

Abstract
Signal transduction histidine kinases (STHK) are key for sensing environmental stresses, crucial for cell survival, and attain their sensing ability using small molecule binding domains. The N-terminal domain in an STHK from Nostoc punctiforme is of unknown function yet is homologous to the central region in soluble guanylyl cyclase (sGC), the main receptor for nitric oxide (NO). This domain is termed H-NOXA (or H-NOBA) since it is often associated with the heme-nitric-oxide/oxygen binding (H-NOX) domain. A structure-function approach was taken to investigate the role of H-NOXA in STHK and sGC. We report the 2.1 Å resolution crystal structure of the dimerized H-NOXA domain of STHK, which reveals a Per-Arnt-Sim (PAS) fold. The H-NOXA monomers dimerize in a parallel arrangement juxtaposing their N-terminal helices and preceding residues. Such PAS-dimerization is similar to that previously observed for EcDOS, AvNifL, and RmFixL. Deletion of 7 N-terminal residues affected dimer organization. Alanine scanning mutagenesis in sGC indicates that the H-NOXA domains of sGC could adopt a similar dimer organization. Although most putative interface mutations did decrease sGCβ1 H-NOXA homodimerization, heterodimerization of full length heterodimeric sGC was mostly unaffected likely due to sGC’s additional dimerization contacts in the coiled-coil and catalytic domains. Exceptions are mutations sGC-α1 F285A and -β1 F217A which each caused a drastic drop in NO stimulated activity and mutations sGCα1 Q368A and -β1 Q309A which resulted in both a complete lack of activity and heterodimerization. Our structural and mutational results provide new insights into sGC and STHK dimerization and overall architecture.
The ability to sense small molecules is key for every life form and provides information about the extra-cellular milieu, monitors intra-cellular physiological status, or establishes cell-cell communication. Sensory signaling proteins are often modular in nature with distinct domains for ligand sensing and for output signals. A number of these domains are conserved in bacteria and animals. A striking example is the heme-nitric-oxide/oxygen-binding H-NOX (previously termed H-NOB) domain which can be a stand-alone protein in Nostoc cyanobacteria, or can be part of a multi-domain protein such as in the mammalian soluble guanylyl cyclase (sGC) (1;2)(Fig. 1A). An additional evolutionary relationship was detected between sGC and 2 other cyanobacterial signaling proteins (1;2): the H-NOX associated H-NOXA (or H-NOBA) domain in sGC is also present at the N-terminus of a cyanobacterial signal transduction histidine kinase (STHK) and 2-component hybrid sensor and regulator (2-CHSR)(Fig. 1) postulated to have a PAS-like fold (1). Both genes of these cyanobacterial proteins are adjacent to genes coding for stand-alone H-NOX domains (Fig. 1A) suggesting they might work in concert. This H-NOXA evolutionary link adds to the already complex and poorly understood regulation of sGC stimulating the need to study this ancient domain.
Figure 1. Structure of H-NOXA domain of NpSTHK and its evolutionary, structural, and sequence relationships.
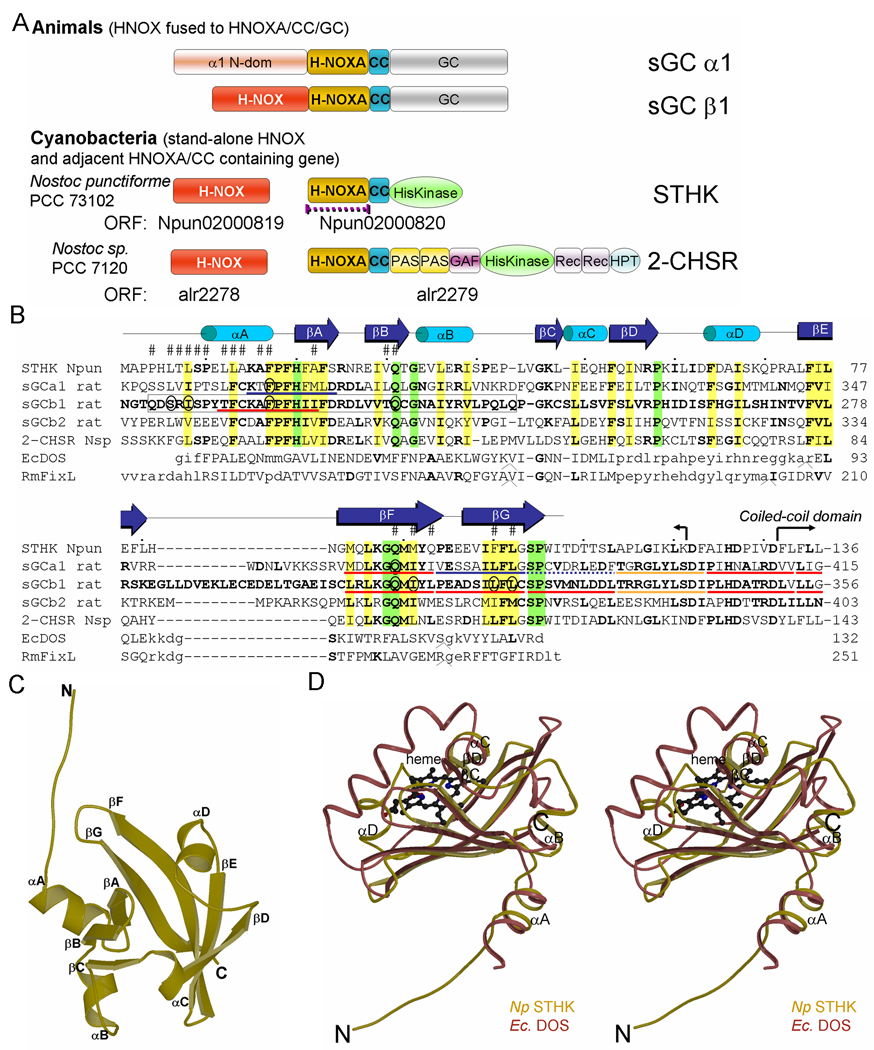
(A) Schematic diagram of the H-NOX and H-NOXA domains present in proteins from animals and Nostoc cyanobacteria. The purple interrupted line indicates the crystallized domain. Domain abbreviations used: CC (coiled-coil), GAF (domain present in cGMP-regulated PDEs, adenylyl cyclases, and the E.coli protein Fh1A), REC (receiver domain), and HPT (histidine phosphotransfer domain).
(B) Structure-based sequence alignment of the H-NOXA domains of STHK, 2-CHSR, sGCα1, -β1, and -β2 subunits, and EcDOS (PDB id 1V9Z) and RmFixL (PDB id 1D06). Residues identical to sGCβ1 are in bold. Semi-conserved hydrophobic residues, excluding heme PAS domains since they have a heme as their hydrophobic core, are highlighted in yellow; fully conserved residues in green. Residues at the NpSTHK dimer interface are labeled with a ‘#’; the putative sGC interface residues probed by mutagenesis are circled. Residues of EcDOS and RmFixL that are in structurally equivalent positions with the H-NOXA structure are in uppercase. The EcDOS and RmFixL sequences contains a few insertions at positions labeled ‘^’ and their insertion sizes are 3, 6, and 2, respectively for both EcDOS and RmFixL. sGCβ1 residues 204–244 have previously been shown to be key for dimerization (17) and are boxed. The expressed construct end (K121) and start of the CC of NpSTHK are shown (hooked arrow). The red underlined stretches of residues highlight deletions that resulted (18) in lack of sGC heterodimerization and concomitant loss of activity; deletion of the orange underlined stretches of residues resulted in sGC heterodimerization but severely compromised cyclase activity; deletion of the blue underlined stretches of residues resulted in increased cyclase activity and decreased EC50 values for BAY41-2272 in combination with nitric oxide; deletion of the interrupted blue underlined stretch of residues had a mixed effect in that this deletion caused a decrease in cyclase activity yet a decrease in EC50 for BAY41-2272 in combination with nitric oxide (18).
(C) Ribbon diagram of the NpSTHK domain monomer structure. Secondary structure elements are labeled.
(D) Stereo-figure depicting superposition of the monomer of the H-NOXA domain of NpSTHK and the EcDOS PAS domain (PDBid 1V9Z). NpSTHK and EcDOS share common αA, αB, and αC helices and even helix αD althought this helix is longer in EcDOS and in a somewhat different orientation. The major difference between the NpSTHK and EcDOS structure is that the EcDOS pocket that harbors the heme (ball-and-stick) flanked by the long helix is collapsed in NpSTHK and filled with the new βD strand.
Upon NO activation, sGC increases its production of the second messenger cGMP (3–5) leading to vasodilation, platelet aggregation, and induction of host defense mechanisms (6;7). sGC is therefore a potential drug target for treating hypertension and erectile dysfunction (8). sGC is comprised of an α and β subunit with the α1/β1 isoform being the most ubiquitous (3). The H-NOXA domain is a central subdomain in both subunits: in sGCβ1, it is flanked by the N-terminal H-NOX domain and C-terminal predicted coiled-coil (CC)(9;10) and catalytic guanylyl cyclase (GC) domain (Fig. 1A). sGCα1 has a similar subunit arrangement except that its N-terminal domain does not bind heme. As in sGCα1 and -β1, the H-NOXA domains in STHK and 2-CHSR are followed by a CC domain further strengthening their evolutionary connection (1)(Fig. 1A).
Structural information on sGC is limited and comes mostly from the structures of the homologous adenylyl cyclase catalytic domain (11–13) and bacterial H-NOX domains (14–16). Even less is structurally known about the H-NOXA/CC region except that it was found to contain two distinct regions key for dimerization: one comprises H-NOXA β1 residues 204–244, and the second contains CC residues 379–408 with the intervening sequences postulated to contribute to a functional binding region (17). Analogous regions in sGCα1 have also been shown to be important for sGC activity via deletion studies (9). These regions in sGCα1 and -β1 have recently been further narrowed down (18). The α1 and β1 H-NOXA domains are 38% sequence identical and also share ~35% sequence identity with the H-NOXA domains in the STHK from Nostoc punctiforme PCC 73102 (NpSTHK) and the 2-CHSR from Nostoc sp. PCC 7120 (Fig. 1B). Unlike the sGC subunits, the H-NOXA domain of the STHK was amendable to crystallographic analysis. We describe here the 2.1 Å crystal structure of the dimerized H-NOXA domain of NpSTHK revealing a PAS fold, a fold commonly found in sensory signaling proteins. We carried out additional structure-function studies in sGC and find that the H-NOXA subdomains of sGC could likely form a similarly arranged PAS-type heterodimer with an important role for functional sGC hetero-dimerization by the H-NOXA domains.
EXPERIMENTAL PROCEDURES
Cloning of the H-NOXA domain of NpSTHK
The genomic DNA of Nostoc punctiforme PCC 73102 was used as the template to PCR the 1–121 H-NOXA fragment of STHK Npun02000820 using the forward primer 5'-ggaattccatatggctcctcctcaccttacg-3' and backward primer 5'-ccggaattctcatttgagtttgatacccaaaggagc-3'. To obtain the 8–121 (Δ7) H-NOXA construct, the 5'-ggaattccatatgctttcaccagagttactggcga-3' forward primer was used. The PCR amplified fragments were inserted into a pET28a (Novagen) expression vector.
Expression and purification of NpSTHK H-NOXA domains
Both the 1–121 and truncated Δ7 H-NOXA domains were expressed as N-terminal his-tagged protein in E.coli BL21(DE3) Star cells (Invitrogen) using IPTG for induction. The cells were pelleted and sonicated for 5 minutes on ice in the following buffer: 20mM Tris pH7.5, 100mM NaCl, 5mM β-mercaptoethanol. The cell lysate was centrifuged at 16,000g for 10 min at 4°C and the supernatant was incubated with Ni-NTA (Qiagen) beads. The beads were washed with the washing buffer: 20mM Tris pH7.5, 100mM NaCl, 5mM β-mercaptoethanol, 20mM imidazole. The protein was released from the beads by thrombin (Enzyme Research labs) digestion. Further purification was performed by gel filtration in a Sephadex75 (Pharmacia) column equilibrated with 5mM Tris pH7.5, 100mM NaCl, and 1mM β-mercaptoethanol.
Subcloning, expression, and purification of the H-NOXA domain of sGCβ1
Residues 202–344 of the H-NOXA domain of rat sGCβ1 were subcloned into pET22b using the forward primer: 5’-ggaaattccatatgggtacccaggactcccgtatc-3’, and backward primer:5’gcgaattctcagtggtggtggtggtggtgtccagggatgtcactcaggtacag-3’. The C-terminally his-tagged sGCβ1 H-NOXA protein was expressed and purified similarly to the homologous NpSTHK counterpart except that the protein was eluted from the Ni-NTA beads by an imidazole gradient instead of thrombin digestion. The Ala-scanning mutants of sGCβ1 H-NOXA domain were introduced into pET22b plasmid using the QuikChange site-directed mutagenesis kit (Quickchange, Stratagene) sequence confirmed.
Mutagenesis of rat sGC and transfection in COS-7 cells
Templates were cDNAs encoding the α1 and β1 subunits of rat sGC cloned into the mammalian expression vector pCMV5 (19). Mutations described in the text were introduced by PCR (Quickchange, Stratagene) and sequenced. COS-7 cells were grown in DMEM supplemented with 10% heat-inactivated fetal bovine serum, penicillin, and streptomycin (100 units/mL). Cells were transfected for 48hrs with Superfect reagent using the protocol of the supplier (Qiagen).
Cytosol preparation and western blot analysis
COS-7 cells were washed twice with ice-cold PBS and then scraped off the plate in cold lysis buffer: PBS buffer containing protease inhibitors, 50mM HEPES pH 8.0, 1mM EDTA, and 150 mM NaCl and 0.1% Triton. Cells were broken by sonication (3 pulses of 3 sec) and centrifuged at 16,000 × g for 10 min at 4°C to collect the soluble fraction (referred to as cytosol in the text). To determine the efficiency of transfection for wt sGC or mutants in COS-7 cells, 15µg of cytosol was resolved on 10% SDS-PAGE and analyzed by immunoblotting with anti-sGC (anti-α1 subunit and anti-β1 subunit; Cayman Chemicals).
sGC activity assay
GC activity was determined by formation of [α-32P]cGMP from [α-32P]GTP, as previously described (20). Reactions were performed for 5 min at 33 °C in a final volume of 100 µl, in a 50 mM HEPES pH 8.0 reaction buffer containing 500µM GTP, 1mM DTT, and 5 mM MgCl2. Typically, 40µg of COS-7 cytosol transfected with either wt or mutants were used in each assay reaction. All assays were done in duplicate and each experiment repeated twice. Enzymatic activity was stimulated with the NO-donor SNAP (Calbiochem) at 100µM. sGC activity is expressed in pmol.min−1.mg−1 and as mean ± S.E.
NpSTHK crystallization
Crystals of the NpSTHK 8–121 protein construct were grown at 4°C by sitting-drop vapor diffusion. Protein was concentrated to 20mg/ml in 5mM Tris pH7.5, 100mM NaCl, 1mM β-mercaptoethanol and was mixed with an equal volume of the reservoir solution: 1.7–1.9M ammonium sulfate, 100mM Tris-HCl (pH 7.7–9.0). Crystals were prepared for data collection by fast transfer into cryoprotectant solution containing 2.0M ammonium sulfate 100mM Tris-HCl with 15% glycerol. The 1–121 NpSTHK protein was concentrated to 15mg/ml after the gel-filtration step and mixed with equal volume of 0.1M HEPES pH7.5 and 1.5M lithium sulfate monohydrate. Crystals appeared after three days at 20°C. For data collection, the crystals were soaked in cryo-protectant containing 25% glycerol in addition to the crystallization solution and dunked into liquid nitrogen prior to data collection. Crystals of selenomethionine substituted 8–121 protein were used for Single-Wavelength Anomalous Dispersion (SAD) phasing.
NpSTHK structure determination and refinement
Due to great difficulty in obtaining diffraction quality crystals for the 1–121 NpSTHK construct, the initial structure determination was carried out using crystals of the shorter 8–121 construct. A native 2.0 Å resolution dataset for the 8–121 NpSTHK protein was collected at ALS (Beamline 4.2.2.) and processed with D*trek (21). The crystals belong to space group P6122, with cell dimensions a=b=72.3 Å, c=169.1 Å and two molecules in the asymmetric unit. To obtain crystallographic phase information, a SeMet crystal of the 8–121 NpSTHK construct was used for SAD phasing. A 2.6 Å SAD data set was collected at the selenium peak wavelength at ALS. SOLVE/RESOLVE (22) was used for phasing and automatic model building. Manual model building was carried out in Coot (23) and REFMAC (24) was used for refinement. The final model includes NpSTHK residues 8–113 of molecule A and residues 10–119 of molecule B and 95 waters and 2 sulfate ions. The final model yielded an R/Rfree of 23.9/27.8%.
For 1–121 H-NOXA, a native 2.1 Å resolution data set was collected at the Advanced Photon Source (Beamline 19ID) belonging to space group C2 with cell dimensions a=95.2 Å, b=44.4 Å, c=59.7 Å and 2 molecules in the asymmetric unit. The structure of this larger construct was determined via Molecular Replacement using PHASER(25) using one of the truncated monomer structures from the P6122 space group. Coot and REFMAC were used for model building and crystallographic refinement yielding a final model includes residues 1–107 of molecule A, 1–105 of molecule B, and 55 waters (R/Rfree are 19.9/26.2%). The stereochemistry was checked using PROCHECK (26)(Table 1). Figures are generated using MOLSCRIPT and Raster3D (27;28) and Pymol (http://pymol.sourceforge.net/).
Table 1.
Data collection, phasing, and refinement statistics for NpSTHK.
| Native (8–121) | Se-peak (8–121) | Native (1–121) | |
|---|---|---|---|
| Datacollection | |||
| Wavelength (Å) | 1.2398 | 0.9790 | 0.97934 |
| Space group | P6122 | P6122 | C2 |
| Cell dimension (Å) | a=b=72.33 | a=b=72.26 | a=95.21 |
| c=169.08 | c=169.11 | b=44.36 | |
| α=β=90° | α=β=90° | c=59.74 | |
| γ=120° | γ=120° | α=γ=90° | |
| β=104.51° | |||
| Resolution (Å) | 2.0 | 2.6 | 2.1 |
| Total observations | 104640 | 150704 | 213337 |
| Unique observations | 18435 | 8621 | 12364 |
| I/SigI | 12.2 (2.5) | 16.2 (6.9) | 10.0 (1.6) |
| Redundancy | 5.68 (4.05) | 17.48 (19.27) | 3.4 (1.9) |
| Completeness (%) | 99.8 (99.2) | 100.0 (100.0) | 87.4 (59.8) |
| Rsym (%) | 8.0 (36.9) | 9.3 (38.3) | 11.9 (45.6) |
| SAD phasing | |||
| No. of sites | 6 | ||
| Figure of merit | 0.63 | ||
| Refinement | |||
| Resolution (Å) | 2.0–42.3 | 2.1–27.8 | |
| No. of protein atoms | 1773 | 1732 | |
| No of waters | 111 | 55 | |
| Sulfate ions | 2 | 0 | |
| Rfactor (%) | 23.9 | 19.9 | |
| Rfree (%) | 27.8 | 26.2 | |
| R.m.s.d. for bond lengths (Å) | 0.012 | 0.011 | |
| R.m.s.d. for bond angles (°) | 1.37 | 1.35 | |
| Ramachandran plot statistics | |||
| Residues in: | |||
| -most favored regions | 88.7 | 88.8 | |
| -additional allowed regions | 11.3 | 11.1 | |
| -in generously allowed regions | 0 | 0 | |
| -disallowed regions | 0 | 0 | |
Values for the highest resolution shell are listed in parenthesis.
RESULTS
NpSTHK structure reveals a PAS fold
The structure of the H-NOXA domain of NpSTHK was solved via SAD of first a smaller Δ7 N-terminally truncated fragment, followed by molecular replacement to solve the full length NpSTHK H-NOXA domain structure (Table 1 and Fig. 1C). Two independent molecules in the asymmetric unit form a dimer. The final refined model of the NpSTHK dimer contains residues 1–107 of molecule A and 1–105 of molecule B.
The NpSTHK monomer structure is comprised of a 7-stranded anti-parallel β-barrel flanked by several α-helices (Fig. 1C). The structure of the NpSTHK monomer is similar to that of the PAS fold: its most similar structural neighbours are two heme containing sensors bjFixL and EcDOS (PDB id 1DRM and 1V9Z), followed by dPER (1WA9), HIF-2α (1P97), and photoactive yellow protein (3PYP) with DALI Z-scores (29) ranging from 8.3 to 7.6, resp. A superposition of the NpSTHK and the EcDOS PAS domain (30)(Fig. 1D) details their close structural similarity (r.m.s.d. of 2.2 Å for 70 Cα atoms). Their structure-based sequence alignment (Fig. 1B) reveals only 10% sequence identity. The canonical PAS fold contains 5 β-strands (31) but both NpSTHK and EcDOS contain an additional short βC strand; NpSTHK contains even an additional 7th (βD) strand (Fig. 1D). The region in between the two N-terminal strands (βA/βB) and three C-terminal strands (βE/βF/βG) usually harbors 4 helices in PAS domains (31). Three of those helices are present in NpSTHK (Fig. 1B) although helix αD is in a somewhat different orientation (Fig. 1D). This multi-helix containing region in between βB and βE is structurally the most variable part of PAS domains, and can have as few as 2 helices such as CitA (32), possibly dictated by whether or not the PAS domain binds a ligand and if so, the type of ligand (31;33) (Fig. 1D). The similarity between NpSTHK and EcDOS is thus remarkable despite little sequence identity and extends beyond the PAS fold since they both contain an additional N-terminal helix, αA (Fig. 1D).
A structure-based sequence analysis of the homologous sGC subunits using the NpSTHK H-NOXA domain structure was used to probe sequence conservation and sites of insertions and deletions (Fig. 1B). Both sGC subunits have a large insertion between the βE and βF strands in their H-NOXA domain: a 9 residue insertion for α1 and 19 for β1. In addition, there are some smaller 1 residue insertions and deletions around the βC strand (Fig. 1B). To test whether the sGC subunit sequences are compatible with the PAS-fold of NpSTHK, MODELLER (34) was used to build homology models for the α1 and β1 H-NOXA domains using the alignment in Fig. 1B. The resulting models yielded no negative VERIFY3D structure validation (35) scores (or even scores with values lower than 0.1) indicating that both subunits can adopt NpSTHK’s PAS fold (the 19 residue insertion in β1 was omitted from the modeling due to its size and lack of template).
NpSTHK dimer organization
The two dimerized NpSTHK molecules make extensive interactions burying 2,395 Å2 of surface (67% hydrophobic; surface calculations done using MSCON (36)) (Figs. 2A–2D). Dimer interface analysis programs ranked the significance of this interface very high yielding a maximal complexation significance score (CSS) of 1.0 and a PITA score of 90 (37;38). Truncated Δ7 NpSTHK forms a flipped dimer (Fig. 2E), buries less surface (1,888 Å2) with many water-mediated interactions and is likely non-physiological as indicative of the lower CCS and PITA dimer evaluation scores of 0.44 and 47.1, respectively, the latter being below the threshold value of 67 (38). The two NpSTHK domains comprising the full-length dimer are related by an approximate 2-fold non-crystallographic axis (178.5°) and are very similar in conformation (r.m.s.d of 0.45Å for 105 Cα atoms). Dimerization involves juxtapositioning of the N-terminal helices and preceding residues and the face of the β-sheet (Fig. 2A–2D). The dimer interface contains a central cluster of hydrophobic residues (including L8, L13, F17, F100, and L102) and hydrogen bonds (involving side chains of residues T7, S9, Q31, Q89, and Q93 and main chain atoms of several other residues)(Figs. 2B and 2C).
Figure 2. Homo-dimeric structure of H-NOXA domains of NpSTHK.
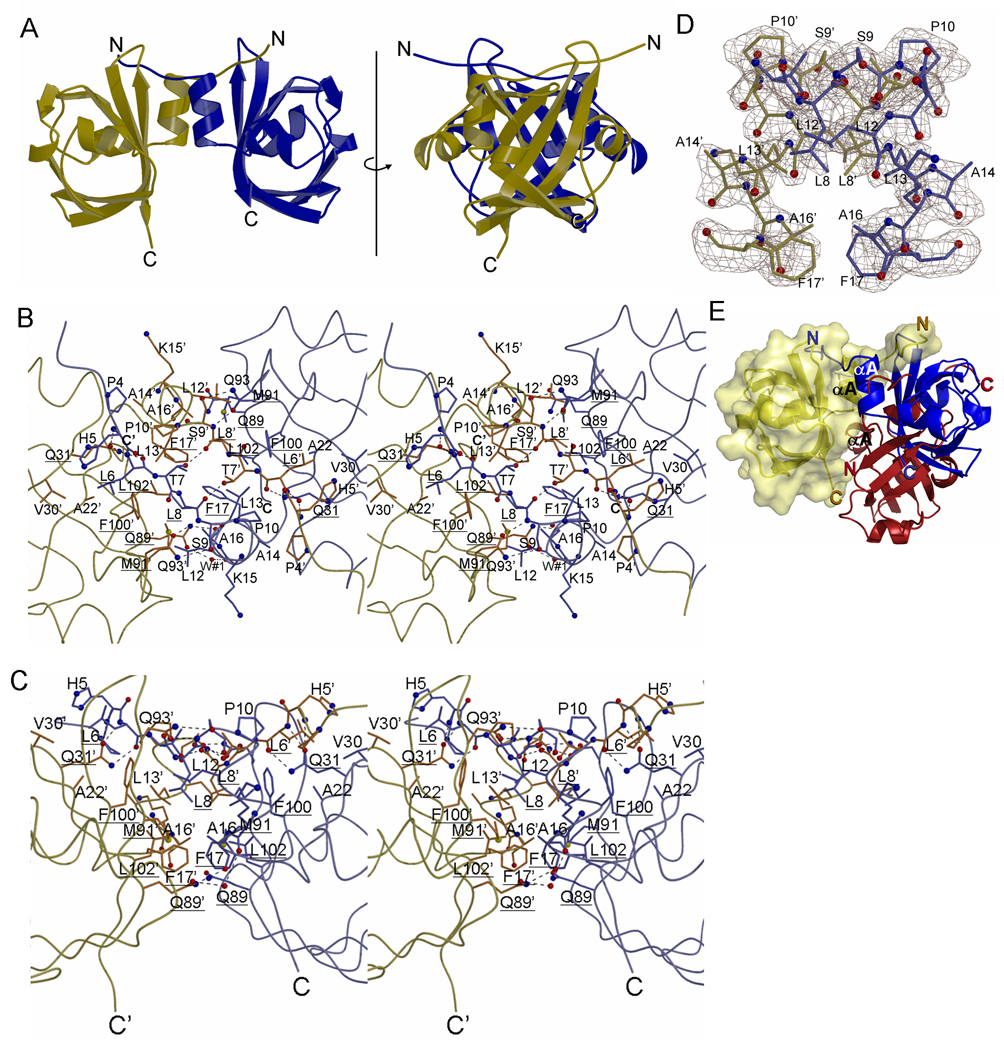
(A) Schematic diagram of the NpSTHK dimer in two side views related to each other by a 90° rotation around the axis shown.
(B) Stereo diagram of the dimer interface. View is that from the top of H-NOXA dimer with the two N-terminal helices perpendicular to the plane of the paper. Hydrogen bonds are depicted as dashed lines. Residues that are equivalent to the ones targeted by Ala scanning mutagenesis in sGC are underlined.
(C) Same as in (B) except rotated 90° along the horizontal axis such that the 2 N-terminal helices are now oriented vertically.
(D) Electron density of the N-terminal helix region of NpSTHK. Depicted are residues 7–17 of both monomers and the omit |Fo|-|Fc| density (contoured at 2.5σ) after these residues were omitted (omitted portion is about 10% of the total structure). Residues 7–17 contains helix αA and a few preceding residues that are involved in part of the dimer interface interactions.
(E) Different dimer configurations of full length and truncated NpSTHK. The full length NpSTHK monomers entailing the crystallized dimer are shown in blue and yellow. A different dimer arrangement can be found in the asymmetric unit of the crystallized truncated Δ1–7 NpSTHK: in keeping one of the H-NOXA monomers in a fixed superimposed position (yellow), the second H-NOXA subunit (red) interacts in an almost flipped orientation compared to the blue H-NOXA monomer yielding a possible novel dimer organization (yellow and red). This results in drastic differences in relative positioning of the N- and C-termini of these different dimer organizations.
The resemblance of NpSTHK with EcDOS extends beyond the monomer fold since their dimer organizations are also similar involving N-terminal helix juxtapositioning (Fig. 3). The dimeric NpSTHK and EcDOS structures can be superimposed with an r.m.s.d. of 3.5 Å (for 2×70 Cα atoms), which is higher than the r.m.s.d. of 2.2 Å for the monomer superposition yet low enough to indicate a similar dimer arrangement. Their similar dimer organization is attained since the hydrophobic nature of several residues at the core of the dimer interface is conserved (L13, F100, and L102 in NpSTHK correspond to L26, L127, and L129, respectively, in EcDOS; see Fig. 1B). The same fold and dimer organization are also present in another heme containing oxygen-sensor, RmFixL(39), also an STHK protein (Fig. 3). Furthermore, such PAS dimerization was also recently observed in AvNifL (40)(Fig. 3) which led to the suggestion that this is a conserved dimerization motif for a subset of PAS domains (40).
Figure 3.
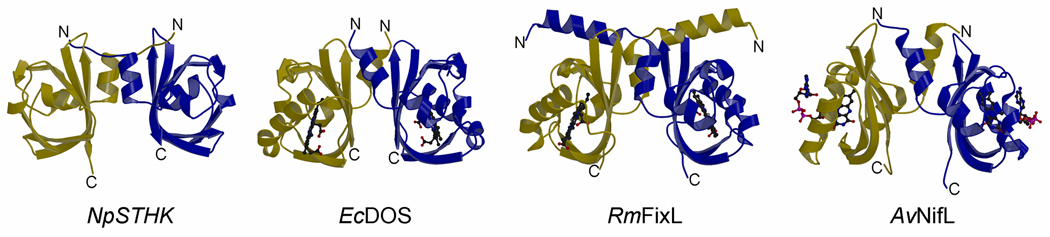
Structural similarity of the NpSTHK dimer and other PAS domain dimer containing proteins. The NpSTHK dimer is depicted along with EcDOS, RmFixL, and AvNifl. The latter has an FAD molecule bound in each monomer whereas both EcDOS and RmFixL each have a heme moietys bound (ligands are shown in ball-and-stick).
Evidence for a NpSTHK-type dimer for the H-NOXA domains of sGC
The N-terminal region of the H-NOXA domain of NpSTHK provides most of the dimer contacts which is in agreement with the corresponding sGCβ1 residues 204–244 found to be important for sGC dimerization (Fig. 1B)(17). Furthermore, the dimer interface region is largely conserved in sGCα1 and -β1 (Fig. 4A), suggestive of a similar dimerization function in sGC. The hydrophobic nature of all 5 residues comprising the central cluster of the dimerization interface (L8, L13, F17, F100, and L102) is conserved as well suggesting that sGCα1 and β1 H-NOXA domains could similarly heterodimerize, or homodimerize as observed in solution for the sGCβ1 H-NOXA domain (Fig. 4B)). To test whether a heterodimeric sGC indeed could adopt a similar NpSTHK dimer arrangement, a homology model was constructed for the α1β1 sGC H-NOXA heterodimer using MODELER which resulted in similar (non-negative) VERIFY3D scores as was observed for the individual monomers. The modeled sGC heterodimer structure was additionally validated by carrying out dimer analysis calculations using PISA resulting in a ΔG for dimer formation of −16.9kcal/mol and 2,156Å2 of buried surface (α2β1 yielded similar results). These values are very similar to that calculated for the experimentally determined NpSTHK dimer itself whereas a MODELLER generated α1β1 heterodimer homology model based on the likely non-physiological Δ7 NpSTHK dimer yielded only a ΔG of −7.9kcal/mol (1,742 Å2 of buried surface). These automated modeling and analysis results suggest that the sGC H-NOXA domain sequences are indeed compatible with formation of a NpSTHK-type heterodimer.
Figure 4. Probing the putative H-NOXA dimer interface in sGC by mutagenesis.
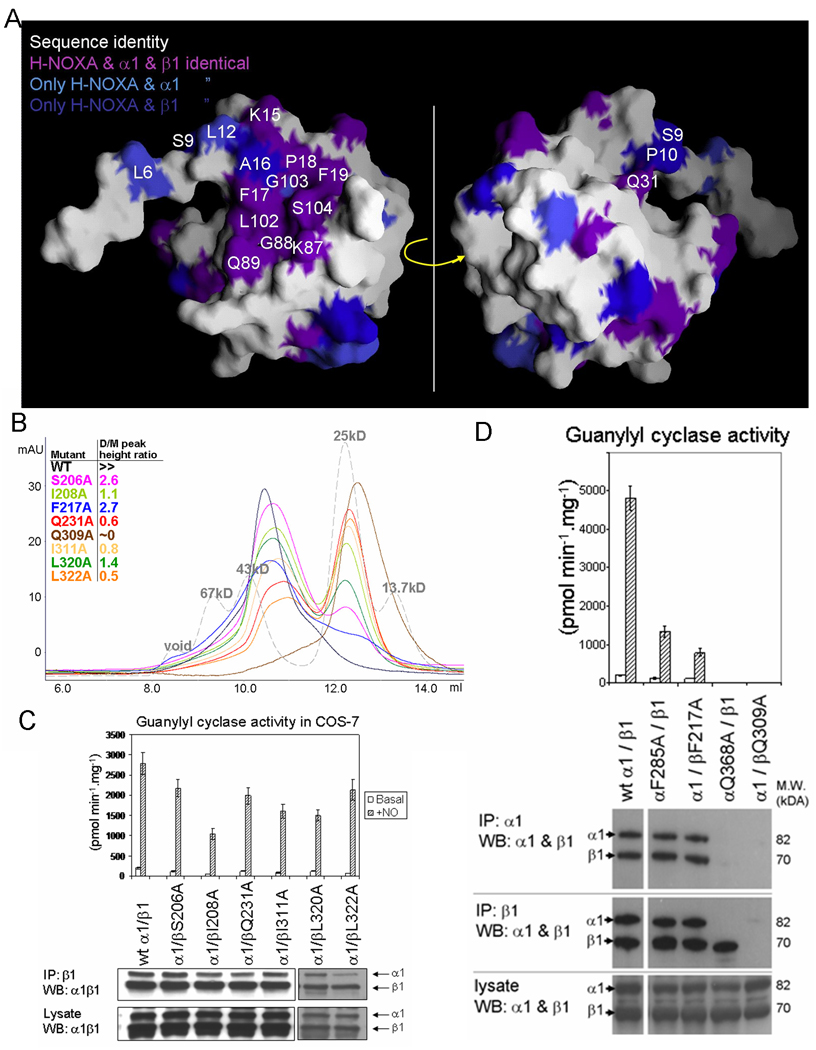
(A) Conservation of surface residues of the NpSTHK H-NOXA monomer with the α1 and β1 sGC subunits. The surface facing the dimer interface (left) and the opposite side (right) are shown. Non-conserved surface H-NOXA residues are shown as white. Conserved residues at the dimer interface or in the vicinity are labeled.
(B) Probing dimerization state of sGCβ1 H-NOXA domain mutants using size exclusion chromatogram. A superdex 75 column (HR10/30, GE Biosciences) was used to test the oligomeric state of mutant H-NOXA proteins in which the following sGCβ1 residues were mutated to Ala (corresponding residues in NpSTHK are in parentheses): S206 (L6), I208 (L8), F217 (F17), Q231 (Q31), Q309 (Q89), I311 (M91), L320 (F100), and L322 (L102). A mixture of molecular weight protein standards was also applied to the column for reference (grey). The theoretic molecular weight for the monomer of wt sGCβ1 H-NOXA domain is 17kD, which includes the His-tag. For comparison, the crystallized NpSTHK, with a theoretical molecular weight of 14kD, elutes at 10.6ml (not shown) which is similar to that of the wt sGCβ1 H-NOXA domain confirming the dimeric state for both proteins. The injecting volume for each run is 100 µl and protein concentration is 1mg/ml.
(C) Guanylyl cyclase activity and Western blot analysis of co-immunoprecipitated wt and 6 sGCβ1 mutants of sGC. Basal and NO-stimulated guanylyl cyclase activity of cell lysates with transfected sGC subunits are shown (upper panel). The Western blot (lower panel) includes co-immunoprecipitated sGC subunits pulled down with anti-sGCβ1 antibody (top gel) as well as the contents of the cell lysates (bottom). The Western blot was probed simultaneously with anti-sGCα1 and anti-sGC β1 antibodies.
(D) Guanylyl cyclase activity measurements and Western blot co-immunoprecipitation analysis of additional mutants of sGC generated in both α1 and β1 subunits. These mutations include the sGCβ1 F217A and Q309A mutations and are also generated at the equivalent positions in sGCα1 (α1 mutations F285A and Q368A, respectively). Data presentation similar as in (C) but includes additional co-IP using an anti-sGCα1 antibody (top gel). This Western blot was also probed similarly as in (C). The difference in specific activity observed between experiments 4C and 4D (as reflected in the wt activity) is probably due the fact that two different batches of COS-7 cells were used in 4C and 4D. For each set of experiments, COS-7 cells were transfected three times and activity assays repeated three times with each measurement done in duplicate.
To further validate the presence of a NpSTHK-type dimer interface in sGC we mutated 8 putative H-NOXA interface residues to alanines (see Figs. 1B, 2B, 2C, and 4 for the NpSTHK interface residue positions that were targeted for mutagenesis of the corresponding residue in sGC). We tested the effect of these mutations on dimerization of the isolated sGCβ1 H-NOXA domain and the dimerization and activity of full length sGC. The 8 Ala mutations were made in sGCβ1; two of the mutations were also generated at the corresponding positions in sGCα1. All mutations had a negative effect, with varying degrees, on dimerization of the β1 H-NOXA domain (Fig. 4B). The β1 mutations that caused the largest effects on H-NOXA dimerization are Q231A, Q309A, and L322A, as more than half of the mutant protein became monomeric at 1mg/ml (Fig. 4B). Even the mutations that were the least disruptive, S206A and F217A, did still have a small but measurable negative effect on dimerization indicative of a role in dimerization (Fig. 4B). The latter F217A mutation caused also a widening of the dimer peak, perhaps suggesting an (additional) larger structural destabilization caused by this mutation (Fig. 4B).
The 8 sGCβ1 and 2 -α1 Ala-scanning mutations had in most cases less of an effect on dimerization of the full length sGC as probed by immuno-precipitation and western blotting (Fig. 4C and 4D). Exceptions are the sGCβ1 mutations Q309A (and equivalent Q368A mutation in sGCα1), L322A, and to a lesser degree Q231 and I208A (Figs, 4C and 4D). The Q309A (and -α1 equivalent) caused a complete loss of sGC hetero-dimerization as partnering subunits could not be pulled down with antibodies directed against either the α or β subunit (Fig. 4D). It is interesting to note that when this Q mutation occurs in the sGCα1 subunit, the wt sGCβ1 subunit can be pulled down using a β-specific antibody at comparable levels, whereas when the mutation is introduced in the sGCβ1 subunit, wt -α1 itself cannot be pulled down with an α-specific antibody despite being present in the cell lysate probably as a result of aggregation. This differential sGC subunit behaviour is similar at physiological receptor concentrations in mice with individual sGC subunits knocked-out since only the sGCβ1 subunit could be detected in sGCα1 knock-outs (41) but not vice versa (42). A number of the mutations had a comparable effect on both sGCβ1 H-NOXA homodimerization and full length sGC heterodimerization. sGCβ1 mutations Q309A, Q231, and L322A caused a significant decrease in dimerization in both experiments whereas F217A and S206A both had only a minor effect on H-NOXA homodimerization and little if any effect on full length sGC dimerization (Fig. 4).
The effect of the Ala sGC mutations on guanylyl cyclase activity was also measured in mutant transfected COS-7 cells. The effect on activity was most pronounced for the Q309A sGCβ1 mutant and the equivalent -α1 Q368A mutant which both resulted in a complete loss of NO-stimulated and basal activity, in agreement with their inability to even form a heterodimer (Fig. 4D), the minimum needed for sGC basal activity (43). The β1 F217A and equivalent α1 F285A mutations caused a 3.6- and 6.1-fold drop in NO-stimulated activity, respectively (Fig. 4D). This result is somewhat unexpected since both F→A mutants had only limited effects on H-NOXA homo- and sGC heterodimerization (Fig. 4B and 4D). Nevertheless, the substantial decrease in NO-stimulated activity by these F→A mutations could perhaps be explained by either a more global destabilization of the H-NOXA domains as mentioned above or perhaps an altering of the relative orientation of the H-NOXA dimer within the mutant sGC (described in more detail below). Except for the mutants shown in Figure 4D, the remaining mutants had only a modest negative effect on guanylyl cyclase (Figs. 4C) yielding a maximal decrease of NO-stimulated activity of just under 3-fold. However, the basal activity of the cell lysate is also lower for these mutants such that the fold activity enhancement upon NO-stimulation above for these latter mutants is similar to wt being around 15. The one exception is the L322A sGCβ1 mutant which harbors a 29-fold increase in stimulated activity over basal activity (Fig 4C).
The Ala-scanning mutagenesis revealed that some of the residues at the putative H-NOXA dimerization interface in sGC are critical for dimerization and/or activity. The most critical residues are F285 and Q368 in sGCα1, and the equivalent F217 and Q309 in sGCβ1, and correspond to F17 and Q89 in NpSTHK (Figs. 1B, 2B, and 2C). Interestingly, F17 and Q89 cluster and are both located at the bottom of the NpSTHK interface (Fig. 2C) yet still partially solvent exposed in proximity of the C-termini of this domain. This proximity suggests that this region could have a key role in perhaps additionally interacting with the CC domain (or GC domain) or perhaps be important for the precise orientation of the H-NOXA domains and its C-terminally located domains. The F→A and Q→A mutations will be discussed in more structural detail below within the context of the NpSTHK structure.
F17 is partly solvent exposed and provides hydrophobic interactions via mostly its tip with the symmetry related pair F17’ and also with L102’ (Fig. 2B) and buries only 40Å2 of surface upon dimerization. Since this residue is near the 2-fold axis, when it is mutated in the isolated β1 H-NOXA homodimer, the amount of buried surface lost is much less than double since this residue is mostly interacting with its symmetry F17’ mate. F17’s limited dimer interface contribution might provide an explanation for the only minor decrease in dimerization seen in the corresponding sGC mutants. However, its potentially critical position along the 2-fold dimer axis situated towards the C-terminal CC+GC domains might be crucial for H-NOXA domain orientation and when mutated might affect CC/GC interactions (or orientation) impeding signal transduction and thus provide an explanation for the strong effects on NO-stimulated activity.
Residue Q89 of molecule A of NpSTHK forms a hydrogen bond and a water-mediated hydrogen bond with the backbone oxygens of A16’ and K15’, respectively, in addition to packing against residue M91 being a key hydrophobic interface residue (Figs. 2B, 2C, and 2D). Note that due to the 1.5° deviation from the perfect 2-fold axis of the crystallized NpSTHK dimer, residue Q89’ of the other subunit is located 1Å more distant from the dimer interface although still involved in packing against M91, a likely key interface residue. Minor deviations from a perfect 2-fold axis have been observed to be as large a 7° in other dimer protein crystal structures (44) and we anticipate a symmetrical dimer when in solution.
Of all the mutants generated in this study, mutations at the equivalent position of STHK:Q89 in sGC (Q368A and Q309A in sGCα1 and -β1, respectively) resulted in drastic loss of sGCβ1 H-NOXA homodimerization, loss of sGC heterodimerization and loss of basal activity. This glutamine residue position is thus clearly crucial and, like F17, is positioned at the edge of the dimer interface towards the C-terminal CC+GC subunits. We speculate that its mutation in the sGCβ1 H-NOXA homodimer affects its homodimerization via loss of 2 direct hydrogen bonds and 2 water mediated hydrogen bonds across the interface, and reorientation of its neighbouring interface residues M91 and M91’. That the corresponding mutations in the full length sGC receptor also lead to complete loss of heterodimerization is somewhat surprising since there are additional dimerization interactions in full length sGC (CC domain and GC domain). However, the protein concentrations of the full length sGC heterodimerization experiments in cell lysates are much lower compared to those in the purified H-NOXA homodimerization experiments which would render the sGC dimerization state more sensitive to mutations even if it were present in only one of the domains. Nevertheless, it is still remarkable effect since only one of the Q residues is mutated per mutant due to the heterodimeric nature of sGC. We hypothesize that the reason that even the single α1Q368A and β1Q309A mutants causes loss of sGC heterodimerization is the cumulative effects of a) loss of a direct hydrogen bond and a water-mediated hydrogen bond across the interface, b) possible reorientation of its neighbouring interface residue M91 equivalent (αI370 and βI311), and c) possible reorientation of the H-NOXA domains and/or loss of interactions with C-terminal domains due to the location of the Q89 interface residue towards these CC+GC domains (Fig. 2C).
In summary, Ala scanning mutagenesis of the putative dimerization interfaces of the H-NOXA domains of sGCβ1 and sGCα1 can lead to loss of dimerization and/or guanylyl cyclase activity pointing to H-NOXA’s role in sGC dimerization and activity. The two mutations that were carried out in both sGCα1 and -β1 subunits resulted in similar behaviour (Fig. 4D) suggesting that they have roughly equally important roles. Together, these results are compatible with the postulate that the H-NOXA domains of sGC arrange themselves in a NpSTHK like fashion. Such heterodimeric arrangement in sGC could be either rigid, required to pivot/reorient, or be more transient during one of the stages of sGC activation. The implications of such a possible arrangement are discussed next.
DISCUSSION
The PAS fold and postulated NpSTHK-type dimer organization for sGC’s H-NOXA domains (Figs. 2, 3, and 4) has interesting consequences regarding preferential sGC heterodimerization, possible allostery, and sGC’s overall domain architecture.
Possible implications for preferential hetero-dimerization of sGC
Our structural results regarding the 1–121 and Δ7 truncated 8–121 NpSTHK constructs point to a critical role for the N-terminus in dimer formation. Deleting the first 7 residues of NpSTHK resulted in a flipped dimer suggesting that these residues are critical for providing specificity for correctly orienting the monomers within the dimer. Within these first 7 residues, residues P4, L6, and T7 are involved in dimer interface interactions via either hydrogen bonds or van der Waals interactions (Figs. 2B and 2C) in the 1–121 NpSTHK dimer structure. Residue L6 has a major role in that both its side chain makes interactions, with a relatively conserved hydrophobic region at the interface involving A22 and V30, and its main chain N and O atoms make interface hydrogen bonds with the conserved Q31 residue. N-terminal H-NOXA interactions thus likely provide the specificity for correct monomer:monomer juxtapositioning in NpSTHK yet could also have a role in preferential sGC heterodimerization as discussed next.
In addition to functional α1β1 and α2β1 heterodimerization, sGC homodimers are also observed for β1(43), β2 (45), and α1 (46), the latter being less stable. Homodimers of sGC are inactive (43), except for the β3 subunit from Manduca sexta (47) and possibly the β2 sGC subunit although its weak activation needs non-physiological manganese thus providing no conclusive evidence that its H-NOXA domains are dimerized under physiological conditions (45). A physiological equilibrium is thought to be present between homo- and heterodimeric sGC (43) although homodimeric β1 (41) and in particular homodimeric α1 are found to be very unstable in vivo (42). This balance is likely influenced by each of the three known interfaces involving the GC, H-NOXA, and CC domains (9;17;48). Such preferential heterodimerization tendency within these sGC domains is likely a consequence of complementary interface differences between α1 (or α2) and β1 subunits. Although speculative, sequence comparisons of interface residues and modeling of homodimeric and heterodimeric H-NOXA dimers suggests that sGC residues corresponding to NpSTHK residues L6 and A22’ and V30’ could be, in part, responsible for preferential heterodimerization of sGC. These residues cluster (Fig. 2B and 2C) and size variations across the interface suggest that a heterodimer might be more sterically compatible. In sGCα1 (and -α2), these residues are L274, M290’, and L298’, respectively, yet are smaller in sGCβ1 being S206, I222’, and T230’, respectively, all within the stretch of residues 204–244 shown to be key for binding to α1 (17). Larger-sized interface residues at all these positions, such as in an α1 (or α2) homodimer, would likely cause steric hindrance between M290’ and L274 such that the latter cannot position itself to form key mainchain hydrogen bonds (equivalent to NpSTHK residue L6). This disruption would disfavor such dimerization perhaps analogous to the disruptive effects of the Δ7 deletion in NpSTHK. sGCβ1 has smaller residues at these 3 positions likely permitting homodimer formation yet a heterodimeric α1β1 could possibly be favored via improved van der Waals packing by complementary combinations of small and larger residues at these 3 interface positions. Although it is interesting that the NpSTHK dimer structure could suggest a structural basis for a possible H-NOXA-contributing role in sGC heterodimerization, it remains speculative. Such a H-NOXA contributing role for sGC heterodimerization awaits future experimental validation especially in light of that residues preceding the N-terminal helix could possibly adopt different conformations such as a α-helical configuration in RmFixL (39).
Possible role for PAS-dimer in signaling or allosteric regulation in sGC and NpSTHK
The possibility of PAS-based allostery in sGC and NpSTHK is intriguing since the PAS domain is an important ancient sensory domain which can for example sense redox potential, small ligands such as oxygen, and light, in addition to maintaining protein-protein interactions (49;50). PAS sensory domains are often linked with output domains such as a histidine-kinase, phosphodiesterase, or adenylyl cyclase domains, and are remarkably abundant in cyanobacteria (51). Our results reveal a PAS-dimer organization for NpSTHK indicating that it has the same subdomain architecture as RmFixL STHK: a parallel dimerized (oxygen-sensing) PAS domain followed by a CC domain and histidine kinase domain. As noted earlier, such parallel oriented PAS domain dimers are also observed in EcDOS and AvNifL (Fig. 3) which lead to its recognition as a conserved PAS domain dimerization motif (40). RmFixL, EcDos, and AvNifL either sense redox potential or oxygen each involving postulated signaling mechanisms that lead to changes at the PAS-domain interface (30;40). The presence of a PAS domain in NpSTHK and sGC and the noted dimerization similarities raises the possibility that these PAS domains in NpSTHK and sGC are also used for signal transduction purposes or perhaps even sensory purposes. Obviously, the major sensory domain in sGC is the NO-sensing H-NOX domain yet it is tempting to speculate that the PAS domain could harbor a second allosteric regulatory module in sGC. This module could be present as an evolutionary remnant, for perhaps heme binding, or as a site for an unknown regulator. Intriguing candidates could be the allosteric sGC regulators YC-1, as well as ATP and GTP, whose binding site, or sites (52), to sGC has not been unambiguously mapped (6;53–56). Alternative to a small molecule, the pocket of a PAS domain is also capable of harboring a tryptophan side chain of a different subunit as observed in PERIOD (57). If such a PAS-mediated signaling or sensory regulation were indeed to occur in sGC or NpSTHK, some opening up of the PAS domain would be necessary for binding. Other possible PAS regulatory mechanisms have been observed as well and include N-terminal PAS helix unfolding (58), a speculated helix-swap event (59), or PAS pocket mediated inter-subunit interactions with a C-terminal helix (57) indicating that PAS-mediated contacts seems to play a key role in a number of these signal transduction events. A regulatory/mechanistic role for the centrally positioned H-NOXA domains is a sincere possibility as discussed next.
NpSTHK-type H-NOXA dimer in sGC and the implications for sGC domain organization and activation
The H-NOXA domain is located in the center of both the α1 and β1 subunits of sGC and the structure of the dimerized homologous NpSTHK H-NOXA domain therefore offers new insights into the arrangement of the flanking H-NOX and CC-GC domains. The NpSTHK dimer positions the two C-termini at ~18 Å distance (for residues I107 and P105’) at the bottom face of the dimer (Figs. 2A and 2C). The N-termini protrude in opposite directions and are located on the side of the NpSTHK dimer, each positioned about ~30 Å from the C-terminus of the other monomer. The relative close proximity of the C-termini of the NpSTHK domain dimer suggests that the succeeding CC domain, present in both NpSTHK and sGC, is in a parallel arrangement in accord with a previous sequence analysis study which had named this CC region a signaling helix or S-helix (10). The locations of the N- and C-termini has interesting consequences for the overall architecture of the heterodimeric sGC when combined with additional constraints in that the GC domains need to form a catalytically active heterodimer and that there is a direct interaction between β1 H-NOX and GC domain (48). The structure of sGC H-NOX and GC domains are not known but we have taken their homologous NsSTHK (16) and adenylyl cyclase crystal structures (11;12). The CC region is depicted as parallel CC segments as previously predicted (10), although we do not rule out other CC arrangements such as a 4 helix bundle. Despite some limitations, we generated a model for the entire sGC heterodimer (Fig. 5). In this modeled composite structure, part of the H-NOX domain is able to reach the GC domain (Fig. 5) with a stretch of 15 residues between the heme domain and the H-NOXA domain allowing such conformational flexibility. Although it is not known where and whether a protein:protein regulatory site is present on the sGC GC domains, such a site is present in the homologous adenylyl cyclase domains. The adenylyl cyclase activity is regulated by Gsα binding to the AC2 domain, which is homologous to the β1 cyclase domain (11). Assuming that sGC uses the same site on its catalytic domain for regulation, the H-NOX domain is oriented such that its region near loop L1 (containing D44–D45, speculated to be near the activation switch (14;60) and shift up activation (16)), is closest to, and can be sensed by, the β1 cyclase domain site that corresponds to the surface where Gsα binds adenylyl cyclase. The above sGC model is speculative, in particular as it is in large part based on the NpSTHK-type PAS dimer organization and PAS domains can be capable of undergoing (mechanistic) reorganization such as in PilT (61). Nevertheless, this is a new global model of sGC domain arrangement that provides a framework for future structure-guided experiments and can aid in the interpretation of a recent deletion mutagenesis study (18) as discussed below.
Figure 5. Possible subunit arrangement of sGC.
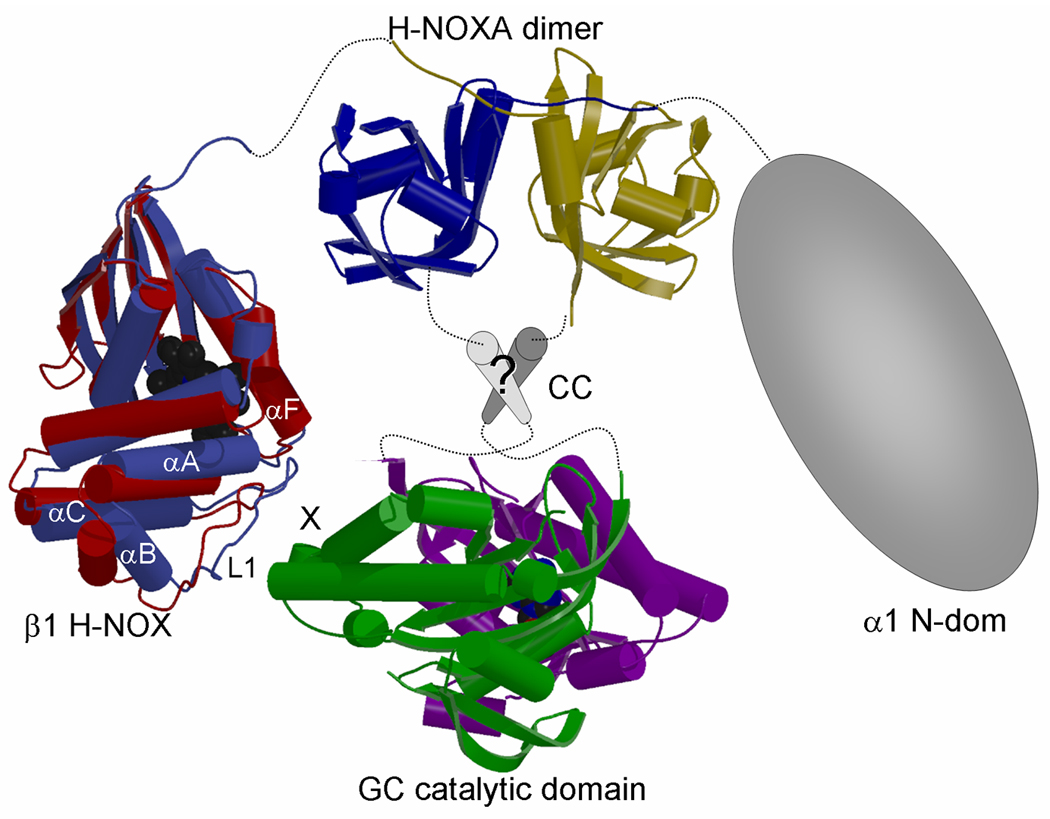
Composite figure depicting possible subunit arrangement of heterodimeric sGC. The structure of the helical putative CC region is not known (labeled ‘?’) as well as the structure of the α1 N-terminal domain. The H-NOX domain is oriented such that helix αF and loop L1 in H-NOX are in proximity to the site that corresponds to where Gsα binds and regulates the homologous adenylyl cyclase (marked ‘X’). In H-NOX, both αF and the N-terminal helical subdomain (αA-αC), which includes the loop L1 containing the potential switch residues D44–D45(14;60), are postulated to shift upon activation (16). To illustrate the N-terminal subdomain shift, we have depicted the superimposed NsH-NOX (red)(16) and TtH-NOX (blue)(14) structures which are postulated to represent the basal and activated state, respectively, of an H-NOX domain. Note that we cannot rule out direct interactions between the H-NOXA domain and the GC catalytic domain, since we do not know the conformation of the intervening sequences and the structure and position of the CC region.
The above noted abundance of inter-subdomain interactions within sGCα1β1 provides the possibility that some of these interfaces are more involved in the sGC activation mechanism than needed for sGC hetero-dimerization. A recent study detailing a systematic deletion mutagenesis analysis (18) in light of our structural results hints at such a possibility. This study first observed a roughly equal importance of the α1 and β1 CC residues for overall sGC heterodimerization (18); the results from this study are also included in Fig. 1B. This study also points to a critical dimerization role for α1:363–372 and β1:304–313 which are both located in H-NOXA (18). This is in agreement with our structure-function results as these homologous stretches of sGC residues include the critical α1:Q368 and β1:Q309 as well as β1:I311 which also has a role in dimerization (Fig. 4B). In addition to the H-NOXA interface, we had noted above that some of these residues might also provide key interactions with the downstream CC region. In further concord with our results, the deletion study found additional stretches of H-NOXA residues that either affected sGC dimerization and/or sGC activity (18)(Fig. 1B); all deletions contain residues that would correspond to the H-NOXA dimer interface or are in between the H-NOXA and CC domains (deletions depicted in Fig. 1B). However, equivalent deletions in α1 and β1 have a different effect (except for the noted α1:363–372 and corresponding β1:304–313). Deletions in β1-H-NOXA caused loss of dimerization, and thus all activity, whereas α1-H-NOXA deletions Δ283–292, Δ373–382 lead to an increase in cyclase activity and decreased EC50 for BAY41-2272 in the presence of 1nM DEA/NO indicating that they are more readily and more potently activated (18). These differences from this deletion study are intriguing but need to be taken with the caveat that their system actually introduced an additional artificial dimerization interface as the α1 and β1 sGC subunits were each fused with a different half of YFP that only fluoresces when the halves come together (18). Nevertheless, these unexpected observed differences when equivalent stretches of α1 or β1 H-NOXA residues are deleted likely provides new insights into the role of the H-NOXA domains for sGC’s activation mechanism. We propose that the CC domains provide the main, although not sole, driving force for sGC dimerization, while the H-NOXA subdomain heterodimer possibly serves as a regulatory interface needed to bring along and position the adjacent NO-regulated H-NOX domain such that it can interact and regulate the catalytic GC subunits (48). This could explain why deletions within the β1-H-NOXA domain are deleterious for hetero-dimerization as they likely disrupt not only the H-NOXA:H-NOXA interface but also indirectly disrupt the H-NOX:GC interface since H-NOX can likely no longer be properly positioned by β1-H-NOXA. However, deletions of most stretches of residues within the α1-H-NOXA domain did not cause loss of dimerization but, unexpectedly, made sGC more active even at lower concentrations of BAY41-2272 + 1nM DEA/NO (18). We therefore speculate that α1-H-NOXA is a likely negative regulatory subdomain that holds β1-H-NOXA, and thus indirectly also the adjacent H-NOX domain, in an inhibitory position/conformation prior to NO activation. Disruption of this interaction by deletions in α1-H-NOXA could possibly relax the β1-H-NOXA/H-NOX subdomains such that it is now more susceptible to BAY41-2272/NO activation yielding also higher cyclase activity. Further studies are needed to determine whether the H-NOXA domains in sGC, or PAS domain in STHK, are indeed used for such regulatory, or even sensory, mechanisms. Nevertheless, this structural interpretation of the Rothkegel et al 2007 deletion data (18) using our structural results suggests that targeting the H-NOXA dimer interface or putative PAS-ligand binding pocket could lead to new therapeutic sGC stimulators and activators. It is worth noting that this approach of targeting a PAS domain with no known ligand was successful for the PAS kinase domain, whose pocket is mainly closed as well, using the SAR-by-NMR approach (62).
Acknowledgments
We thank Dr. J. Meeks (UC Davis) for the genomic DNA of Nostoc punctiforme PCC 73102. We thank Drs. R. Bonomo, E. Jankowski, S. Misra, P. Pattanaik, J. Qin, M. Snider, and V. Yee for helpful comments and Drs. D. Lodowski and K. Palczewski (CWRU) for sharing beamtime. We thank beam line support personnel at APS and ALS. This work is supported by grants from the NIH to FvdA (R01 HL075329) and AB (R01 GM067640) and the AHA (SDG 0335159N) to FvdA. Coordinates and structure factors for 1–121 and 8–121 NpSTHK domains have been deposited with the PDB (PDB identifiers 2P04 and 2P08).
Abbreviations used
- H-NOXA
heme nitric oxide and oxygen binding associated
- PAS
an acronym formed from three proteins: Per, Arnt, Sim
- STHK
signal transduction histidine kinase
- 2-CHSR
two-component hybrid sensor and regulator
- sGC
soluble guanylyl cyclase
Reference List
- 1.Iyer LM, Anantharaman V, Aravind L. BMC.Genomics. 2003;4:5. doi: 10.1186/1471-2164-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ohmori M, Ikeuchi M, Sato N, Wolk P, Kaneko T, Ogawa T, Kanehisa M, Goto S, Kawashima S, Okamoto S, Yoshimura H, Katoh H, Fujisawa T, Ehira S, Kamei A, Yoshihara S, Narikawa R, Tabat S. DNA Res. 2001;8:271–284. doi: 10.1093/dnares/8.6.271. [DOI] [PubMed] [Google Scholar]
- 3.Koesling D, Russwurm M, Mergia E, Mullershausen F, Friebe A. Neurochem.Int. 2004;45:813–819. doi: 10.1016/j.neuint.2004.03.011. [DOI] [PubMed] [Google Scholar]
- 4.Pyriochou A, Papapetropoulos A. Cell Signal. 2005;17:407–413. doi: 10.1016/j.cellsig.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 5.Padayatti PS, Pattanaik P, Ma X, van den Akker F. Pharmacol.Ther. 2004;104:83–99. doi: 10.1016/j.pharmthera.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 6.Cary SP, Winger JA, Derbyshire ER, Marletta MA. Trends Biochem.Sci. 2006;31:231–239. doi: 10.1016/j.tibs.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 7.Ignarro LJ. J.Physiol Pharmacol. 2002;53:503–514. [PubMed] [Google Scholar]
- 8.Behrends S. Curr.Med.Chem. 2003;10:291–301. doi: 10.2174/0929867033368286. [DOI] [PubMed] [Google Scholar]
- 9.Shiga T, Suzuki N. Zoolog.Sci. 2005;22:735–742. doi: 10.2108/zsj.22.735. [DOI] [PubMed] [Google Scholar]
- 10.Anantharaman V, Balaji S, Aravind L. Biol.Direct. 2006;1:25. doi: 10.1186/1745-6150-1-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tesmer JJ, Sunahara RK, Gilman AG, Sprang SR. Science. 1997;278:1907–1916. doi: 10.1126/science.278.5345.1907. [DOI] [PubMed] [Google Scholar]
- 12.Zhang G, Liu Y, Ruoho AE, Hurley JH. Nature. 1997;386:247–253. doi: 10.1038/386247a0. [DOI] [PubMed] [Google Scholar]
- 13.Tews I, Findeisen F, Sinning I, Schultz A, Schultz JE, Linder JU. Science. 2005;308:1020–1023. doi: 10.1126/science.1107642. [DOI] [PubMed] [Google Scholar]
- 14.Pellicena P, Karow DS, Boon EM, Marletta MA, Kuriyan J. Proc.Natl.Acad.Sci.U.S.A. 2004;101:12854–12859. doi: 10.1073/pnas.0405188101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nioche P, Berka V, Vipond J, Minton N, Tsai AL, Raman CS. Science. 2004;306:1550–1553. doi: 10.1126/science.1103596. [DOI] [PubMed] [Google Scholar]
- 16.Ma X, Sayed N, Beuve A, van den Akker F. EMBO J. 2007;26:578–588. doi: 10.1038/sj.emboj.7601521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou Z, Gross S, Roussos C, Meurer S, Muller-Esterl W, Papapetropoulos A. J.Biol.Chem. 2004;279:24935–24943. doi: 10.1074/jbc.M402105200. [DOI] [PubMed] [Google Scholar]
- 18.Rothkegel C, Schmidt PM, Atkins DJ, Hoffmann LS, Schmidt HH, Schroder H, Stasch JP. Mol.Pharmacol. 2007;72:1181–1190. doi: 10.1124/mol.107.036368. [DOI] [PubMed] [Google Scholar]
- 19.Yuen PS, Doolittle LK, Garbers DL. J.Biol.Chem. 1994;269:791–793. [PubMed] [Google Scholar]
- 20.Lamothe M, Chang FJ, Balashova N, Shirokov R, Beuve A. Biochemistry. 2004;43:3039–3048. doi: 10.1021/bi0360051. [DOI] [PubMed] [Google Scholar]
- 21.Pflugrath JW. Acta Crystallogr.D.Biol.Crystallogr. 1999;55:1718–1725. doi: 10.1107/s090744499900935x. [DOI] [PubMed] [Google Scholar]
- 22.Terwilliger TC, Berendzen J. Acta Crystallogr.D Biol.Crystallogr. 1999;55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Emsley P, Cowtan K. Acta Crystallogr.D.Biol.Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 24.Murshudov GN, Vagin AA, Dodson EJ. Acta Crystallogr.D.Biol.Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 25.McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Acta Crystallogr.D.Biol.Crystallogr. 2005;61:458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 26.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. J.Appl.Cryst. 2001;26:283–291. [Google Scholar]
- 27.Kraulis PJ. J.Appl.Cryst. 1991;24:946–950. [Google Scholar]
- 28.Merritt EA, Bacon DJ. Methods Enzymol. 1997;277:505–524. doi: 10.1016/s0076-6879(97)77028-9. [DOI] [PubMed] [Google Scholar]
- 29.Holm L, Sander C. Trends Biochem.Sci. 1995;20:478–480. doi: 10.1016/s0968-0004(00)89105-7. [DOI] [PubMed] [Google Scholar]
- 30.Kurokawa H, Lee DS, Watanabe M, Sagami I, Mikami B, Raman CS, Shimizu T. J.Biol.Chem. 2004;279:20186–20193. doi: 10.1074/jbc.M314199200. [DOI] [PubMed] [Google Scholar]
- 31.Hefti MH, Francoijs KJ, de Vries SC, Dixon R, Vervoort J. Eur.J.Biochem. 2004;271:1198–1208. doi: 10.1111/j.1432-1033.2004.04023.x. [DOI] [PubMed] [Google Scholar]
- 32.Reinelt S, Hofmann E, Gerharz T, Bott M, Madden DR. J.Biol.Chem. 2003;278:39189–39196. doi: 10.1074/jbc.M305864200. [DOI] [PubMed] [Google Scholar]
- 33.Pandini A, Denison MS, Song Y, Soshilov AA, Bonati L. Biochemistry. 2007;46:696–708. doi: 10.1021/bi061460t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.John B, Sali A. Nucleic Acids Res. 2003;31:3982–3992. doi: 10.1093/nar/gkg460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eisenberg D, Luthy R, Bowie JU. Methods Enzymol. 1997;277:396–404. doi: 10.1016/s0076-6879(97)77022-8. [DOI] [PubMed] [Google Scholar]
- 36.Connolly ML. J.Mol.Graph. 1993;11:139–141. doi: 10.1016/0263-7855(93)87010-3. [DOI] [PubMed] [Google Scholar]
- 37.Krissinel E, Henrick K. Detection of Protein Assemblies in Crystals. In: Berthold MR, editor. Computational Life Sciences. Berlin Heidelberg: Springer-Verlag; 2005. [Google Scholar]
- 38.Ponstingl H, Kabir T, Thornton JM. J.Appl.Cryst. 2003;36:1116–1122. [Google Scholar]
- 39.Miyatake H, Mukai M, Park SY, Adachi S, Tamura K, Nakamura H, Nakamura K, Tsuchiya T, Iizuka T, Shiro Y. J.Mol.Biol. 2000;301:415–431. doi: 10.1006/jmbi.2000.3954. [DOI] [PubMed] [Google Scholar]
- 40.Key J, Hefti M, Purcell EB, Moffat K. Biochemistry. 2007;46:3614–3623. doi: 10.1021/bi0620407. [DOI] [PubMed] [Google Scholar]
- 41.Mergia E, Friebe A, Dangel O, Russwurm M, Koesling D. J.Clin.Invest. 2006;116:1731–1737. doi: 10.1172/JCI27657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Friebe A, Mergia E, Dangel O, Lange A, Koesling D. Proc.Natl.Acad.Sci.U.S.A. 2007;104:7699–7704. doi: 10.1073/pnas.0609778104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zabel U, Hausler C, Weeger M, Schmidt HH. J.Biol.Chem. 1999;274:18149–18152. doi: 10.1074/jbc.274.26.18149. [DOI] [PubMed] [Google Scholar]
- 44.Birktoft JJ, Rhodes G, Banaszak LJ. Biochemistry. 1989;28:6065–6081. doi: 10.1021/bi00440a051. [DOI] [PubMed] [Google Scholar]
- 45.Koglin M, Vehse K, Budaeus L, Scholz H, Behrends S. J.Biol.Chem. 2001;276:30737–30743. doi: 10.1074/jbc.M102549200. [DOI] [PubMed] [Google Scholar]
- 46.Wagner C, Russwurm M, Jager R, Friebe A, Koesling D. J.Biol.Chem. 2005;280:17687–17693. doi: 10.1074/jbc.M412099200. [DOI] [PubMed] [Google Scholar]
- 47.Morton DB, Anderson EJ. J.Exp.Biol. 2003;206:937–947. doi: 10.1242/jeb.00160. [DOI] [PubMed] [Google Scholar]
- 48.Winger JA, Marletta MA. Biochemistry. 2005;44:4083–4090. doi: 10.1021/bi047601d. [DOI] [PubMed] [Google Scholar]
- 49.Zhulin IB, Taylor BL, Dixon R. Trends Biochem.Sci. 1997;22:331–333. doi: 10.1016/s0968-0004(97)01110-9. [DOI] [PubMed] [Google Scholar]
- 50.Galperin MY. Environ.Microbiol. 2004;6:552–567. doi: 10.1111/j.1462-2920.2004.00633.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Narikawa R, Okamoto S, Ikeuchi M, Ohmori M. DNA Res. 2004;11:69–81. doi: 10.1093/dnares/11.2.69. [DOI] [PubMed] [Google Scholar]
- 52.Hering KW, Artz JD, Pearson WH, Marletta MA. Bioorg.Med.Chem.Lett. 2006;16:618–621. doi: 10.1016/j.bmcl.2005.10.093. [DOI] [PubMed] [Google Scholar]
- 53.Friebe A, Russwurm M, Mergia E, Koesling D. Biochemistry. 1999;38:15253–15257. doi: 10.1021/bi9908944. [DOI] [PubMed] [Google Scholar]
- 54.Li Z, Pal B, Takenaka S, Tsuyama S, Kitagawa T. Biochemistry. 2005;44:939–946. doi: 10.1021/bi0489208. [DOI] [PubMed] [Google Scholar]
- 55.Stasch JP, Becker EM, Alonso-Alija C, Apeler H, Dembowsky K, Feurer A, Gerzer R, Minuth T, Perzborn E, Pleiss U, Schroder H, Schroeder W, Stahl E, Steinke W, Straub A, Schramm M. Nature. 2001;410:212–215. doi: 10.1038/35065611. [DOI] [PubMed] [Google Scholar]
- 56.Chang FJ, Lemme S, Sun Q, Sunahara RK, Beuve A. J.Biol.Chem. 2005;280:11513–11519. doi: 10.1074/jbc.M412203200. [DOI] [PubMed] [Google Scholar]
- 57.Yildiz O, Doi M, Yujnovsky I, Cardone L, Berndt A, Hennig S, Schulze S, Urbanke C, Sassone-Corsi P, Wolf E. Mol.Cell. 2005;17:69–82. doi: 10.1016/j.molcel.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 58.Zhao JM, Lee H, Nome RA, Majid S, Scherer NF, Hoff WD. Proc.Natl.Acad.Sci.U.S.A. 2006;103:11561–11566. doi: 10.1073/pnas.0601567103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gilles-Gonzalez MA, Gonzalez G. J.Inorg.Biochem. 2005;99:1–22. doi: 10.1016/j.jinorgbio.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 60.Rothkegel C, Schmidt PM, Stoll F, Schroder H, Schmidt HH, Stasch JP. FEBS Lett. 2006;580:4205–4213. doi: 10.1016/j.febslet.2006.06.079. [DOI] [PubMed] [Google Scholar]
- 61.Satyshur KA, Worzalla GA, Meyer LS, Heiniger EK, Aukema KG, Misic AM, Forest KT. Structure. 2007;15:363–376. doi: 10.1016/j.str.2007.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Amezcua CA, Harper SM, Rutter J, Gardner KH. Structure. 2002;10:1349–1361. doi: 10.1016/s0969-2126(02)00857-2. [DOI] [PubMed] [Google Scholar]