

Abstract
The report summarizes studies on the redox behavior of synthetic models for the [FeFe]-hydrogenases, consisting of diiron dithiolato carbonyl complexes bearing the amine cofactor and its N-benzyl derivative. Of specific interest are the causes of the low reactivity of oxidized models toward H2, which contrasts with the high activity of these enzymes for H2 oxidation. The redox and acid-base properties of the model complexes [Fe2[(SCH2)2NR](CO)3(dppv)(PMe3)]+ ([2]+ for R = H and [2′]+ for R = CH2C6H5, dppv = cis-1,2-bis(diphenylphosphino)ethylene)) indicate that addition of H2 and followed by deprotonation are (i) endothermic for the mixed valence (FeIIFeI) state and (ii) exothermic for the diferrous (FeIIFeII) state. The diferrous state is shown to be unstable with respect to coordination of the amine to Fe, a derivative of which was characterized crystallographically. The redox and acid-base properties for the mixed valence models differ strongly for those containing the amine cofactor versus those derived from propanedithiolate. Protonation of [2′]+ induces disproportionation to a 1:1 mixture of the ammonium-FeIFeI and the dication [2′]2+ (FeIIFeII). This effect is consistent with substantial enhancement of the basicity of the amine in the FeIFeI state vs the FeIIFeI state. The FeIFeI ammonium compounds are rapid and efficient H-atom donors toward the nitroxyl compound TEMPO. The atom transfer is proposed to proceed via the hydride, as indicated by the reaction of [HFe2[(SCH2)2NH](CO)2(dppv)2]+ with TEMPO. Collectively, the results suggest that proton-coupled electron-transfer pathways should be considered for H2 activation by the [FeFe]-hydrogenases.
Introduction
The catalytic chemistry of hydrogen evolution and oxidation is topical because H2 is a versatile reagent and a promising carrier of energy.1, 2 New approaches to this area of catalysis have been inspired by the hydrogenase enzymes, and studies on the [FeFe]-hydrogenases have proven especially influential.3 We and others have proposed that catalysis occurs at a single coordination site on one Fe center of the diiron subunit (Figure 1),4,5 with participation of various cofactors.
Figure 1.

Active site of the oxidized state (Hox) of the [FeFe]-hydrogenase (left),7, 8 showing the azadithiolate cofactor and a vacant site on the distal iron center.9 Model complex for the Hox state of the active site, with organophosphorus ligands in place of the cyanide and 4Fe-4S cofactors.
Ongoing research in this area focuses on elucidating the electronic features of the bimetallic site – oxidation state, redox potentials, asymmetry - and equipping models with the cofactors required for efficient catalysis. Thus, models have evolved from the simple Fe2(SR)2(CO)6 to substituted derivatives Fe2(SR)2(CO)6-xLx which exhibit the two essential attributes of hydrogenases – the acid-base and redox behavior. Two cofactors are of functional significance since they enhance the redox or acid-base properties inherent in the diiron center. First, the redox-active 4Fe-4S cluster allows the diiron subsite, which operates via a 1e− couple, to effect 2e− redox reactions, as required by the H2/2H+ redox couple. Second, and very relevant to this report, the amine-containing dithiolate cofactor4, 5 is proposed to relay protons to and from the distal Fe.6
Synthetic models differ from the diiron site in the protein in three important ways. First, most models feature organophosphorus ligands such as cis-1,2-C2H2(PPh2)2 (dppv) and PMe3 in place of the cyanide cofactors. This change enables mechanistic studies without the complications of reactions at FeCN. Second, with a single exception,10 models omit the 4Fe-4S cofactor that is found in the 6-Fe “H-cluster” (Figure 1). Finally, the model complexes differ from the active site in the stereochemistry of one of the iron centers. In the protein, the distal Fe center adopts an “inverted” (also described as “rotated”11) structure in which a CO ligand occupies a semi-bridging position between the two Fe centers. This stereochemistry exposes a coordination site adjacent to the amine of the dithiolate cofactor. The inverted structure is observed in the oxidized models of the type [Fe2(SR)2(CO)4L2]+ and [Fe2(SR)2(CO)3L3]+,12–15 but such inverted structures are rarely observed in reduced diiron models.16, 17 Theoretical studies indicate that rotated structures are destabilized by about 10 kcal in model complexes.11
More recent models incorporate the amine cofactor, which we call azadithiolate (adt = (SCH2)2NH2−). N-Protonation of such compounds shifts the reduction potential of the diiron center by ~0.5 V, and the oxidation potential by ~0.2 V.18, 19 Of relevance to the enymatic mechanism, the complex Fe2(adt)(CO)2(dppv)2 electrocatalyzes hydrogen evolution from weaker acids than is possible for the related complexes lacking the amine, e.g. Fe2(pdt)(CO)2(dppv)2 (pdt = S2(CH2)32−).6 Catalysis by these electron-rich models proceeds via the intermediacy of an iron hydride, for which the rates of formation and deprotonation are modified by the amine. Such amine-complemented models for the Hox state are ideal systems to explore factors relevant to H2 oxidation. In the present report, we examine the interplay between the acid-base behavior of the amine and the redox properties of the Fe2 centers in Fe2(adt)(CO)3(dppv)(PMe3).9 Protonation of Fe2(adt)(CO)3(dppv)(PMe3) gives the ammonium derivative as the only spectroscopically detectable tautomer. Via a first order pathway, the ammonium compound tautomerizes slowly at room temperature in CH2Cl2 solution to the isomeric “μ-hydride,” which feature H− bound to both Fe centers (Scheme 1).9, 20
Scheme 1.

Regiochemistry of protonation of tri- and tetrasubstituted diiron azadithiolato carbonyl complexes.6, 9
In contrast to the “trisphosphine”, the tetrasubstituted diiron dithiolates form spectroscopically detectable terminal hydrides. The enzyme is thought to operate via such terminal hydrides, not μ-hydrides. For [HFe2(adt)(CO)2(dppv)2]BF4, the pKa CD2Cl2 is between 5.7 and 8.2.6 The terminal hydride and ammonium tautomers of this complex coexist in comparable amounts in CH2Cl2 solution (Scheme 1). Indicative of the subtleties of this system is the finding that high concentrations of BF4− shift the equilibrium from the hydride toward the ammonium tautomer.6
The process by which H2 is activated by diiron dithiolato complexes came into focus with the finding that H2 is only slowly oxidized by [Fe2(adt)(CO)3(dppv)(PMe3)]+ (at 2000 psi H2, rate ~ 10−4 s−1)9, whereas the enzyme from D. gigas oxidizes H2 at 105 s−1.21 The low reactivity of mixed valence models toward H2 is generally understandable because the heterolytic scission of H2 by iron characteristically requires ferrous centers.1 Theoretical studies show that the diferrous state of the diiron subsite is well suited for activation of H2.22 Diferrous μ-hydride complexes catalyzes H-D exchange between H2 and D2O, albeit only photochemically.23 Diferrous compounds that contain a vacant coordination site are rare,24, 25 and mononuclear ferrous ν2-H2 complexes have much precedence.26, 27 Furthermore, amine-complemented mononuclear ferrous phosphine complexes have been shown to under redox-triggered tautomerization (eq 1).28
 |
(1) |
Results
Synthesis and Characterization of Diferrous κ3-Azadithiolato Complexes
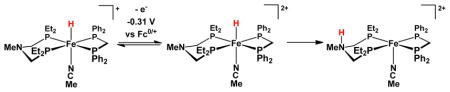
On a preparative scale and consistent with ample precedent,12–15 oxidation of Fe2[(SCH2)2X](CO)3(dppv)(PMe3) with Fc+ was found to yield the mixed valence FeIIFeI derivatives (“Hox models”), where X = CH2, NH, NBn, O, for 1, 2, 2′, 3, respectively (Bn = CH2C6H5).9, 12 These cationic derivatives are stabilized by bulky arylborate counteranions, which are essential for the stabilization of the oxidized azadithiolates.9 The azadithiolates were found to undergo a second oxidation with FcBArF4, and in this way we generated [2′](BArF24)2 (FcBArF4 = [Fe(C5H5)2]B(C6H3-3,5-(CF3)2)4). The 31P NMR spectrum indicates that[2′](BArF4)2 is diamagnetic and Cs-symmetric. The nu;CO bands are shifted to higher energy by ~40 cm−1 from the position for [2′]+, and all three bands occur in the terminal carbonyl region (2066, 2008, and 1977 cm−1). When we instead employed the oxidant FcBF4, a spectroscopically distinct compound was observed (Supporting Information).
Treatment of a CD2Cl2 solution of [2′]2+ with MeCN was found to afford stable adducts, regardless of the counteranion, of the formula [2′(NCMe)]2+. Although the BArF4− salts afforded tacky oils, the BF4− salts readily crystallized. The crystallographic analysis of this salt revealed a diiron dithiolate as expected, but that unlike all previously reported azadithiolato complexes,29–31 the amine is coordinated to Fe (Figure 2). The Fe---Fe distance of 3.447(2) Å is nonbonding, which is also uncommon.24, 32–34 The two iron centers, which are still linked by a pair of thiolates, are each octahedral. The coordination spheres of the Fe subsites are described as Fe(CO)2(PMe3)(NCMe)(SR)2 and Fe(CO)(dppv)(amine)(SR)2. The S-Fe-N angles are acute at ~73º, but the other Fe-ligand bond lengths and angles are within the normal range (Table I).
Figure 2.
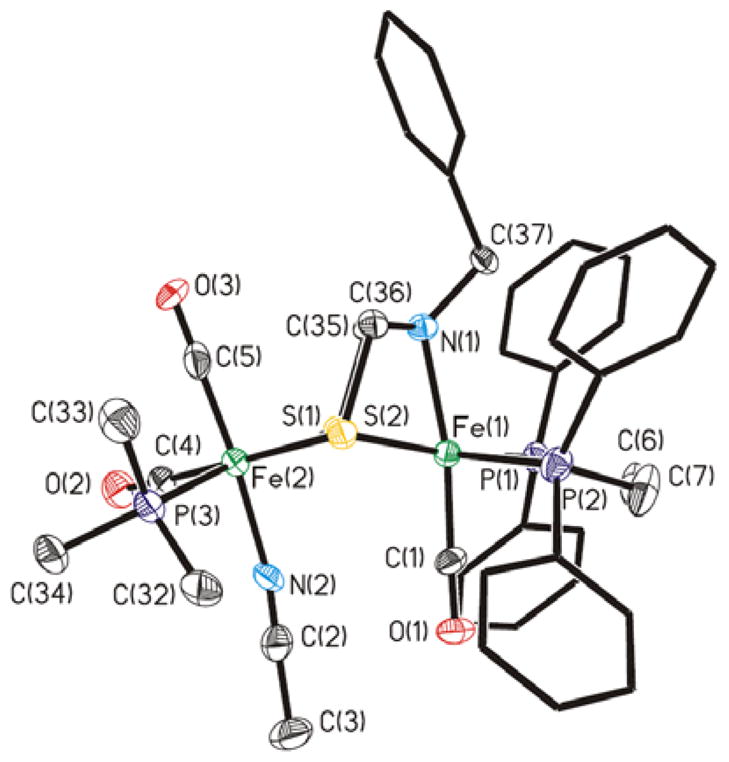
Structure of the dication in [2′(NCMe)](BF4)2. Thermal ellipsoids set at the 30% probability level and hydrogen atoms are not shown.
Table 1.
Selected Bond Distances (Å) and Angles (°) for [2′(MeCN)](BF4)2
| bond | distance | bonds | angles |
|---|---|---|---|
| Fe(1)-N(1) | 2.098(8) | N(1)-Fe(1)-S(2) | 72.9(2) |
| Fe(1)-Fe(2) | 3.477(2) | Fe(1)-S(2)-C(36) | 80.9(3) |
| Fe(1)-C(1) | 1.753(11) | Fe(2)-S(2)-C(36) | 107.7(4) |
| Fe(2)-N(2) | 1.965(10) | S(1)-Fe(1)-S(2) | 80.97(11) |
| Fe(1)-S(2) | 2.336(3) | S(1)-Fe(2)-S(2) | 81.45(11) |
| Fe(2)-S(2) | 2.323(3) | P(1)-Fe(1)-P(2) | 87.24(12) |
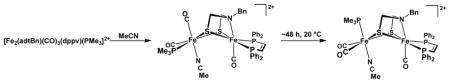
The solution properties of [2′(NCMe)]2+ were examined by variable temperature 31P NMR spectroscopy. Dissolution of the crystalline [2′(NCMe)](BF4)2 at −70 °C gave a compound with the same spectroscopic signature as was obtained by addition of MeCN to a CD2Cl2 solution of [2′](BArF4)2 generated at −70 °C. Over the course of ~24 h at 20 °C, this species isomerized to a second symmetrical isomer (two 31P NMR signals) (eq 2, Figure 3). Solutions of [2′](BArF4)2 were found to rapidly and irreversibly form an adduct upon treatment with 1 atm of CO. In contrast, monocation [2′]+ binds CO reversibly, and the adducts are only observable at low temperatures.9, 13 Solutions of [2′](BArF4)2 were found to be unreactive towards H2 at −78 °C, although more forcing conditions were precluded by the thermal sensitivity of this salt. These dications, e.g. [2′]2+, react at low temperatures with PhSiH3 to give hydrides such as [(μ-H)2′]+.
Figure 3.

31P NMR spectra (CD2Cl2) of a fresh solution of [2′](BArF4)2 at −193 K (top), after treatment with CD3CN (middle), and after allowing the same solution to stand at 293 K for 48 h. Signals at > δ70 are assigned to dppv and those absorbing < δ40 are assigned to PMe3. The former signals are more indicative of symmetry and the latter signals are useful indicators of the number of isomers.
 |
(2) |
Electrochemical Properties of Diiron Azadithiolates
Diiron dithiolates of the formula Fe2[(SCH2)2X](CO)3(dppv)(PMe3) undergo a reversible 1e− oxidation for X = CH2, NH, NCH2C6H5, and O (for 1, 2, 2′, 3; see Figures 4, eq 3, and Table 2). For 2′ the ratios of the oxidation and reduction currents (ipc/ipa) are >0.9 at a scan rate of 0.100 V/s in noncoordinating solvents (CH2Cl2). The linear dependence of ip on (scan rate)0.5 also indicates a diffusion-controlled process.
Figure 4.

Cyclic voltammograms of a CH2Cl2 solution of 1 (black) and 2′ (red) illustrative of the effect of the azadithiolate on the second anodic event. Conditions: 0.001 M 2′, 0.300 M [(C4H9)4N]PF6, CH2Cl2, 20 °C, 0.1 V/s scan rate.
Table 2.
Half-wave Potentials (V) for the [Fe2(SR)2]0/+ and [Fe2(SR)2]+/2+ Couples for Fe2[(SCH2)2X](CO)3(dppv)(PMe3). Conditions: 1 mM diiron complex, 100 mM [Bu4N]PF6, CH2Cl2 Solution, vs Fc0/+, 0.1 V/s scan rate. Under our experimental conditions, an internal standard of Fc (1 mM) displayed ΔEp ≈ 100 mV. The ipa/ipa values for E1 were recorded under conditions where the scan range did not extend to E2
| Dithiolate | Electrolyte | E1 for [Fe2(SR)2]0/+ (ΔEp, V) [ipc/ipa] | E2 for [Fe2(SR)2]+/2+ (ΔEp) [ipc/ipa] | E2 – E1 |
|---|---|---|---|---|
| (SCH2)2NBn (2′) | [(C4H9)4N]PF6 | −0.643 0.118) [>0.9] | −0.128 (0.147) [>0.9] | 0.515 |
| (SCH2)2NBn (2′) | [(C4H9)4N]BArF4 | −0.715 (0.111) [0.9] | −0.070 (0.222) [0.7]b | 0.645 |
| (SCH2)2NH (2) | [(C4H9)4N]PF6 | −0.561 (0.095) not determineda | −0.363 (0.101) not determineda | 0.198 |
| (SCH2)2NH (2) | [(C4H9)4N]BArF4 | −0.624 (0.075) [>0.9] | −0.249 (0.175) [0.1]b | 0.375 |
| (SCH2)2CH2 (1) | [(C4H9)4N]PF6 | −0.609 (0.126) [>0.9] | 0.356 (0.160) [0.2] | 0.965 |
| (S2C2H4) | [(C4H9)4N]PF6 | −0.469 (0.121) [>0.9] | 0.345 (0.143) [0.5] | 0.930 |
| (SCH2)2O (3) | [(C4H9)4N]PF6 | −0.528 (0.139) [>0.9] | 0.353 (0.233) [0.1] | 0.840 |
| [(SCH2)2NBn(H)]+ ([H2′]+) | [(C4H9)4N]PF6 | Ep/2 = 0.040 (irrev.) | --- | --- |
| [(SCH2)2NH2]+ ([H2]+) | [(C4H9)4N]PF6 | Ep/2 = 0.050 (irrev.) | --- | --- |
The close separation of the two oxidation steps precludes accurate measurement of this current ratio.
At fast scan rates the cathodic return wave of E2 becomes broadened making the ipc/ipa value an inaccurate representation of reversibility. At slow scan rates E2 is fully reversible.
Oxidations corresponding to the [Fe2(SR)2]+/2+ (FeIIFeI/FeIIFeII) couple proved highly dependent on the dithiolate (Figure 4 and eq 3). For the propane- and oxadithiolato compounds, but not the azadithiolates, a poorly reversible second oxidation is observed at 0.890±0.040 V more anodic than the [Fe2(SR)2]o/+ couple. Unlike the amine-free derivatives, the [Fe2(SR)2]+/2+ couple for the azadithiolates 2 and 2′ occurs at mild potentials and displayed full reversibility. Interestingly ΔE for 2 is significantly smaller than that for 2′. When non-coordinating [(C4H9)4N]BArF4 electrolyte was employed, ΔE increased by 130 and 177 mV for 2′ and 2 respectively. This separation of electrochemical events is commonly observed in non-coordinating electrolytes,35 and the larger value of ΔE for 2 versus 2′ is attributable to the ability of smaller fluoroanions such as PF6− to engage in hydrogen-bonding.36 Upon addition of MeCN to CH2Cl2 solutions of 2′, E2 becomes irreversible and two closely spaced cathodic waves are observed at −0.80 and −0.90 V. The strong effect of MeCN is consistent with the formation of the adduct [2′(MeCN)]2+.
 |
(3) |
Acid-Base Reactions
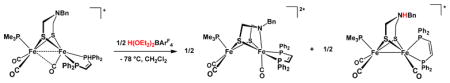
The pKa CD2Cl2 values of the ammonium compounds [H2′]+ and [H2]+ were measured as 3.3 and 3.2, respectively, by titration of 2′ and 2 with [HPMe2Ph]BF4 (pKa CD2Cl2 = 5.7).37 We could not directly determine the corresponding pKa of [H2′]2+ because [2′]+ undergoes quasi-disproportionation upon treatment with acids, even in solution at −78 °C. The product mixture consists of equal amounts of the reduced ammonium species [H2′]+ and the dication [2′]2+ (eq 4, Figure 5).
Figure 5.

IR spectra (CH2Cl2 solution) for [2′]+ (top), the products of its reaction with 0.5 equiv of H(OEt2)2BArF4 (middle top), [2′](BArF24)2 (middle bottom), [H2′]+ generated by protonation of 2′ with H(OEt2)2BArF24 (bottom). Component IR bands (CH2Cl2) are [2′]+: 2017, 1965, 1867 cm−1; [2′]2+: 2065, 2009, 1977; [H2′]+: 1960, 1925, 1898 cm−1.
 |
(4) |
The proton-induced quasi-disproportionation of [2′]+ (eq 4) is driven by the strong oxidizing ability of [H2′]2+. The requirement of 0.5 equiv of acid for the reaction in eq 2 was confirmed. Furthermore, >0.5 equiv H(OEt2)2BArF4 was found not to affect the product distribution, a result consistent with the low basicity of [2′]2+, wherein the amine is coordinated to Fe. In contrast to this behavior, mixed-valence Fe(I)Fe(II) complexes lacking azadithiolates are unreactive towards acid. For example, a CH2Cl2 solution of [Fe2(S2C3H6)(CO)3(dppv)(PMe3)]BArF4 was unaffected by treatment with H(OEt2)2BArF4 at 20 °C.
Together with the pKa for [H2]+, E1/2 for the [H2]+/2+ couple would allow us to calculate the acidity of the mixed valence ammonium compound, i.e. pKa for [H2]2+. The [H2]+/2+ couple is irreversible, thus precluding accurate determination of E1/2. Nonetheless, estimating E1/2 as the potential of the anodic wave at half-height would indicate that 1e− oxidation of [H2]+ decreases the basicity of the amine by as much as 109 (CH2Cl2 solution, Scheme 2).
Scheme 2.

Bordwell calculation at 20 °C of the pKaCD2Cl2 of [H2′]2+ assuming E1/2 of the [H2′]+/2+ couple as 0.04 V.
H-Atom Transfer Reactions
The electron-rich N-protonated azadithiolato complexes were found to serve as H-atom donors. Thus, treatment of [H2′]+ with one equiv of 2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPO), an H-atom abstracting agent,38 immediately and quantitatively yielded [2′]+ at 293 °C (eq 5, R2NO· = TEMPO). This reaction is conveniently monitored by IR spectroscopy in the nu;CO region.
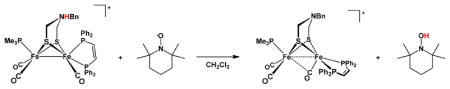 |
(5) |
When the reaction was monitored by in-situ IR spectroscopy at low temperatures, a transient build-up of 2′ was detected. This species results from the reversible deprotonation of [H2′]+ by TEMPOH, the product of eq 5.39 At 199 K, the rate of disappearance of [H2′]+ proceeds at 8.13 × 10−3 s−1M−1. By measuring the temperature dependence of the rate constant over the range 199–229 K, we determined that ΔG‡ is 13.5 kcal/mol (199 K). The secondary ammonium [H2]+ was observed to react with TEMPO faster than did [H2′]+ under similar conditions: the reaction was complete in minutes vs ~60 min for the Bn derivative at 199 K.
Insights into a possible mechanism of the hydrogen-atom transfer reactions were provided by experiments with the terminal hydrides [HFe2[(SCH2)2NR](CO)2(dppv)2]+ and [HFe2[(S2C3H6)(CO)2(dppv)2]+.6 In CH2Cl2 solution, both species were found to react (t1/2 ≈ 10 min, −199 K) with TEMPO to give the corresponding mixed-valence Fe(II)Fe(I) derivatives.25 The IR signatures for the diferrous hydride starting materials and mixed valence products overlap, thus we conducted these oxidations under an atmosphere of CO, which rapidly affords the CO adducts that display distinctive IR signatures.25
Conclusions
The hydrogenases function by coupling or combining acid-base and redox properties. The present study examined the interplay of these properties in a diiron model that contains both a base and a redox center. We report three unusual findings:
The mildness and reversibility of the FeIIFeI/FeIIFeII couple in models containing the amine cofactor arises from the formation of an Fe-N bond.*40, 41 Our measurements suggest that coordination of the amine stabilizes the diferrous state by ~11 kcal/mol as indicated by ΔEFeIIFeI/FeIIFeII.
The FeIFeI ammonium center serves as an efficient H-atom donor, with concomitant Fe-centered redox. This finding demonstrates the ability of hydrogenase models and, by implication, the enzyme to participate in PCET.
The basicity of the amine is highly sensitive to the oxidation state of the underlying diiron centers. The behavior of the mixed valence ammonium centers in models and in proteins may be quite different owing to the effects of site isolation provided by the protein.
The [FeFe]-hydrogenases are characterized by reduced and oxidized states, respectively Hred and Hox that differ by 1e−. In terms of enzyme mechanism, the reduced state activates protons and the oxidized state activates H2. The oxidation state for the diiron center in Hred remains uncertain, but is likely either a diferrous hydride or a disubferrous ammonium which would be readily interconverted.6 As we demonstrated in this work, Hred and Hox are separated formally as well as operationally by H·.
Our measurements bear on the mechanism for activation of H2. In the heterolytic pathway which is assumed for all hydrogenases,26 H2 is a source of H−, invariably bound to a Fe(II), and H+, which is usually bound to an organic base. We show that the binding of hydride to Fe(II)Fe(I) complexes is far less favorable than to Fe(II)Fe(II) derivatives (Figure 6). The hydride acceptor strength of [2′]+, −48 kcal/mol, is insufficient to compensate for the energy required for the heterolysis of H2. Thus H2 activation by [2′]+ is predicted to be unfavorable, even when coupled to the neutralization of the proton by a moderately strong base.
Figure 6.

Free energy changes (vs E(Fc0/+) = 0 V) for the hydrogenation of mixed-valence and diferrous dithiolato complexes using data obtained for [Fe2[(SCH2)2NR](CO)3(dppv)(PMe3)]n (n = +, 2+). The calculation assumes that the proton binds to a base of pKaMeCN = 10.
The hydride acceptor strength of the diferrous center, e.g., [2′]2+ (assuming κ2-adt), −88 kcal/mol, is sufficiently exergonic that H2 heterolysis is favorable. No biophysical evidence exists, however, for unsaturated diferrous state for the enzyme. In fact, Nature might avoid this state because it would be unstable with respect to coordination of the amine. The 2e− change required for exergonic H2 activation can however proceed via a PCET pathway,42 avoiding formation of the Lewis acidic diferrous state. In this scenario, the appended 4Fe-4S cluster is poised to provide the oxidizing equivalent (eq 6). PCET has recently been proposed for other hydrogen redox reactions.3
 |
(6) |
The relevance of PCET pathways is reinforced by the facile transfer of H· from the ammonium and hydride tautomers of the diiron dithiolates, [Fe2[(SCH2)2NHR)](CO)3(dppv)(PMe3)]+ and [HFe2(SR)2(CO)2(dppv)2]+, respectively.
The κ3-aminodithiolate ligand may be relevant to recent observations on the [FeFe]-hydrogenases. Upon being oxidized at ~0 V (vs SHE), the enzyme from D. desulfuricans reversibly deactivates to a state that is protected against oxidative (aerobic) damage.43, 44 It is assumed that this protection is provided by a ligand that occupies the apical site on the distal Fe. Although this blocking ligand might be water or hydroxide,2, 43 the amine could also serve this protective role. To the best of our knowledge, there are no reports that have addressed the feasibility of Fe-N bond formation within the enzyme or whether the cavity in which the H-cluster resides could accommodate an the elongation of the Fe-Fe distance. Although we observe the Fe-Fe distance to increase by ~1 Å in [2′(MeCN)]2+ relative to standard Hox models,12, 13 this may simply be to accommodate the size of the exogenous ligand, MeCN. Other unsaturated diferrous complexes show Fe-Fe distance elongation of only ~0.3 Å.40
Materials and Methods
Synthetic methods for Fe2[(SCH2)2(NBn)(CO)3(dppv)(PMe3)] have been recently described.9 In situ IR measurements employed a React-IR 4000 (Mettler-Toledo). Compounds 245, 2′,9 FcBArF4,46 and H(OEt2)2BArF447 were prepared as previously reported. 2,2,6,6-Tetramethylpiperidine 1-oxyl (TEMPO) and Cp*2Co were purchased from Sigma Aldrich and sublimed before use. Rate constants were obtained by simulation of experimental data using the program Kintecus.48
Protonation of [Fe2[(SCH2)2NBn](CO)3(dppv)(PMe3)]BArF4
To a solution of 0.025 g (0.029 mmol) of 2′ in 5 mL of CH2Cl2 was added a solution of 0.030 g (0.029 mmol) FcBArF24 in 5 mL of CH2Cl2. The resulting purple solution was thermally equilibrated in an acetone/CO2 bath for 10 min.. A solution of 0.014 g (0.015 mmol) H(OEt2)2BArF4 in 2 mL of CH2Cl2 was added to the reaction mixture. The solution immediately became orange in color and the IR spectrum indicated the presence of only [2′](BArF4)2 and [H2′]BArF4 (see Figure 4). The presence of [H2′]+ was confirmed by allowing it to isomerize to its μ-H counterpart. Upon warming to 20 °C, the reaction mixture was filtered through Celite, and the high-field 1H NMR spectrum (CD2Cl2) confirmed the presence of [2′(μ-H)]+. Under analogous conditions, a solution of [1]BArF4 in CH2Cl2 was shown by IR spectroscopy to be unaffected by the addition of H(OEt2)2BArF4, even after warming to 25 °C.
[Fe2[(SCH2)2NBn](CO)3(dppv)(PMe3)](BArF4)2
Into a J.Young NMR tube containing 0.010 g (0.012 mmol) of 2′ and 0.025 g (0.023 mmol) of FcBArF4, immersed in liquid N2, was distilled 1 mL of CD2Cl2. The frozen mixture was thawed in a acetone/CO2 bath and mixed, with care not to let the contents leave the cold bath. The tube was then quickly inserted into a spectrometer probe pre-cooled to −70 °C. Several hundred scans were necessary for a well-resolved spectrum. 31P NMR (CD2Cl2, −70 °C): δ 77.0 (s, dppv), 44.1 (s, PMe3).
[Fe2[(SCH2)NBn](CO)3(dppv)(PMe3)(CD3CN)](BArF4)2
A solution of [2′](BArF24)2 in CD2Cl2 was generated in a J. Young NMR tube as described above. The solution was frozen, re-evacuated, and onto it was distilled 0.1 mL of CD3CN. The tube was thawed in an acetone/CO2 bath and re-inserted into the probe, which was pre-cooled to −70 °C. 31P NMR (CD2Cl2, −70 °C): δ 73.5 (d, dppv, JP-P = 11 Hz, isomer A), 73.0 (s, dppv, isomer A), 20.8 (d, J = 11 Hz, PMe3, isomer A). Chemical shifts vary slightly from the isolated complex due to change in counterion (BF4− vs BArF4−). ESI-MS: m/z 432.9 ([Fe2[(SCH2)2(NBn)(CO)3(dppv)(PMe3)]2+), 454.9, 619.4 ([FeCl(CO)2(dppv)(PMe3)]+), ([Fe2[(SCH2)2NBn](CO)3(dppv)(PMe3)(CD3CN)]2+), 900.7 ([Fe2[(SCH2)2NBn]Cl(CO)3(dppv)(PMe3)]+).
Fe2[(SCH2)2NBn](CO)4(dppv)(PMe3)](BArF4)2 ([2′CO](BArF4)2)
A solution of [2′](BArF4)2 in CD2Cl2 was generated in a J. Young valve NMR tube as described above. The solution was frozen, re-evacuated, and pressurized with 1 atm of CO. The tube was thawed in an acetone/CO2 bath and then slowly warmed to 20 °C. 31P NMR (CD2Cl2, 20 °C): δ 68.4 (m, dppv), 68.2 (m, dppv), 15.7 (d, J = 11 Hz, PMe3). ESI-MS: m/z 446.9 ([Fe2[(SCH2)2NBn](CO)3(dppv)(PMe3)(CO)]2+), 619.4 ([FeCl(CO)2(dppv)(PMe3)]+), 900.7 ([Fe2[(SCH2)2NBn]Cl(CO)3(dppv)(PMe3)]+).
Treatment of [2′](BArF4)2 with PhSiH3
A mixture of 0.050 g (0.058 mmol) [2′](BArF24)2 and 0.121 g (0.116 mmol) FcBArF24 was cooled in an acetone/CO2 bath, and to it was added 2 mL of CH2Cl2. In situ IR spectra indicated the presence of [2′](BArF24)2. IR (CH2Cl2): 2066, 2008, 1977. To this mixture was added 0.2 mL of PhSiH3. After ~30 min, the [2′](BArF4)2 had been consumed. After warming to 20 °C, the sample was found to be spectroscopically (31P, 1H, IR, ESI-MS) identical to [Fe2(μ-H)[(SCH2)2NBn](CO)3(dppv)(PMe3)(CO)]BArF4.9
Oxidation of [Fe2[(SCH2)2N(H)R](CO)3(dppv)(PMe3)]BArF4 with TEMPO (R = H, Bn)
A solution of [2′H]BArF4 was generated at −78 °C by the addition of 0.8 mL of CH2Cl2 to a mixture of 0.023 g (0.027 mmol) of 2′ and 0.027 g (0.027 mmol) of H(OEt2)2BArF4. Treatment of this solution with 0.103 g (0.66 mmol) of TEMPO gave [2′]+. Similar spectra were obtained using [2H]+ in place of [2′H]+. We independently confirmed by IR spectroscopy that [2′H]+ was fully deprotonated by 1 equiv of TEMPOH, the organic product of the H-atom transfer reaction (see eq 3). In a related experiment, we found that exposure of a solution of TEMPOH and [2′H]+ to air rapidly gave [2]+. Precautions were taken to avoid this facile aerobic oxidation pathway.
[Fe2[(SCH2)2NBn](CO)3(dppv)(PMe3)(MeCN)](BF4)2
Obtaining single crystals of salts derived from [2′]2+ proved challenging. Various counterions (BF4−, SbF6−, BArF4−, and BPh4−) and various solvent combinations (slow diffusion at −30 °C of hexanes, Et2O, or toluene into CH2Cl2 solutions of the respective salts of [2′]2+) all afforded amorphous tacky solids. We thus turned to the adduct, [2′(MeCN)]2+. To a Schlenk tube containing a mixture of 0.050g (0.06 mmol) 2′ and 0.032 g (0.12 mmol) of FcBF4, cooled to −78 °C, was added 8 mL of CH2Cl2. The solution was stirred vigorously for 5 min, and then 0.1 mL of MeCN was added. Stirring was stopped, and 40 mL of hexane was carefully layered on top of the reaction mixture and allowed to diffuse at −30 °C. After ~4 days, red crystals had formed. The supernatant was filtered off to remove ferrocene and then the crystalline solid was scrapped from the flask. Finally, this material was dried en vacuo, extracted into 5 mL of CH2Cl2, filtered through Celite, and precipitated as an orange-colored powder upon the addition of 20 mL of hexanes. IR (CH2Cl2, cm−1): νCO = 2065, 2042, 2001, 1974. 31P NMR (CD2Cl2, 20 °C): δ 73.0 (s, dppv, isomer A), 72.9 (d, dppv, JP-P = 13 Hz, isomer A), 21.3 (d, J = 13 Hz, PMe3, isomer A. 31P NMR (CD2Cl2, 48 h at 20 °C): δ 79.9 (s, dppv, isomer B), 25.8 (s, PMe3, isomer B). MS ESI: m/z = 453.2 ([Fe2[(SCH2)2NBn](CO)3(dppv)(PMe3)(MeCN)]2+). The dppv P-Fe-P coupling is not resolved (J<5 Hz), instead the dppv signal is broadened (FWHH = 14 Hz). Anal. Calcd (Found) for C43H45B2F8Fe2N2O3P3S2: C, 47.81 (47.09); H, 4.20 (4.41); N, 2.59 (2.40).
Single crystals were obtained from 8 mL of CH2Cl2 solution of 7 mM [2′](BF4)2, which was generated at −78 °C as described above and then treated with 5 drops of MeCN. The solution was then layered with 50 mL of hexane and stored at −30 °C. After 1 week, several red crystals had appeared. Alternatively, a 7 mM solution of [2′]BF4 was treated with 1 drop of MeCN and then layered with 50 mL of hexane. After 1 week, a single cluster of red crystals had formed and were separated from the dark brown solution.
Crystallography
Structure was phased by dual space methods. Systematic conditions suggested the ambiguous space group P1-. The space group choice was confirmed by successful convergence of the full-matrix least-squares refinement on F2. The highest peak in the final difference Fourier map was located 2.6 Å from the nearest aromatic H atom. This residual density located in a void suggests the possibility of a partially occupied water solvate. The final map had no other significant features. A final analysis of variance between observed and calculated structure factors showed no dependence on amplitude or resolution. The proposed model includes two disordered positions for phenyl ring C20–25 of the host cation and two disordered positions for one of two CH2Cl2 solvate molecules. Phenyl rings were refined as rigid, idealized groups. A common geometry was imposed on the disordered CH2Cl2 solvates using effective standard deviations of 0.01 and 0.02 Å for bond lengths and bond angles, respectively. Rigid-bond restraints (esd 0.01) were imposed on displacement parameters for all disordered sites and similar displacement amplitudes (esd 0.01) were imposed on disordered sites overlapping by less than the sum of van der Waals radii. Methyl H atom positions were optimized by rotation about R-C bonds with idealized C-H, R-H and H---H distances. Remaining H atoms were included as riding idealized contributors. Methyl H atom U’s were assigned as 1.5 times Ueq of the carrier atom; remaining H atom U’s were assigned as 1.2 times carrier Ueq.
Electrochemistry
Cyclic voltammetry experiments were carried out in a ~10 mL scintillation vial inside of an anaerobic dry glove box. The working electrode was a glassy carbon disk (0.3 cm in diameter), the pseudo-reference electrode an Ag wire, and the counter electrode a Pt wire. Under our experimental conditions (CH2Cl2 solution), we generally observed that ΔEp was ~0.12 V for the [Fe2]0/+ couple, whereas an equimolar internal standard of Fc displayed ΔEp as ~0.1 V. All potentials were referenced versus the 0.001 M internal Fc standard.
Supplementary Material
Table II.
Estimated Affinities of [2′]+ for H+, H·, and H− (kcal/mol) in MeCN Solution (20 °C)
| ΔG(H+) | ΔG(H·) | ΔG(H−) |
|---|---|---|
| 15 | −56 | −48 |
Acknowledgments
This research was sponsored by NIH. MTO thanks the NIH for a CBI-Fellowship.
Footnotes
In the absence of the amine, 32e− diferrous dithiolates, e.g. analogues of [Fe2(S2C3H6)(CO)3(dppv)(PMe3)]2+, are predicted to be stabilized by agostic interactions with the central methylene of the dithiolate. We expect that amine binding would be stronger than an agostic interaction, thus stabilizing this oxidation.
Supporting Information Available: Crystallographic information file (cif) for [2′(MeCN)](BF4)2, thermodynamic calculations, and associated spectra.
References
- 1.Kubas GJ. Metal Dihydrogen and σ-Bond Complexes. Kluwer Academic/Plenum; New York: 2001. p. 472. [Google Scholar]
- 2.Vincent KA, Parkin A, Armstrong FA. Chem Rev. 2007;107:4366–4413. doi: 10.1021/cr050191u. [DOI] [PubMed] [Google Scholar]
- 3.Rakowski DuBois M, DuBois DL. Chem Soc Rev. 2009;38:62–72. doi: 10.1039/b801197b. [DOI] [PubMed] [Google Scholar]
- 4.Lubitz W, Reijerse E, van Gastel M. Chem Rev. 2007;107:4331–4365. doi: 10.1021/cr050186q. [DOI] [PubMed] [Google Scholar]
- 5.Silakov A, Wenk B, Reijerse EJ, Lubitz W. Phys Chem Chem Phys. 2009;11:6592–6599. doi: 10.1039/b905841a. [DOI] [PubMed] [Google Scholar]
- 6.Barton BE, Olsen MT, Rauchfuss TB. J Am Chem Soc. 2008;130:16834–16835. doi: 10.1021/ja8057666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peters JW, Lanzilotta WN, Lemon BJ, Seefeldt LC. Science. 1998;282:1853–1858. doi: 10.1126/science.282.5395.1853. [DOI] [PubMed] [Google Scholar]
- 8.Nicolet Y, de Lacey AL, Vernede X, Fernandez VM, Hatchikian EC, Fontecilla-Camps JC. J Am Chem Soc. 2001;123:1596–1601. doi: 10.1021/ja0020963. [DOI] [PubMed] [Google Scholar]
- 9.Olsen MT, Barton BE, Rauchfuss TB. Inorg Chem. 2009;48:7507–7509. doi: 10.1021/ic900850u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tard C, Liu X, Ibrahim SK, Bruschi M, De Gioia L, Davies SC, Yang X, Wang LS, Sawers G, Pickett C. J Nature. 2005;433:610–614. doi: 10.1038/nature03298. [DOI] [PubMed] [Google Scholar]
- 11.Tye JW, Darensbourg MY, Hall MB. Inorg Chem. 2006;45:1552–1559. doi: 10.1021/ic051231f. [DOI] [PubMed] [Google Scholar]
- 12.Justice AK, Rauchfuss TB, Wilson SR. Angew Chem Int Ed. 2007;46:6152–6154. doi: 10.1002/anie.200702224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Justice AK, De Gioia L, Nilges MJ, Rauchfuss TB, Wilson SR, Zampella G. Inorg Chem. 2008;47:7405–7414. doi: 10.1021/ic8007552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu T, Darensbourg MY. J Am Chem Soc. 2007;129:7008–7009. doi: 10.1021/ja071851a. [DOI] [PubMed] [Google Scholar]
- 15.Thomas CM, Liu T, Hall MB, Darensbourg MY. Inorg Chem. 2008;47:7009–7024. doi: 10.1021/ic800654a. [DOI] [PubMed] [Google Scholar]
- 16.Olsen MT, Bruschi M, De Gioia L, Rauchfuss TB, Wilson SR. J Am Chem Soc. 2008;130:12021–12030. doi: 10.1021/ja802268p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheah MH, Tard C, Borg SJ, Liu X, Ibrahim SK, Pickett CJ, Best SP. J Am Chem Soc. 2007;129:11085–11092. doi: 10.1021/ja071331f. [DOI] [PubMed] [Google Scholar]
- 18.Eilers G, Schwartz L, Stein M, Zampella G, De Gioia L, Ott S, Lomoth R. Chem Eur J. 2007;13:7075–7084. doi: 10.1002/chem.200700019. [DOI] [PubMed] [Google Scholar]
- 19.Ezzaher S, Orain P, Capon J, Gloaguen F, Petillon F, Roisnel T, Schollhammer P, Talarmin J. Chem Comm. 2008:2547–2549. doi: 10.1039/b801373j. [DOI] [PubMed] [Google Scholar]
- 20.Barton BE, Zampella G, Justice AK, De Gioia L, Rauchfuss TB, Wilson SR. Dalt Trans. 2009 doi: 10.1039/b910147k. Advance Article. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fontecilla-Camps JC, Volbeda A, Cavazza C, Nicolet Y. Chem Rev. 2007;107:4273–4303. doi: 10.1021/cr050195z. [DOI] [PubMed] [Google Scholar]
- 22.Siegbahn PEM, Tye JW, Hall MB. Chem Rev. 2007;107:4414–4435. doi: 10.1021/cr050185y. [DOI] [PubMed] [Google Scholar]
- 23.Zhao X, Chiang CY, Miller ML, Rampersad MV, Darensbourg MY. J Am Chem Soc. 2003;125:518–524. doi: 10.1021/ja0280168. [DOI] [PubMed] [Google Scholar]
- 24.van der Vlugt JI, Rauchfuss TB, Wilson SR. Chem Eur J. 2005;12:90–98. doi: 10.1002/chem.200500752. [DOI] [PubMed] [Google Scholar]
- 25.Justice AK, Nilges MJ, Rauchfuss TB, Wilson SR, De Gioia L, Zampella G. J Am Chem Soc. 2008;130:5293–5301. doi: 10.1021/ja7113008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kubas GJ. Chem Rev. 2007;107:4152–4205. doi: 10.1021/cr050197j. [DOI] [PubMed] [Google Scholar]
- 27.Landau SE, Morris RH, Lough AJ. Inorg Chem. 1999;38:6060–6068. doi: 10.1021/ic990876a. [DOI] [PubMed] [Google Scholar]
- 28.Henry RM, Shoemaker RK, Dubois DL, Rakowski Dubois M. J Am Chem Soc. 2006;128:3002–3010. doi: 10.1021/ja057242p. [DOI] [PubMed] [Google Scholar]
- 29.Lawrence JD, Li H, Rauchfuss TB, Bénard M, Rohmer MM. Angew Chem, Int Ed. 2001;40:1768–1771. doi: 10.1002/1521-3773(20010504)40:9<1768::aid-anie17680>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 30.Ott S, Kritikos M, Åkermark B, Sun L, Lomoth R. Angew Chem, Int Ed. 2004;43:1006–1009. doi: 10.1002/anie.200353190. [DOI] [PubMed] [Google Scholar]
- 31.Dong W, Wang M, Liu X, Jin K, Li G, Wang F, Sun L. Chem Commun. 2006:305–7. doi: 10.1039/b513270c. [DOI] [PubMed] [Google Scholar]
- 32.Boyke CA, Rauchfuss TB, Wilson SR, Rohmer MM, Bénard M. J Am Chem Soc. 2004;126:15151–15160. doi: 10.1021/ja049050k. [DOI] [PubMed] [Google Scholar]
- 33.Boyke CA, van der Vlugt JI, Rauchfuss TB, Wilson SR, Zampella G, De Gioia L. J Am Chem Soc. 2005;127:11010–11018. doi: 10.1021/ja051584d. [DOI] [PubMed] [Google Scholar]
- 34.van der Vlugt JI, Rauchfuss TB, Whaley CM, Wilson SR. J Am Chem Soc. 2005;127:16012–16013. doi: 10.1021/ja055475a. [DOI] [PubMed] [Google Scholar]
- 35.Geiger WE, Barriere F. Acc Chem Res. 2010;43:1030–1039. doi: 10.1021/ar1000023. [DOI] [PubMed] [Google Scholar]
- 36.Heiden ZM, Rauchfuss TB. J Am Chem Soc. 2009;131:3593–3600. doi: 10.1021/ja807277s. [DOI] [PubMed] [Google Scholar]
- 37.Li T, Lough AJ, Morris RH. Chem Eur J. 2007;13:3796–3803. doi: 10.1002/chem.200601484. [DOI] [PubMed] [Google Scholar]
- 38.Anelli PL, Biffi C, Montanari F, Quici S. J Org Chem. 1987;52:2559–2562. [Google Scholar]
- 39.Sen VD, Golubev VA. J Phys Org Chem. 2008;22:138–143. [Google Scholar]
- 40.Bruschi M, Fantucci P, De Gioia L. Inorg Chem. 2003;42:4773–4781. doi: 10.1021/ic0262132. [DOI] [PubMed] [Google Scholar]
- 41.Geiger WE, Ohrenberg NC, Yeomans B, Connelly NG, Emslie DJH. J Am Chem Soc. 2005;125:8680–8688. doi: 10.1021/ja0302073. [DOI] [PubMed] [Google Scholar]
- 42.Mayer JM. Annual Review of Physical Chemistry. 2004;55:363–390. doi: 10.1146/annurev.physchem.55.091602.094446. [DOI] [PubMed] [Google Scholar]
- 43.Vincent KA, Parkin A, Lenz O, Albracht SPJ, Fontecilla-Camps JC, Cammack R, Friedrich B, Armstrong FA. J Am Chem Soc. 2005;127:18179–18189. doi: 10.1021/ja055160v. [DOI] [PubMed] [Google Scholar]
- 44.van Dijk C, van Berkel-Arts A, Veeger C. FEBS Lett. 1983;156:340–344. [Google Scholar]
- 45.Justice AK, Zampella G, De Gioia L, Rauchfuss TB, van der Vlugt JI, Wilson SR. Inorg Chem. 2007;46:1655–1664. doi: 10.1021/ic0618706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Le Bras J, Jiao H, Meyer WE, Hampel F, Gladysz JA. J Organomet Chem. 2000;616:54–66. [Google Scholar]
- 47.Brookhart M, Grant B, Volpe AF., Jr Organometallics. 1992;11:3920–3922. [Google Scholar]
- 48.Ianni J. Kintecus. 3.962 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.