

Abstract
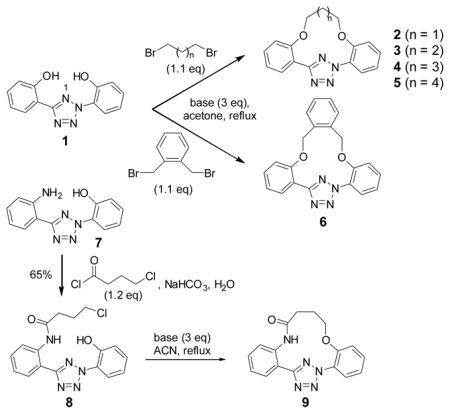
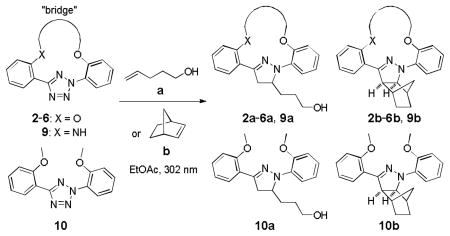
A series of conformationally constrained, macrocyclic tetrazoles were expediently prepared by double alkylation of a bis(o-phenolyl)tetrazole precursor. Several of them showed excellent reactivity toward norbornene in a photoinduced tetrazole-alkene cycloaddition reaction in organic solvent as well as toward a norbornene-modified protein in PBS buffer.
Keywords: bioorthogonal chemistry, tetrazole, alkene, cycloaddition, biotechnology
There is increasing interest to develop robust bioorthogonal reactions for site-specific modification of biomolecules within living systems.[1] A prominent example is the copper-catalyzed azide-alkyne cycloaddition reaction (“CuAAC”).[2] Since copper is toxic to cells, there have been substantial efforts directed toward developing copper-free alternatives for applications in live cells. One strategy involves activation of dipolarophiles by constraining them in macrocyclic rings: several cyclooctynes have been developed for rapid 1,3-dipolar cycloadditions with azide.[3] However, the use of ring structures containing strained dipoles for rapid bioorthogonal 1,3-dipolar cycloaddition reactions was extremely rare,[4] despite the fact that many cyclic dipoles have been used successfully in organic synthesis, e.g., cyclic azomethine imine,[5] cyclic carbonyl ylide,[6] cyclic azomethine ylide,[7] and cyclic nitrones[8] (Scheme 1). To our knowledge, cyclic nitrile imines—an important class of 1,3-dipoles[9] for the preparation of pyrazolines—has not been reported in the literature.
Scheme 1.

Representative cyclic 1,3-dipoles.
We have recently reported the use of diaryltetrazoles as photoactivatable precursors to the reactive nitrile imine dipoles for a photoinduced, bioorthogonal cycloaddition reaction with both the electron-deficient alkenes[10] and the unactivated terminal alkenes.[11] In a photocrystallographic study, we found that upon photoirradiation a diaryltetrazole generated in situ a bent nitrile imine,[12] the geometry postulated to exhibit reduced distortion energy and thus higher reactivity based on recent computational studies.[13] To reinforce the nitrile imine geometry in this reactive conformation, we hypothesized that installing a short “bridge” between the ortho-positions of two flanking phenyl rings to form a macrocyclic diphenyltetrazole should generate a cyclic nitrile imine upon photoirradiation with an overall reduced rotational freedom. Toward this goal, here we report the synthesis of a series of conformationally constrained macrocyclic tetrazoles and the characterization of their reactivity toward both a terminal alkene and a strained alkene in organic solvents as well as a norbornene-modified lysozyme in phosphate buffered saline (PBS).
To create the attachment sites for the installation of a chemical “bridge”, we synthesized two linear tetrazoles, 2,5-bis(o-phenolyl)-tetrazole (1) and 2-o-phenolyl-5-o-anilinyltetrazole (7) using the Kakehi method.[14] To effect macrocyclization via bis-alkylation, we treated tetrazole 1 with 1.1 equiv of dibromoalkanes with variable carbon length and screened four alkali metal bases: LiOH, Na2CO3, K2CO3 and Cs2CO3, anticipating that alkali metal ions with the right size may facilitate ring closure by bringing two phenol-O to the N1-side of tetrazole 1 through chelation (Table 1). Gratifyingly, we found that bases with larger alkali metals such as Cs+ and K+ in general afforded higher yields of the desired macrocyclic products (entries 7–8, 11–12, 15–16, and 19–20). Yields were lowest for tetrazole 2 (entries 3–4), presumably due to its smallest ring size (12-membered ring). Li+ and Na+ bases generally produced only trace amounts of the desired macrocyclic products together with large amounts of intractable oligomers. This metal ion size-dependency suggests that the larger ions Cs+ and K+ may form more stable, tridentate chelates with tetrazole 1 than the smaller ions. We then designed tetrazole 9 (Table 1), suspecting that amido-H may form an internal hydrogen bond with tetrazole-N1 which should reinforce the bent nitrile imine geometry. Thus, tetrazole 9 was readily prepared through amidation of tetrazole 7 with 4-chloro-butanoyl chloride followed by an intramolecular O-alkylation. Notably, Na2CO3 was found to be the optimal base for the ring closure reaction (compare entries 21–24).
Table 1.
Synthesis of the macrocyclic tetrazoles[a].
 | |||
|---|---|---|---|
| entry | base | product | yield (%)[b] |
| 1 | LiOH•H2O | 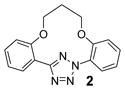 |
trace |
| 2 | Na2CO3 | <5[c] | |
| 3 | K2CO3 | <30[c] | |
| 4 | Cs2CO3 | 38 | |
| 5 | LiOH•H2O | 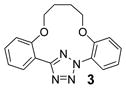 |
trace |
| 6 | Na2CO3 | trace | |
| 7 | K2CO3 | 51 | |
| 8 | Cs2CO3 | 78 | |
| 9 | LiOH•H2O | 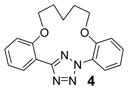 |
trace |
| 10 | Na2CO3 | trace | |
| 11 | K2CO3 | <35[c] | |
| 12 | Cs2CO3 | 45 | |
| 13 | LiOH•H2O | 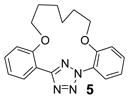 |
trace |
| 14 | Na2CO3 | trace | |
| 15 | K2CO3 | <60[c] | |
| 16 | Cs2CO3 | 67 | |
| 17 | LiOH•H2O | 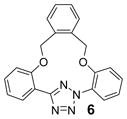 |
trace |
| 18 | Na2CO3 | trace | |
| 19 | K2CO3 | <50[c] | |
| 20 | Cs2CO3 | 61 | |
| 21 | LiOH•H2O | 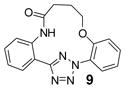 |
trace |
| 22 | Na2CO3 | 70 | |
| 23 | K2CO3 | <20[c] | |
| 24 | Cs2CO3 | trace | |
For bis-O-alkylation, reactions were carried out by refluxing tetrazole 1 with 1.1 equiv of dibromide and 3 equiv of base in acetone overnight. For intramolecular O-alkylation, tetrazole 8 was refluxed in acetonitrile with 3 equiv of base.
Isolated yields were reported unless noted otherwise.
Estimated yields based on TLC.
The structures of macrocyclic tetrazoles 6 and 9 were confirmed by X-ray crystallography (Figure 1). To our surprise, the amide bond in 9 was engaged in a pair of intermolecular H-bonds between N5-H and N1 of the neighboring tetrazole instead of an internal one. The three conjoining aromatic rings are slightly twisted out of the tetrazole plane with the torsional angles of N1-N2-C2-C3 and N1-C1-C8-C13 to be 45.12° and −44.38°, respectively (for tetrazole 6), and −62.46° and 37.17°, respectively (for tetrazole 9). These distortions might be due to lone-pair repulsion between N1 and O1 (distance: 2.815 Å for 6, 2.848 Å for 9) and between N1 and O2 (distance: 2.857 Å for 6, 3.249 Å for 9).
Figure 1.
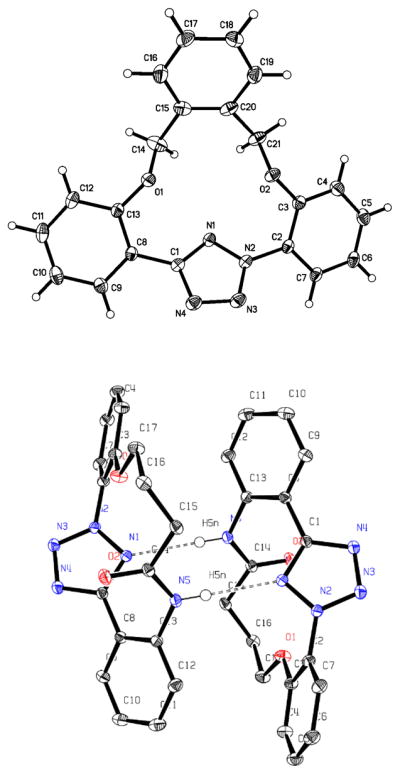
ORTEP diagrams of macrocyclic tetrazole 6 (top) and the dimer of macrocyclic tetrazole 9 (bottom) shown at 50% probability.
To assess the reactivity of macrocyclic tetrazoles, we carried out the photoinduced cycloaddition reactions with either 4-penten-1-ol, an analogue of a bioorthogonal alkene reporter homoallylglycine,[15] or a strained alkene reporter norbornene[16] in ethyl acetate (Table 2). Because the reaction proceeds through two distinct steps: ring opening step to generate the cyclic nitrile imine in situ and the subsequent [3+2] cycloaddition reaction,[10] we monitored the reactions by thin layer chromatography and discontinued the reactions once the tetrazole starting materials were consumed. We found that in general macrocyclic tetrazoles gave higher yields of the pyrazoline cycloadducts (with exceptions of entries 5, 6, 11) compared to their acyclic counterparts (entries 7, 14). As expected, norbornene serves as a more reactive dipolarophile as compared with 4-penten-1-ol, affording the corresponding pyrazolines in higher yields (compare entries 8–14 to 1–7). Among the macrocyclic tetrazoles, the 3- and 4-carbon linked tetrazoles 2 and 3 showed markedly higher reactivity with shorter reaction times and higher yields (entries 2, 8, 9), presumably due to their smaller ring sizes and thus reduced conformational flexibility. It is interesting to note that O-xylylene-linked tetrazole 6 gave a moderate yield with 4-penten-1-ol (entry 5) but a good yield with norbornene (entry 12), which can be attributed to a com-pact exo TS stabilized by attractive edge-to-face C—H•••π interaction between norbornene bridge C—H and xylyl-π crowd.[17] Gratifyingly, the X-ray structure of pyrazoline 3b was obtained, showing that the two phenyl rings are mostly co-planar with the pyrazoline ring with the torsional angles of N1-N2-C15-C16 and N1-C1-C9-C14 to be 30.8° and −26.6°, respectively (Figure 2).
Table 2.
Photoinduced 1,3-Dipolar Cycloaddition Reactions of Macrocyclic and Acylic Tetrazoles with Alkenes[a].
 | |||||
|---|---|---|---|---|---|
| entry | alkene | tetrazole | pyrazoline | time[b] (min) | yield[c] (%) |
| 1 | 2 | 2a | 120 | 59 | |
| 2 | 3 | 3a | 75 | 71 | |
| 3 | 4 | 4a | 84 | 60 | |
| 4 | 5 | 5a | 180 | 70 | |
| 5 | 6 | 6a | 120 | 46[d] | |
| 6 | 9 | 9a | 300 | 43 | |
| 7 | 10 | 10a | 120 | 58 | |
| 8 |  |
2 | 2b | 108 | 95 |
| 9 | 3 | 3b | 54 | 91 | |
| 10 | 4 | 4b | 120 | 81 | |
| 11 | 5 | 5b | 240 | 60 | |
| 12 | 6 | 6b | 90 | 84 | |
| 13 | 9 | 9b | 120 | 84 | |
| 14 | 10 | 10b | 60 | 76 | |
Reactions were conducted by irradiating 0.1 mmol of tetrazole and 10 mmol of alkene dipolarophile in 250 mL EtOAc with a 302-nm UV lamp in quartz flask.
Time was determined by tracing the disappearance of the starting materials on TLC.
Isolated yields.
The pyrazoline adduct was unstable upon standing.
Figure 2.

ORTEP diagram of the macrocyclic pyrazoline 3b shown at 50% probability.
The excellent reactivity of the macrocyclic tetrazoles toward norbornene prompted us to measure the photophysical properties of the resulting macrocyclic pyrazolines (Table 3). Relative to the pyrazoline control 10b (entry 7), four macrocyclic pyrazolines showed the bathochromic shifts in λabs (entries 1–2, 5–6) while two pyrazolines showed the hypsochromic shifts (entries 3–4), presumably due to their twisted macrocyclic geometries. Compared to 10b, macrocyclic pyrazolines exhibited invariable hypsochromic shifts in the maximum emission wavelength (λem) along with increases in the fluorescence quantum yield (φF).
Table 3.
Photophysical properties of macrocyclic pyrazolines[a].
| entry | pyrazoline | λabs (nm) | ε (M−1 cm−1) | λem (nm)[b] | φF[c] |
|---|---|---|---|---|---|
| 1 | 2b | 358 | 20,356 | 456 | 0.30 |
| 2 | 3b | 364 | 10,190 | 466 | 0.35 |
| 3 | 4b[d] | 348 | 1,702 | 468 | 0.35 |
| 4 | 5b[d] | 348 | 830 | 464 | 0.17 |
| 5 | 6b | 358 | 13,852 | 451 | 0.32 |
| 6 | 9b | 376 | 19,440 | 477 | 0.25 |
| 7 | 10b | 350 | 12,534 | 502 | 0.15 |
Compounds were dissolved in PBS:ACN (1:1) at the concentrations of 5 μM unless noted otherwise.
λex = 348 nm.
Quantum yields were determined using DAPI as the standard (φF = 0.58 in DMSO).
20 μM concentrations were used.
To examine whether the macrocyclic tetrazoles can be used to label a norbornene-containing protein, we incubated tetrazoles 2–6 (200 μM) with a norbornene-modified lysozyme (10 μM) in PBS buffer. The mixtures were photoirradiated with a handheld 302-nm UV lamp for 1 min prior to quenching with SDS sample buffer. In-gel fluorescence analysis (Figure 3) indicated that tetrazoles 2, 3 and 6, along with acyclic tetrazole 10, showed specific pyrazoline cycloadducts, in good agreement with their generally higher reactivity (entries 8, 9 and 12 in Table 2) and their superior photophysical properties (entries 1, 2 and 5 in Table 3). In particular, tetrazole 6 showed the strongest fluorescent band, presumably due to rate enhancement in water as a result of tighter hydrophobic packing in the TS. However, initial efforts to determine the reaction yields using mass spectrometry were unsuccessful due to the poor solubility of the pyrazoline-cycloadducts in PBS buffer.
Figure 3.

Photoinduced cycloaddition reaction of tetrazoles with a norbornene-modified lysozyme (m) or wild-type lysozyme (w) at 302 nm: top panel, inverted in-gel fluorescence with λex = 365 nm; bottom panel, Coomassie blue staining.
In summary, we have synthesized a series of structurally novel macrocyclic diphenyl tetrazoles by installing a short “bridge” between the two flanking diphenyl rings. Compared to the acyclic tetrazole, several macrocyclic tetrazoles showed improved reactivity toward a strained alkene both in organic solvent. One macrocyclic tetrazole showed rapid (~ 1 min) functionalization of a norbornene-modified protein in PBS buffer. These macrocyclic tetrazoles should offer a new class of photoactivatable tetrazole reagents for the bioorthogonal tetrazole-alkene cycloaddition reaction in living systems.
Experimental Section
Photoinduced Cycloaddition of Lyso-Norbornene with Macrocyclic Tetrazoles: To a 20-μL solution of either Lyso or Lyso-Nor (10 μM) in PBS buffer, pH 7.4, in a 96-well microtiter plate was added 1 μL of the various tetrazoles (4 mM in DMSO, final concentration is 200 μM). The mixtures were irradiated with a handheld 302-nm UV lamp for 1 min before quenching by addition of 5 μL of 6× SDS sample buffer, boiled for 5 min at 95 °C, loaded onto a NuPAGE 12% Bis-Tris gel (Invitrogen), and then subjected to protein electrophoresis. The Lyso-Pyr cycloadducts in the gel were visualized by illuminating the gel with a handheld 365-nm UV lamp and the resulting image was captured by a digital camera. After the image acquisition, the same gel was stained with Coomassie blue to confirm the size and equal loading of proteins.
Supplementary Material
Figure S1. UV-Vis spectra of the norbornene-derived pyrazolines: Compounds were dissolved in a mixed ACN/PBS buffer (1: 1) solvent to derive the final concentrations of 5 μM for 2b, 3b, 6b, 9b–10b, and 20 μM for 4b, 5b.
Figure S2. Normalized fluorescence spectra of the norbornene-derived pyrazolines: Compounds were dissolved in a mixed ACN/PBS buffer (1:1) solvent to derive final concentrations of 5 μM for 2b, 3b, 6b, 9b–10b, and 20 μM for 4b, 5b. The pyrazolines fluorescent intensities were normalized against DAPI.
Figure S3. Cycloaddition reaction of Lyso-Norbornene with the macrocyclic tetrazole 3: top panel, Coomassie blue staining of the SDS-PAGE gel; bottom panel, inverted fluorescence imaging of the same gel with λex = 365 nm.
Table S1. Crystal data and structure refinement for tetrazole 6.
Table S2. Crystal data and structure refinement for tetrazole 9.
Table S3. Crystal data and structure refinement for pyrazoline 3b.
Acknowledgments
We gratefully acknowledge the National Institutes of Health (GM 85092) for financial support. We thank William Brennessel at University of Rochester for X-ray structural determination. The crystal structures of 6, 9, and 3b have been deposited into the Cambridge Crystallographic Data Centre with the deposit numbers of CCDC 783273, 783275 and 783274, respectively..
Footnotes
Supporting information for this article is available on the WWW under http://www.chemeurj.org/ or from the author.
References
- 1.a) Sletten EM, Bertozzi CR. Angew Chem Int Ed. 2009;48:6974–6998. doi: 10.1002/anie.200900942. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lim RKV, Lin Q. Chem Comm. 2010;46:1489–1600. [Google Scholar]; c) Jewett JC, Bertozzi CR. Chem Soc Rev. 2010;39:1272–1279. doi: 10.1039/b901970g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew Chem Int Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]; b) Torne CW, Christensen C, Meldal M. J Org Chem. 2002;67:3057–3064. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 3.For an excellent review, see: Debets MF, van der Doelen CW, Rutjes FP, van Delft FL. Chembiochem. 2010;11:1168–1184. doi: 10.1002/cbic.201000064.Agard NJ, Prescher JA, Bertozzi CR. J Am Chem Soc. 2004;126:15046–15047. doi: 10.1021/ja044996f.Agard NJ, Baskin JB, Prescher JA, Lo A, Bertozzi CR. ACS Chem Biol. 2006;1:644–648. doi: 10.1021/cb6003228.Baskin JM, Prescher JA, Laughlin ST, Agard NJ, Chang PV, Miller IA, Lo A, Codelli JA, Bertozzi CR. Proc Natl Acad Sci USA. 2007;104:16793–16797. doi: 10.1073/pnas.0707090104.Ning X, Guo J, Wolfert MA, Boons GJ. Angew Chem Int Ed. 2008;47:2253–2255. doi: 10.1002/anie.200705456.Debets MF, van Berkel SS, Schoffelen S, Rutjes FP, van Hest JC, van Delft FL. Chem Comm. 2010;46:97–99. doi: 10.1039/b917797c.Jewett JC, Sletten EM, Bertozzi CR. J Am Chem Soc. 2010;132:3688–3690. doi: 10.1021/ja100014q.
- 4.McKay CS, Moran J, Pezacki JP. Chem Comm. 2010;46:931–933. doi: 10.1039/b921630h. [DOI] [PubMed] [Google Scholar]
- 5.a) Dorn H, Otto A. Angew Chem Int Ed. 1968;7:214–215. [Google Scholar]; b) Truce WE, Allison JR. J Org Chem. 1975;40:2260–2261. [Google Scholar]; c) Hashimoto T, Maeda Y, Omote M, Nakatsu H, Maruoka K. J Am Chem Soc. 2010;132:4076–4077. doi: 10.1021/ja100787a. [DOI] [PubMed] [Google Scholar]
- 6.a) Padwa A, Carter SP, Nimmesgern H. J Org Chem. 1986;51:1157–1158. [Google Scholar]; b) Padwa A, Carter SP, Nimmesgern H, Stull PD. J Am Chem Soc. 1988;110:2894–2900. [Google Scholar]; c) Padwa A, Fryxell GE, Zhi L. J Am Chem Soc. 1990;112:3100–3109. [Google Scholar]
- 7.a) Pandey G, Bagul TD, Sahoo AK. J Org Chem. 1998;63:760–768. doi: 10.1021/jo971728+. [DOI] [PubMed] [Google Scholar]; b) Pearson WH, Stoy P, Mi Y. J Org Chem. 2004;69:1919–1939. doi: 10.1021/jo030334h. [DOI] [PubMed] [Google Scholar]; c) Coldham I, Burrell AJM, White LE, Adams H, Oram N. Angew Chem Int Ed. 2007;46:6159–6162. doi: 10.1002/anie.200701943. [DOI] [PubMed] [Google Scholar]; d) Coldham I, Jana S, Watson L, Pilgram CD. Tetrahedron Lett. 2008;49:5408–5410. [Google Scholar]
- 8.For a recent review, see: Revuelta J, Cicchi S, Goti A, Brandi A. Synthesis. 2007:485–504.Cardona F, Valenza S, Picasso S, Goti A, Brandi A. J Org Chem. 1998;63:7311–7318. doi: 10.1021/jo980809i.Thiverny M, Philouze C, Chavant PY, Blandin V. Org Biomol Chem. 2010;8:864–872. doi: 10.1039/b918612c.Ghini G, Luconi L, Rossin A, Bianchini C, Giambastiani G, Cicchi S, Lascialfari L, Brandi A, Giannasi A. Chem Comm. 2010;46:252–254. doi: 10.1039/b917016b.
- 9.a) Huisgen R. Angew Chem Int Ed. 1963;2:565–598. [Google Scholar]; b) Huisgen R. Angew Chem Int Ed. 1963;2:633–645. [Google Scholar]; c) Bianchi G, Gandolfi R. In: 1,3-Dipolar Cycloaddition Chemistry. Padwa A, editor. Wiley; New York: 1984. pp. 470–477. [Google Scholar]
- 10.a) Wang Y, Vera CR, Lin Q. Org Lett. 2007;9:4155–4158. doi: 10.1021/ol7017328. [DOI] [PubMed] [Google Scholar]; b) Song W, Wang Y, Qu J, Madden MM, Lin Q. Angew Chem Int Ed. 2008;47:2832–2835. doi: 10.1002/anie.200705805. [DOI] [PubMed] [Google Scholar]; c) Wang Y, Hu WJ, Song W, Lim RKV, Lin Q. Org Lett. 2008;10:3725–3728. doi: 10.1021/ol801350r. [DOI] [PubMed] [Google Scholar]; d) Song W, Yu Z, Madden MM, Lin Q. Mol Biosys. 2010;6:1576–1578. doi: 10.1039/c003470c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Song W, Wang Y, Qu J, Lin Q. J Am Chem Soc. 2008;130:9654–9655. doi: 10.1021/ja803598e. [DOI] [PubMed] [Google Scholar]; b) Wang Y, Song W, Hu WJ, Lin Q. Angew Chem Int Ed. 2009;48:5330–5333. doi: 10.1002/anie.200901220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zheng SL, Wang Y, Yu Z, Lin Q, Coppens P. J Am Chem Soc. 2009;131:18036–18037. doi: 10.1021/ja9094523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a) Ess DH, Houk KN. J Am Chem Soc. 2007;129:10646–10647. doi: 10.1021/ja0734086. [DOI] [PubMed] [Google Scholar]; b) Ess DH, Houk KN. J Am Chem Soc. 2008;130:10187–10198. doi: 10.1021/ja800009z. [DOI] [PubMed] [Google Scholar]; c) Schoenebeck F, Ess DH, Jones GO, Houk KN. J Am Chem Soc. 2009;131:8121–8133. doi: 10.1021/ja9003624. [DOI] [PubMed] [Google Scholar]
- 14.Ito S, Tanaka Y, Kakehi A, Kondo K. Bull Chem Soc Jpn. 1976;49:1920–1923. [Google Scholar]
- 15.Song W, Wang Y, Yu Z, Rivera Vera CI, Qu J, Lin Q. ACS Chem Biol. 2010;5 doi: 10.1021/cb100193h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.a) Devaraj NK, Weissleder R, Hilderbrand SA. Bioconjugate Chem. 2008;19:2297–2299. doi: 10.1021/bc8004446. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gutsmiedl K, Wirges CT, Ehmke V, Carell T. Org Lett. 2009;11:2405–2408. doi: 10.1021/ol9005322. [DOI] [PubMed] [Google Scholar]; c) Han HS, Devaraj NK, Lee J, Hilderbrand SA, Weissleder R, Bawendi MG. J Am Chem Soc. 2010;132:7838–7839. doi: 10.1021/ja101677r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paliwal S, Geib S, Wilcox CS. J Am Chem Soc. 1994;116:4497–4498. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. UV-Vis spectra of the norbornene-derived pyrazolines: Compounds were dissolved in a mixed ACN/PBS buffer (1: 1) solvent to derive the final concentrations of 5 μM for 2b, 3b, 6b, 9b–10b, and 20 μM for 4b, 5b.
Figure S2. Normalized fluorescence spectra of the norbornene-derived pyrazolines: Compounds were dissolved in a mixed ACN/PBS buffer (1:1) solvent to derive final concentrations of 5 μM for 2b, 3b, 6b, 9b–10b, and 20 μM for 4b, 5b. The pyrazolines fluorescent intensities were normalized against DAPI.
Figure S3. Cycloaddition reaction of Lyso-Norbornene with the macrocyclic tetrazole 3: top panel, Coomassie blue staining of the SDS-PAGE gel; bottom panel, inverted fluorescence imaging of the same gel with λex = 365 nm.
Table S1. Crystal data and structure refinement for tetrazole 6.
Table S2. Crystal data and structure refinement for tetrazole 9.
Table S3. Crystal data and structure refinement for pyrazoline 3b.