the second model (Fig. 7B,C) the two drugs simply operate at different points in linear conversion of monomeric FPIX to Hz that must proceed in some fashion via μ-oxo dimer. CQ again stabilizes the μ-oxo dimer, slowing conversion to HT dimer and Hz, similar to earlier proposals (e.g. see [7]). In contrast, QN slows conversion of monomer to μ-oxo, but not μ-oxo to HT. This model is easily modified to include a π – π stabilized heme dimer [41, 42] between monomer and μ-oxo dimer, wherein CQ still pulls equilibrium towards the μ-oxo dimer and QN pulls equilibrium towards the monomer, with both binding phenomena preventing formation of a non-covalent π – π stabilized heme dimer.
As shown in Table 4, AQ complexation with FPIX yields an intermediate FPIX magnetic moment (4.48), as well as intermediate effects on changes in the chemical shifts of AQ protons (e.g. −0.11 for the proton at position 2, relative to −0.20 for CQ and −0.03 for QN). These data, along with recalling that AQ has slightly higher potency in parasite proliferation assays relative to either CQ or QN, lead us to suggest that AQ is capable of both types of HT conversion inhibition. Meaning, we predict that AQ: μ-oxo dimer complexes and acid aggregated AQ: monomer complexes are both relevant for AQ inhibition of Hz formation. The overall point is that any expectation that different quinoline antimalarial drugs all show antimalarial activity via essentially the same molecular mechanism is a simplification that neglects distinct molecular preferences for different FPIX chemical forms. A caveat of this conclusion is that different patterns of resistance to these drugs could be due, at least in part, to the parasite manipulating bioavailability of different FPIX chemical forms. This can be done rather easily in solution via subtle changes in pH, ionic environment, or lipid composition.
Lastly, strong fluorescence observed for the QN-FPIX adduct (Fig. S1) is an interesting property for QN-heme complexes not previously observed to our knowledge. It is at least 7-fold higher than QN fluorescence (heme is not fluorescent at this wavelength) and is likely due to transition dipole ordering in the complex. We suggest this fluorescence represents a convenient marker for further optimization of quinoline drug activity.
Supplementary Material
Acknowledgments
We thank Drs. Bahram Moasser and Shibu Abraham (Georgetown) for insightful discussions regarding the mass spectrometry results, Dr. Dan McEleheny (UIC) for help with the solid-state NMR, and Daniel P. Iwaniuk and Dr. Christian Wolf for advice and help with quinine analogue synthesis. L.B.C. gratefully acknowledges the support of the AAUW Postdoctoral Fellowship.
5. Abbreviations
- AQ
Amodiaquine
- BHIA
β-hematin Inhibitory Activity
- CN
Cinchonine
- CQ
Chloroquine
- CQR
Chloroquine Resistant
- CQS
Chloroquine Sensitive
- DADQQN
Deamino-dequinuclidylquinine
- DHQN
Dehydroxyquinine
- DQQN
Dequinuclidylquinine
- DV
Digestive Vacuole
- DVpH
Steady state digestive vacuolar pH
- ESI-MS
electro spray ionization – mass spectrometry
- FPIX
Iron Protoporphyrin IX
- ZnPIX
Zinc Protoporphyrin IX
- Hb
Hemoglobin
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HF
Halofantrine
- Hz
Hemozoin
- PfCRT
Plasmodium falciparum chloroquine resistance transporter
- PfMDR
Plasmodium falciparum multi drug resistance protein 1
- PfNHE
Plasmodium falciparum Sodium proton exchange
- QD
Quinidine
- QN
Quinine
- QNR
Quinine Resistant
- QNS
Quinine Sensitive
- THF
tetrahydrofuran
- VAR
Vacuolar Accumulation Ratio
Footnotes
Supported by NIH grants AI045957 (PDR) and AI060792 (AdD, PDR and Christian Wolf, Georgetown University)
Supporting Information Available:
Fluorescence excitation and emission spectra for QN, FPIX, and QN-FPIX adduct, synthetic methods, 1H NMR, 13C NMR, and HPLC spectra for 2-((6-methoxyquinolin-4-yl)methyl)-8-vinylquinuclidine (2, DHQN), 1-(6-methoxyquinolin-4-yl)propan-1-ol (5, DADQQN), and 2-(diethylamino)-1-(6-methoxyquinolin-4-yl)ethanol (8, DQQN).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.World Health Organization WHO. 2008 report. http://apps.who.int/malaria/wmr2008.
- 2.Noedl H, Se Y, Schaecher K, Smith BL, Socheat D. M M Fukuda N Engl J Med. 2008;359:2619–20. doi: 10.1056/NEJMc0805011. [DOI] [PubMed] [Google Scholar]
- 3.Dondorp AM, Nosten F, Yi P, Das D, Phyo AP, Tarning J, Lwin KM, Ariey F, Hanpithakpong W, Lee SJ, Ringwald P, Silamut K, Imwong M, Chotivanich K, Lim P, Herdman T, An SS, Yeung S, Singhasivanon P, Day NP, Lindegardh N, Socheat D, White N. J N Engl J Med. 2009;361:455–67. doi: 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meshnick S, Dobson M. In: Antimalarial chemotherapy: Mechanism of action, Resistance and New directions in drug discovery. Rosenthal P, editor. Humana, Totowa, New Jersey: 2001. pp. 15–26. [Google Scholar]
- 5.Rathore D, McCutchan TF, Sullivan M, Kumar S. Expert Opin Investig Drugs. 2005;14:871–83. doi: 10.1517/13543784.14.7.871. [DOI] [PubMed] [Google Scholar]
- 6.Egan TJ, Ncokazi KK. J Inorg Biochem. 2005;99:1532–1539. doi: 10.1016/j.jinorgbio.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 7.Dorn A, Vippagunta SR, Matile H, Jaquet C, Vennerstrom JL, Ridley RG. Biochem Pharmacol. 1998;55:727–36. doi: 10.1016/s0006-2952(97)00510-8. [DOI] [PubMed] [Google Scholar]
- 8.Slater AF, Cerami A. Nature. 1992;355:167–9. doi: 10.1038/355167a0. [DOI] [PubMed] [Google Scholar]
- 9.Egan TJ, Ross DC, Adams PA. FEBS Lett. 1994;352:54–7. doi: 10.1016/0014-5793(94)00921-x. [DOI] [PubMed] [Google Scholar]
- 10.Cooper RA, Lane KD, Deng B, Mu J, Patel JJ, Wellems TE, Su X, Ferdig MT. Mol Microbiol. 2007;63:270–82. doi: 10.1111/j.1365-2958.2006.05511.x. [DOI] [PubMed] [Google Scholar]
- 11.Cooper RA, Ferdig MT, Su XZ, Ursos LM, Mu J, Nomura T, Fujioka H, Fidock DA, Roepe PD, Wellems TE. Mol Pharmacol. 2002;61:35–42. doi: 10.1124/mol.61.1.35. [DOI] [PubMed] [Google Scholar]
- 12.Sanchez CP, Stein WD, Lanzer M. Mol Microbiol. 2008;67:1081–93. doi: 10.1111/j.1365-2958.2008.06108.x. [DOI] [PubMed] [Google Scholar]
- 13.Peel SA, Bright P, Yount B, Handy J, Baric RS. Am J Trop Med Hyg. 1994;51:648–58. doi: 10.4269/ajtmh.1994.51.648. [DOI] [PubMed] [Google Scholar]
- 14.Reed MB, Saliba KJ, Caruana SR, Kirk K, Cowman AF. Nature. 2000;403:906–9. doi: 10.1038/35002615. [DOI] [PubMed] [Google Scholar]
- 15.Sidhu AB, Valderramos SG, Fidock DA. Mol Microbiol. 2005;57:913–26. doi: 10.1111/j.1365-2958.2005.04729.x. [DOI] [PubMed] [Google Scholar]
- 16.Bennett TN, Patel J, Ferdig MT, Roepe PD. Mol Biochem Parasitol. 2007;153:48–58. doi: 10.1016/j.molbiopara.2007.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ferdig MT, Cooper RA, Mu J, Deng B, Joy DA, Su XZ, Wellems TE. Mol Microbiol. 2004;52:985–97. doi: 10.1111/j.1365-2958.2004.04035.x. [DOI] [PubMed] [Google Scholar]
- 18.Leed AK, DuBay K, Sears D, De Dios AC, Roepe PD. Biochemistry. 2002;41:10245–10255. doi: 10.1021/bi020195i. [DOI] [PubMed] [Google Scholar]
- 19.Pagola S, Stephens PW, Bohle DS, Kosar AD, Madsen SK. Nature. 2000;404:307–310. doi: 10.1038/35005132. [DOI] [PubMed] [Google Scholar]
- 20.Casabianca LB, Kallgren JB, Natarajan JK, Alumasa JN, Roepe PD, Wolf C, de Dios AC. J Inorg Biochem. 2009;103:745–748. doi: 10.1016/j.jinorgbio.2009.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Casabianca LB, An D, Natarajan JK, Alumasa JN, Roepe PD, Wolf C, de Dios AC. Inorg Chem. 2008;47:6077–81. doi: 10.1021/ic800440d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Behere DV, Goff HM. J Am Chem Soc. 1984;106:4945–50. [Google Scholar]
- 23.Constantinidis I, Satterlee JD. J Am Chem Soc. 1988;110:927–32. [Google Scholar]
- 24.Karle JM, Karle IL, Gerena L, Milhous WK. Antimicrob Agents Chemother. 1992;36:1538–44. doi: 10.1128/aac.36.7.1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karle JM, Bhattacharjee AK. Bioorg Med Chem. 1999;7:1769–74. doi: 10.1016/s0968-0896(99)00120-0. [DOI] [PubMed] [Google Scholar]
- 26.Warhurst DC, Craig JC, Adagu IS, Meyer DJ, Lee SY. Malar J. 2003;2:26. doi: 10.1186/1475-2875-2-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Villiers KA, Marques HM, Egan TJ. J Inorg Biochem. 2008;102:1660–7. doi: 10.1016/j.jinorgbio.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 28.Trager W, Jensen JB. Science. 1976;193:673–675. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- 29.Bennett TN, Paguio M, Gligorijevic B, Seudieu C, Kosar AD, Davidson E, Roepe PD. Antimicrob Agents Chemother. 2004;48:1807–10. doi: 10.1128/AAC.48.5.1807-1810.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lambros C, Vanderberg JP. J Parasitol. 1979;65:418–20. [PubMed] [Google Scholar]
- 31.Alumasa JN, Nickley KB, Brower JB, de Dios AC, Roepe PD. 2010 Submitted. [Google Scholar]
- 32.Evans DF. J Chem Soc. 1959:2003–05. [Google Scholar]
- 33.Kimura H, Nakamura K, Eguchi A, Sugisawa H, Deguchi K, Ebisawa K, Suzuki I, Shoji A. J Mol Struc. 1998;447:247–55. [Google Scholar]
- 34.Krogstad DJ, Schlesinger PH. Biochem Pharmacol. 1998;35:793–8. doi: 10.1016/0006-2952(86)90345-x. [DOI] [PubMed] [Google Scholar]
- 35.Hawley SR, Bray PG, O’Neill PM, Park BK, Ward SA. Biochem Pharmacol. 1996;52:723–33. doi: 10.1016/0006-2952(96)00354-1. [DOI] [PubMed] [Google Scholar]
- 36.Bennett TN, Kosar AD, Ursos LMB, Dzekunov S, Sidhu ABS, Fidock DA, Roepe PD. Mol Biochem Parasitol. 2004;133:99–114. doi: 10.1016/j.molbiopara.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 37.Asher C, de Villiers KA, Egan TJ. Inorg Chem. 2009;48:7994–8003. doi: 10.1021/ic900647y. [DOI] [PubMed] [Google Scholar]
- 38.de Dios AC, Tycko R, Ursos LMB, Roepe PD. J Phys Chem A. 2003;107:5821–5825. [Google Scholar]
- 39.Pisciotta JM, Coppens I, Tripathi AK, Scholl PF, Shuman J, Bajad S, Shulaev V, Sullivan DJ., Jr Biochem J. 2007;402:197–204. doi: 10.1042/BJ20060986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.De Dios AC, Cassabianca L, Kosar A, Roepe PD. Inorg Chem. 2004;43:8078–8084. doi: 10.1021/ic0489948. [DOI] [PubMed] [Google Scholar]
- 41.de Villiers KA, Kaschula CH, Egan TJ, Marques HM. J Biol Inorg Chem. 2007;12:101–17. doi: 10.1007/s00775-006-0170-1. [DOI] [PubMed] [Google Scholar]
- 42.Crespo MP, Tilley L, Klonis N. J Biol Inorg Chem. 2010 Apr 29; doi: 10.1007/s00775-010-0661-y. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.