

Abstract
Retroviral vectors based on human foamy virus (HFV) have been developed and show promise as gene therapy vehicles. Here we describe a method for the production of HFV vector stocks free of detectable helper virus. The helper and vector plasmid constructs used both lack the HFV bel genes, so recombination between these constructs cannot create a wild-type virus. A fusion promoter that combines portions of the cytomegalovirus (CMV) immediate-early and HFV long terminal repeat (LTR) promoters was used to drive expression of both the helper and vector constructs. The CMV–LTR fusion promoter allows for HFV vector production in the absence of the Bel-1 trans-activator protein, which would otherwise be necessary for efficient transcription from the HFV LTR. Vector stocks containing either neomycin phosphotransferase or alkaline phosphatase reporter genes were produced by transient transfection at titers greater than 105 transducing units/ml. G418-resistant BHK-21 cells obtained by transduction with neo vectors contained randomly integrated HFV vector proviruses without detectable deletions or rearrangements. The vector stocks generated were free of replication-competent retrovirus (RCR), as determined by assays for LTR trans-activation and a marker rescue assay developed here for the detection of Bel-independent RCR.
OVERVIEW SUMMARY
Vectors based on human foamy virus have been developed but low titers and the presence of replication-competent retrovirus (RCR) in vector stocks have prevented their use in preclinical animal experiments. We have developed a transient transfection method that can be used to produce replication-incompetent HFV vector stocks at titers greater than 105/ml, and that does not produce contaminating RCR. The use of CMV-HFV LTR fusion promoters in the helper and vector constructs has circumvented the requirement for the HFV Bel-1 trans-activator protein. Consequently, the potential for generating wild-type HFV by recombination between helper and vector constructs during vector production has been eliminated. Here we describe HFV vector production using this Bel-independent system.
INTRODUCTION
HUMAN FOAMY VIRUS (HFV) is a complex retrovirus of the spumavirus family that is not known to cause disease in humans. The virus was originally isolated from a human nasopharyngeal carcinoma cell line (Achong et al., 1971), but a more recent study failed to detect HFV in humans, and the virus is now considered a chimpanzee virus variant (Schweizer et al., 1995). The foamy viruses have several unique properties that set them apart from other retroviruses. In addition to gag, pol, and env genes, HFV contains the bel genes (between the envelope and the 3′ LTR [long terminal repeat]) (Flugel et al., 1987). The bel1 gene is transcribed from an internal promoter in the env gene (Lochelt et al., 1993a), and encodes a transcriptional trans-activator protein that induces transcription from both the LTR and the internal promoter (Keller et al., 1991; Rethwilm et al., 1991; Kang et al., 1998). Bel1 is essential for viral replication (Lochelt et al., 1991), while the functions of the bel2 and bel3 genes are unknown and these genes are dispensable for viral replication in vitro (Baunach et al., 1993; Yu and Linial, 1993). Other unique features of foamy viruses include a Pol protein that is produced independently of Gag from a spliced mRNA transcript (Enssle et al., 1996; Jordan et al., 1996; Lochelt and Flugel, 1996), and the presence of significant amounts of full-length cDNA molecules in gradient-purified extracellular virions (Yu et al., 1996).
Viral vectors based on HFV have been developed (Schmidt and Rethwilm, 1995; Hirata et al., 1996; Russell and Miller, 1996; Bieniasz et al., 1997; Nestler et al., 1997) and have several potential advantages. HFV vectors are able to transduce a wide variety of vertebrate cells (Russell and Miller, 1996; Nestler et al., 1997) including hematopoietic progenitor cells (Hirata et al., 1996), and they also transduce quiescent cells more efficiently than do murine leukemia virus (MLV) vectors (Russell and Miller, 1996). These vectors transduce by integration into the host genome and have a larger packaging capacity than other retroviral vectors. Furthermore, HFV vectors are not inactivated by human serum and do not require polycations for efficient transduction (Russell and Miller, 1996), so they may be well suited for in vivo gene delivery.
Previous HFV vector production methods either relied on the presence of replication-competent retrovirus (RCR) or had the potential to generate wild-type (wt) HFV by recombination. Replication-incompetent HFV vectors were produced by transient transfection of BHK-21 cells at titers in excess of 104/ml with a helper construct that contained a wt HFV provirus (Hirata et al., 1996; Russell and Miller, 1996). Vector titers were approximately 10-fold lower when a helper construct with a deleted provirus was used in the same system, and recombination between vector and helper constructs led to contaminating RCR production (Russell and Miller, 1996). Replication-competent HFV vectors have also been produced by transient transfection and subsequent vector amplification, with titers in excess of 105/ml (Schmidt and Rethwilm, 1995). These vector production methods have proven useful for HFV vector studies; however, for most gene therapy applications it would be preferable to eliminate RCR from vector stocks. Producer cell lines that rely on Bel1 expression in trans and produce more than 105 transducing units/ml have been described by Bieniasz et al. (1997). However, these authors noted that in some cases RCR was still generated, presumably by recombination between vector constructs and the integrated bel1 gene present in the producer cells. We describe here a method for the production, in the absence of the Bel proteins, of helper-free HFV vector stocks with titers greater than 105/ml.
MATERIALS AND METHODS
Cell culture and virus production
The cell lines used included human embryonic kidney 293 cells (ATCC CRL-1573), simian virus 40 (SV40) T antigen-transformed 293T cells (DuBridge et al., 1987), NIH 3T3 TK− mouse fibroblasts (Wei et al., 1981), human fibrosarcoma HT-1080 cells (Rasheed et al., 1974), SV40-transformed African green monkey kidney COS-1 cells (ATCC CRL-1650), baby hamster kidney BHK-21 cells (Macpherson and Stoker, 1962), normal human foreskin fibroblasts (Palmer et al., 1987), and FAB hamster HFV β-galactosidase (β-Gal) indicator cells (Yu and Linial, 1993). All cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum (FBS). FBS was heat inactivated at 56°C. The wt HFV used as a positive control in the marker rescue assay was generated by CaPO4 transfection of BHK-21 cells with the pHSRV13 infectious clone (Lochelt et al., 1991). Virus-containing supernatants were harvested 72 hr after transfection and passed through 0.45-μ m pore size filters.
Plasmid constructs
The HFV Bel-independent plasmids (see Fig. 1) were constructed by standard techniques and were based on the pH-SRV13 foamy virus infectious clone (Lochelt et al., 1991). The pCGPES helper construct contains the following genetic elements; a ColE1-type plasmid backbone and cytomegalovirus (CMV) promoter (nucleotides [nt] 2208–4455 and nt 1–782 of GenBank U89929), the R-U5-gag-pol-env region of HFV (nt 765–9483 of GenBank U21247), and an SV40 polyadenylation signal (complement of nt 2683–2533 of GenBank J02400). The pCGPMAPΔBel vector construct was engineered so that the HFV LTR transcription start site (Maurer et al., 1988) was situated the same distance from the CMV TATA box as it normally is from the HFV LTR TATA box. This was achieved by inserting an XbaI site 9 bp downstream from the CMV promoter TATA box by using polymerase chain reaction (PCR) primer mutagenesis. The pCGPMAPΔBel construct contains the following regions: a ColE1-type plasmid backbone and CMV promoter (nt 2390–4255 and nt 1–672 of GenBank U89929, respectively), the HFV R-U5-gag-pol and partial env region (nt 765–6957 of GenBank U21247), a fragment of the MLV LTR (nt 7847–8296 of GenBank J02255), the human placental alkaline phosphatase (AP) gene (nt 1–1806 of GenBank M12551), and the HFV partial bel3 and LTR region (nt 10744–11954 of GenBank U21247). pCGPSNΔBel is derived from pCGPMAPΔBel with an SV40 early promoter (complement of nt 164–1 and nt 5243–5170 of GenBank J02400) and a Tn5-derived neo gene (nt 505–1668 of GenBank U32991) in place of the MLV LTR-AP cassette.
FIG. 1.

Bel-independent HFV vector constructs. (A) Map of the wt HFV provirus pHSRV13, indicating sites of transcriptional transactivation by the Bel1 protein. The positions of the LTRs and of the gag, pol, env, and bel genes are indicated. (B) Maps of the Bel-independent vector constructs engineered with fusion promoters consisting of the CMV promoter in place of the HFV LTR 5′ U3 region. Transcription is initiated at the R region of the LTR, independently of Bel1. The engineered XbaI site used to ligate the CMV and HFV promoter elements is underlined and the reported HFV start site is indicated by an asterisk above the pCGPMAPΔBel construct. The positions of the CMV fusion promoter, MLV LTR promoter (M), alkaline phosphatase gene (AP), SV40 early promoter (S), and neo gene are indicated. (C) Map of the pCGPES helper construct, showing the locations of the CMV–HFV LTR fusion promoter (sequence slightly different than that of pCGPMAPΔBel), HFV gag, pol, and env genes, and the SV40 polyadenylation site (SpA). (D) Structure of integrated vector provirus resulting from transduction of Bel-independent vectors. The wt HFV 5′ LTR is regenerated by reverse transcription prior to integration. The locations of relevant restriction sites and the probe used for Southern analysis are shown. Transcription start sites are indicated by arrows.
Vector production
All vector stocks were prepared by CaPO4-mediated transfection of 293T cells unless otherwise noted. Cells were plated at a density of 2.5 ×105 cells in 2.5 ml per 35-mm well in six-well plates and allowed to grow overnight. Transfection mixes consisting of 10 μ g of total DNA in 50 μ l of 2.0 M CaCl2 and 350 μ l of 0.1×TE (10 mM Tris [pH 8.0], 1 mM EDTA) added dropwise to 400 μ l of 2×HEPES–saline (280 mM NaCl, 50 mM HEPES [pH 7.1]) containing 4 μ l of 0.15 M Na2HPO4 (pH 7.1) were prepared, and these transfection mixes were allowed to stand for 20 min at room temperature. Eight hundred microliters of the final mixture was added dropwise to the plated cells. Seven hours later, the medium was changed to fresh DMEM with 10% FBS and supernatants containing HFV vectors were collected 65 hr later. Supernatants (vector-containing media) were collected and passed through 0.45-μ m pore size cellulose acetate filters before being assayed. All experiments included a control consisting of untransfected cells. In addition, a control consisting of cells transfected with the vector construct alone was used to confirm that AP focus-forming units (FFU) or G418-resistant colonies were not due to uptake of reporter proteins present in the stock or to CaPO4-mediated transfer of residual vector construct DNA.
Transduction assays
AP vector stocks were titered on the FAB cell line (Yu and Linial, 1993), which allows quantitation of potential wt contamination and AP transducing units in the same well. Some stocks were also titered on normal human fibroblasts and HT-1080 cells. Vector stocks were added to 35-mm-diameter wells that had been plated the day before with 1.5 ×105 cells. Forty hours later the cells were fixed and transduction was measured by histochemical staining for both β-Gal and AP (Fields-Berry et al., 1992) and by counting of individual stained cell foci. neo vector stocks were titered as follows. BHK-21 cells were plated at 1.5 ×105 cells in a 35-mm-diameter well and cultured overnight. Serial dilutions of vector supernatants were added to the cells the following morning and 24 hr later the cells were treated with trypsin and plated at 1:20 and 1:100 dilutions into 10-cm dishes. These cells were cultured for an additional 24 hr, and then G418 was added to a final concentration of 0.7 mg/ml. Surviving colonies were counted 8 days later after staining with Coomassie Brilliant Blue G, when all cells in control cultures were dead.
Helper virus assays
The FAB assay (Yu and Linial, 1993) was performed by plating 1.5 ×105 FAB cells in a 35-mm-diameter well, adding dilutions of vector stocks or viral supernatants the following day, and determining the number of β-Gal focus-forming units (BFFU) 40 hr later by histochemical staining. The FMR (foamy marker rescue) cell line was generated by CaPO4-mediated transfection of BHK-21 cells with the pCGPSN-ΔBel construct followed by selection of G418-resistant colonies in G418 (0.7 mg/ml). To screen these G418-resistant clones for rescuable neo vector, 10 independent colonies were expanded to 1 ×105 cells and infected with wt HFV at a multiplicity of infection (MOI) of 0.1. Forty-eight hours after infection supernatants were harvested, passed through 0.45-μ m pore size filters, and rescued neo vectors were titered on BHK-21 cells. One cell line (named FMR) contained rescuable neo vector and was used for marker rescue assays. The marker rescue assay was performed by plating 1 ×105 FMR cells in a 12-well dish, and then adding serial dilutions of wt HFV (positive control), AP vector supernatants, or wt HFV spiked with AP vector supernatant the following morning. The serial dilutions of wt HFV were also concurrently added to FAB cells to determine the titer. After 5 days, supernatants from the infected FMR cells were passed through 0.45-μ m pore size filters, and 1.5 ml was transferred to BHK-21 cells. Twenty-four hours later the cells were treated with trypsin and 9:10 and 1:10 dilutions were plated into 10-cm dishes. These dilutions were cultured for a further 24 hr, and then G418 was added to a final concentration of 0.7 mg/ml and surviving colonies were counted 8 days later.
Analysis of integrated HFV vectors
Supernatants containing pCGPSNΔBel neo vectors were used to transduce BHK-21 cells at an MOI of 0.01. Transduced cells were selected in G418 (0.7 mg/ml) and 10 independent G418-resistant colonies were expanded to approximately 107 cells in a 10-cm dish. High molecular weight genomic DNA was extracted (Gross-Bellard et al., 1973) and digested with either EcoNI or EcoRI. Approximately 5 μ g of digested DNA was analyzed by Southern blot (Maniatis et al., 1989), using a 1.4-kb probe containing portions of the SV40 promoter and the neo gene.
RESULTS
Production of Bel-independent HFV vectors
To increase HFV vector titers over those produced by previous methods, we designed helper and vector constructs with a CMV-HFV LTR fusion promoter. The strong CMV promoter is especially potent in the 293T cell line, and CMV-driven expression of MLV vectors has resulted in vector titers in excess of 106 transducing units/ml by transient transfection of 293T cells (Pear et al., 1993; Soneoka et al., 1995). Use of the CMV promoter also made possible the removal of the HFV bel genes from both the helper and vector constructs, so that vector production was Bel-independent (see Fig. 1). The pCGPES helper construct expresses Gag, Pol, and Env under control of the CMV promoter and provides these proteins in trans for vector assembly. This construct has a deletion of the bel genes and cannot complete reverse transcription owing to a deletion of the 3′ LTR, which is replaced by an SV40 polyadenylation signal. The pCGPMAPΔBel vector construct is also under the control of the CMV promoter, and was designed such that transcription initiates from the HFV transcription start site. During reverse transcription of the vector genome the 5′ HFV LTR is regenerated from the 3′ LTR, resulting in a vector provirus with two HFV LTRs and no CMV promoter sequences. The pCGPMAPΔBel vector construct has deletions of the env, bel1, bel2, and bel3 genes, and contains a human placental alkaline phosphatase (AP) reporter gene under the control of an internal MLV LTR promoter. The absence of the bel genes in this system means that recombination between the helper and vector construct cannot regenerate wt HFV. In addition, integrated Bel-independent vectors should have only minimal expression from the HFV LTRs, which are not efficiently transcribed in the absence of Bel1 (Rethwilm et al., 1991; Lochelt et al., 1993a).
The pCGPES and pCGPMAPΔBel constructs were used to optimize the transient transfection protocol for generating vector stocks. Stocks were prepared by cotransfecting the pCGPES and pCGPMAPΔBel plasmids into 293T cells and harvesting cell-free supernatants containing HFV AP vector virions. Vector supernatants were used to infect FAB indicator cells and 40 hr later the vector titer was determined by counting AP foci after histochemical staining. The FAB cell line was used because it allows for a convenient determination of vector titer and potential wt contamination in the same cell monolayer (see below). Production of Bel-independent AP vectors by transient transfection was compared in several cell lines (Fig. 2) and AP titers were determined. Transfections performed in the 293T cell line produced the highest titers, which were in excess of 1 ×105 AP transducing units/ml; this cell line was therefore used in subsequent experiments. The amounts of helper and vector constructs present in the transfection mix, and the density of the transfected cells, were varied to optimize the transfection protocol. A cell density of 2.5 ×105 293T cells/35-mm well transfected 1 day later with 5 μ g each of plasmids pCGPES and pCGPMAPΔBel produced the highest vector titers (data not shown). There was considerable toxicity evident 48 hr posttransfection, as evidenced by syncytia and “balloon-like” cell clusters, which were associated with higher vector titers. A time course of vector production was performed and the highest titers were obtained at 72 hr posttransfection (Fig. 3). The average titer of 29 independent vector stocks generated by this optimized procedure was 1.7 ×105 ± 3.0 ×104 AP FFU/ml (standard error) on FAB cells. Three vector stocks were also titered on normal human fibroblasts and HT-1080 human fibrosarcoma cells. The titer on these human cells was two- and three-fold higher, respectively, than on FAB cells, consistent with a previous study comparing HFV vector transduction rates on different cell types (Russell and Miller, 1996).
FIG. 2.
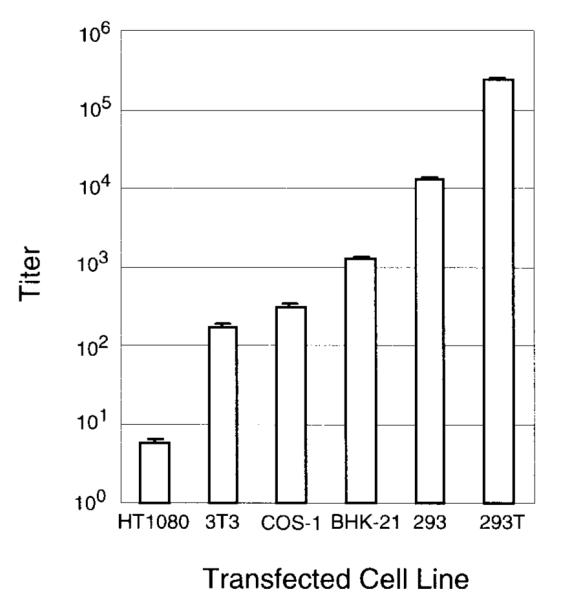
Vector production in various cell lines. The helper plasmid pCGPES and vector plasmid pCGPMAPΔBel were cotransfected into the indicated cell lines and cell-free supernatants were harvested 72 hr later, filtered, and titered on FAB cells. Values shown are titers (AP FFU/ml) with means and standard errors from three independent measurements for each cell line. In some cases, standard errors were too small to be displayed in the figure. Controls of untransfected cells and cells transfected with vector plasmid pCGPMAPΔBel alone produced no AP transducing units (<1 AP FFU/ml). Helper virus contamination was not detected by FAB assay (<1 BFFU/ml).
FIG. 3.
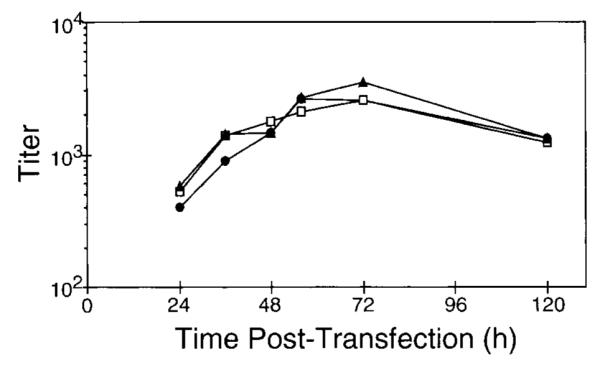
Time course of vector production by transient transfection of 293T cells. Three independent transfections were performed with plasmids pCGPES and pCGPMAPΔBel, and at 24, 36, 48, 56, 72, and 120 hr 100 μ l of vector-containing medium was removed from each well and frozen at −80°C until assayed. Samples were then thawed together at room temperature and titered on FAB cells. Controls of cells transfected with vector plasmid alone were negative (<1 AP FFU/ml). Values shown are titers (AP FFU/ml), with each symbol type representing a different transfection.
In an attempt to improve the yield of functional vector particles various harvesting methods were compared (Fig. 4). In each case the cell-free vector stocks were passed through a 0.45-μ m pore size filter before titration. Freshly harvested supernatants contained the highest vector titers, and freeze–thawing of stocks reduced titers significantly. Freeze–thawing vector-containing culture medium in the presence of the transfected cells reduced the loss of vector titer due to freezing, presumably owing to a release of intracellular virions that compensated for the damage to vector virions caused by freezing and thawing. Previous studies with wt HFV have shown that the bulk of the virus is cell associated and can be released after multiple freeze–thaw cycles (Yu and Linial, 1993). Three freeze–thaw cycles did not significantly improve the recovery of vector in this transient transfection system, nor did the addition of 40% glycerol.
FIG. 4.
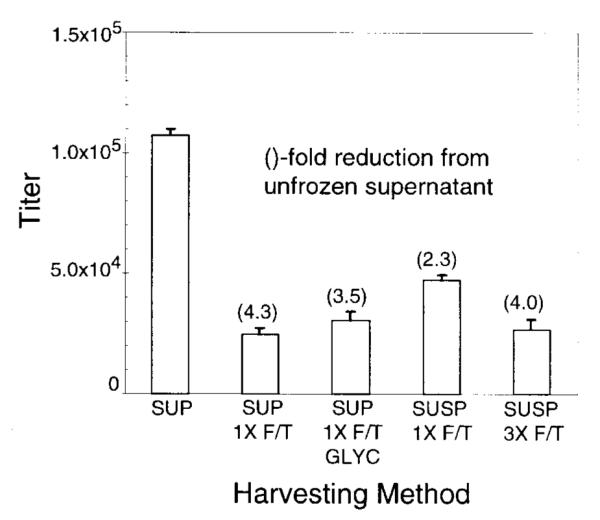
Comparison of vector harvest methods. 293T cells were transfected with both the helper construct pCGPES and vector construct pCGPMAPΔBel, and 72 hr later supernatants (vector-containing medium) or suspensions of cells and supernatants were harvested as follows and titered on FAB cells. Harvest conditions included supernatant filtered without freezing (SUP); filtered supernatant that was frozen at −80°C for 30 min, then thawed at room temperature (SUP 1×F/T); filtered supernatant with glycerol added to 40% (v/v) that was frozen at −80°C for 30 min, then thawed at room temperature (SUP 1×F/T GLYC); supernatant combined with a suspension of the transfected cells that was frozen at −80°C for 30 min, thawed at room temperature, then filtered (SUSP 1×F/T); or supernatant combined with a suspension of the transfected cells that was frozen in an ethanol–dry ice bath and thawed at 37°C three times, then filtered (SUSP 3×F/T). Controls of untransfected cells or cells transfected with vector plasmid pCGPMAPΔBel alone produced no AP FFU (<1/ml). Supernatant from a control well transfected with the same transfection mixture used for all treatments was tested by FAB assay and did not contain Bel-dependent RCR (<1 BFFU/ml). Mean titer values (AP FFU/ml) with standard errors from three independent measurements are plotted. Values in parentheses indicate the fold reduction as compared with unfrozen supernatants.
Bel-independent vector stocks are free of replication-competent helper virus
Removal of the bel genes from the helper and vector constructs makes it impossible for a wt virus to be generated by recombination between the helper and vector plasmids. We used the FAB cell line (Yu and Linial, 1993) to confirm that the Bel-independent vector stocks did not contain contaminating wt HFV. This cell line has an integrated HFV LTR upstream of a nuclear-localizing β-gal reporter gene, so after infection by wt HFV the integrated HFV LTR is activated by the Bel1 protein and β-Gal is produced. β-Gal-producing cells can be detected by histochemical staining 40 hr postinfection (BFFU). We performed the FAB assay on more than 40 different vector stocks produced by transfection of pCGPES and pCGPMAPΔBel, and no BFFU were detected (<1/ml of vector stock). This was the expected result from a Bel-free vector production system. It is possible that a new foamy-like RCR could have been generated during vector production even in the absence of Bel1 by capture of a cellular trans-activator gene that acts on the HFV LTR. The negative FAB assay also demonstrates that this did not occur, as the integrated HFV LTR in the FAB cell line would have led to β-Gal production in response to any potential exogenous trans-activator.
Another possibility is that a novel LTR could be generated during stock production that can be transcribed independently of Bel1, thus generating a new type of replication-competent virus. This novel LTR could take the form of a mutated HFV LTR, or a non-HFV promoter such as a new LTR based on the CMV promoter. These Bel-independent RCRs would not be detected by the FAB assay. To test for this possibility we developed a marker rescue assay that could detect Bel-independent RCR. A cell line was generated that contains an integrated HFV vector genome under the control of the constitutive CMV promoter and a neo reporter gene driven by the SV40 early promoter. This integrated vector genome also contains functional HFV gag and pol genes, but does not express the bel or env genes. A potential Bel-independent RCR that infected this cell line would supply Gag, Pol, and Env in trans and package the CMV-driven neo vector transcripts into virions that could then be detected by a transduction assay (Fig. 5). In theory, a replication-incompetent virion expressing only Env should also lead to rescue (since Gag and Pol are encoded by the vector construct). To generate this cell line, the pCGPSNΔBel plasmid (see Fig. 1) was transfected into BHK-21 cells and stable integrants were selected in medium containing G418 and tested to determine if neo-expressing vectors could be rescued by infecting with wt HFV (see Materials and Methods). One cell line named FMR contained neo vector integrants that could be efficiently rescued by wt HFV. The sensitivity of this cell line was tested using wt HFV as a positive control in three separate experiments (Table 1), which produced positive assays with 92, 3, or 82 BFFU of wt HFV, respectively. In experiment 3 an additional control of wt HFV spiked with vector was performed to determine if vector particles would interfere with RCR detection. Some variation in the sensitivity of the assay was expected as amplification of RCR during the 5-day culture period could occur at variable rates, and the FAB assay used to titer the wt HFV stock could overestimate the true RCR titer as it only measures transfer of a functional bel1 gene and not virus replication. Using this FMR assay, rescue of potential RCR was not detected from 1 ml each of nine independently generated AP vector stocks (average titer, 4.5 ×104 AP FFU/ml). The addition of vector stocks to the wt HFV positive control for RCR (experiment 3, Table 1) did not decrease the sensitivity of the assay. These experiments show that the Bel-independent vector stocks described here are free of detectable RCR that can package an HFV vector transcript.
FIG. 5.

Foamy marker rescue assay. An FMR cell is depicted on the left with its integrated, env-deficient neo vector provirus. On the right an FMR cell is shown with both RCR and vector proviruses after infection by an env+ RCR virion. This cell produces additional RCR particles that can spread through the culture, and neo vector particles that can transduce BHK-21 cells. The positions of gag, pol, env, and neo genes in the proviruses are indicated. The CMV-HFV LTR fusion promoter (C|F), the HFV LTR (F), and a novel Bel-independent LTR (X) are indicated.
Table 1.
Testing Of Vector Stocks For Replication-Competent Retrovirus, Using The Foamy Marker Rescue Cell Line
| Exp. No. | Vector titer (AP foci/ml)a |
wt HFV dose (BFFU)b |
RCR (+/−) |
G418-resistant coloniesc |
|---|---|---|---|---|
| 1 | 3.0 × 104 | NA | − | 0 |
| 6.3 × 104 | NA | − | 0 | |
| 3.0 × 104 | NA | − | 0 | |
| NA | 920 | + | 20 | |
| NA | 92 | + | 1 | |
| NA | 9 | − | 0 | |
| NA | 1 | − | 0 | |
| NA | 0 | − | 0 | |
| 2 | 8.1 × 104 | NA | − | 0 |
| 6.3 × 104 | NA | − | 0 | |
| 3.2 × 104 | NA | − | 0 | |
| NA | 280 | + | >300 | |
| NA | 28 | + | 55 | |
| NA | 3 | + | 1 | |
| NA | 0 | − | 0 | |
| 3 | 3.6 × 104 | NA | − | 0 |
| 2.3 × 104 | NA | − | 0 | |
| 4.4 × 104 | NA | − | 0 | |
| NA | 820 | + | 17 | |
| NA | 82 | + | 5 | |
| NA | 8 | − | 0 | |
| NA | 1 | − | 0 | |
| NA | 0 | − | 0 | |
| 3.4 × 104 | 820 | + | 44 | |
| 3.4 × 104 | 82 | + | 6 | |
| 3.4 × 104 | 8 | − | 0 | |
| 3.4 × 104 | 1 | − | 0 | |
| 3.4 × 104 | 0 | − | 0 |
Each assay was performed on 1 ml of vector stock with the indicated titer.
Doses were 10-fold serial dilutions made from wt HFV stocks that were titered at the same time on FAB cells. Numbers shown are based on the calculated FAB titer determined at that time.
Total number of G418-resistant colonies produced from the inoculum assayed.
Abbreviation: NA, Not applicable.
Analysis of Bel-independent vector integrants
It was shown previously that HFV vectors integrate randomly into genomic DNA (Russell and Miller, 1996). Although the bel2 and bel3 genes are not necessary for viral replication (Baunach et al., 1993; Yu and Linial, 1993) and the Bel1 protein can be supplied in trans, we wanted to confirm that Bel-independent vectors also integrate and determine whether the vector proviruses were deleted or rearranged. Vector stocks were generated by transient transfection of 293T cells with the pCGPSNΔBel plasmid (see Fig. 1) and the helper construct pCGPES at a titer of 1.5 ×105 G418-resistance transducing units/ml. This pCGPSNΔBel vector stock was used to transduce BHK-21 cells at an MOI of 0.01 and 10 independent G418-resistant colonies were isolated and expanded. Genomic DNA from each clone was extracted and digested with either EcoNI or EcoRI. EcoNI cleaves twice in the vector construct, releasing a 7.7-kb fragment (see Fig. 1D), while EcoRI cleaves once in the vector and produces a fragment that varies in size in relation to the nearest flanking chromosomal EcoRI site. An SV40-neo probe (see Fig. 1) was used to detect proviral fragments by Southern analysis. All 10 clones contained integrated proviruses of the predicted size, indicating that gross vector rearrangements or deletions did not occur at a significant frequency (Fig. 6). The 10 proviruses were integrated randomly as shown by the variation in size of the neo-containing EcoRI fragment.
FIG. 6.

Southern analysis of integrated Bel-independent vector genomes. High molecular weight DNAs isolated from 10 independent G418-resistant clones of BHK-21 cells transduced by pCGPSNΔBel (lanes 1–10) or untransduced BHK-21 cells were digested with the indicated enzymes and probed for neo sequences. Samples were digested with either EcoNI, which produces a 7.7-kb fragment from an intact provirus (A) or with EcoRI, which cuts once within the vector (B). Uneven loading decreased the vector signal in lanes 1 and 3 (A) and lane 10 (B). See Fig. 1 for restriction sites and probe map.
DISCUSSION
We previously generated HFV vectors at titers in excess of 104 transducing units/ml and demonstrated that they can transduce many cell types including hematopoietic progenitor cells (Hirata et al., 1996; Russell and Miller, 1996). These first-generation HFV vector stocks contained relatively high amounts of contaminating helper virus. Bieniasz et al. (1997) reported the production of replication-defective HFV vectors in Bel1-expressing producer cell lines at titers exceeding 105 transducing units/ml. However, bel1-containing RCR was generated in some producer cell clones, which presumably arose from recombination between vector sequences and the integrated bel1 gene in the producer cell line. Here we have described the use of CMV-HFV LTR fusion promoters in both the helper and vector constructs, which allowed us to remove completely the bel1 gene from the vector production system and express helper and vector transcripts from the strong CMV promoter in transiently transfected 293T cells. We have now produced high-titer HFV vector stocks that are free of Bel-dependent and Bel-independent RCR.
Previous investigators obtained high titers of MLV vectors using analogous CMV-MLV fusion promoters in 293-based cell lines (Pear et al., 1993; Soneoka et al., 1995; Ory et al., 1996). The use of a CMV-HFV LTR fusion promoter has also been reported for production of wt HFV and HFV vector constructs; however, transduction by Bel-independent vectors was not investigated. Moebes et al. (1997) reported the use of a CMV-LTR fusion promoter to produce a replication-competent HFV by transient transfection of 293T cells. In this system a CMV-driven HFV construct containing the bel genes was used to generate a wt HFV stock at 103 BFFU/ml as determined by FAB assay. It is not clear why the titers of our CMV-driven vector stocks were higher than those reported previously, but small differences in the sequence of the fusion promoter could play a role. In addition, a report by Fischer et al. (1998) showed that bel-deleted vectors containing the same CMV-HFV LTR fusion promoter reported by Moebes et al. (1997) could produce HFV particles in 293T cells as determined by electron microscopy (EM). However, vector titers were not examined in this study. We have measured the titers of vectors produced by our Bel-independent system, and found that both neo and AP vectors could be generated at titers in excess of 105/ml.
The transient transfection protocol for producing Bel-independent vectors was optimized and the highest titers were obtained at 72 hr posttransfection, when a relatively low density of 293T cells was transfected and large transfection volumes were used. This results in considerable cytotoxicity to the transfected cell monlayer, presumably owing to a combination of cell death from the calcium phosphate precipitate and cell fusion due to HFV Env expression. Pear et al. (1993) also noted that doubling the volume of transfection reagents increased the titer of MLV vectors produced using a CMV-MLV LTR fusion promoter in 293T cells. The cell-free HFV vector stocks produced by this transient transfection method are sensitive to freeze–thaw damage, with an average 4.3-fold drop in titer. Freezing in the presence of transfected cells and filtering the stocks after thawing reduced the loss of titer, presumably owing to the release of intracellular vector virions.
The pCGPSNΔBel vector integrated randomly in BHK-21 cells and no rearrangements of the vector construct were detected in 10 of 10 G418-resistant transductants analyzed. This contrasts with our previous integration analysis of vector stocks produced with helper constructs containing a simple deletion in the gag and pol genes (Russell and Miller, 1996). These first-generation vector production methods frequently resulted in rearranged vector proviruses due to recombination between helper and vector constructs. Presumably these rearrangements were avoided in the Bel-independent system described here because the removal of the 3′ LTR in the pCGPES helper construct prevented the completion of reverse transcription from recombined genomes, even if they were packaged into virions. Previous studies have shown that the bel2 and bel3 genes are dispensable for virus production in vitro (Baunach et al., 1993; Yu and Linial, 1993; Schmidt and Rethwilm, 1995), and that mature foamy virus particles can be detected by electron microscopy in the absence of all bel genes (Fischer et al., 1998). Our results further demonstrate that the bel genes are not required for integration of viral genomes.
The removal of the Bel1 transcriptional trans-activator from this vector system eliminates RCR contamination and leads to improved transduction properties. Both the FAB assay (Yu and Linial, 1993) and a marker rescue assay failed to detect RCR in Bel-independent vector stocks. The FMR assay was sensitive to 3, 92, or 82 BFFU/ml in three separate experiments using wt HFV as a positive control, and should have detected both RCR and replication-incompetent virions expressing Env, including Bel-independent RCRs. Thus we conclude that the cotransfection method described here reproducibly generates helper-free stocks. The ability to generate high-titer vector stocks free of RCR should simplify the testing of HFV vectors in animal gene transfer studies, since transduction can be measured in the absence of viral replication. Although foamy viruses are not known to be pathogenic, the absence of RCR eliminates any unknown risks that might be associated with foamy virus infection. The removal of the bel genes adds an additional level of safety, since transcription should not occur from the LTRs of integrated vector proviruses, thereby preventing activation of downstream cellular genes. There could also be sites in cellular chromosomes where the Bel1 protein binds and influences transcription, which will not occur in a Bel-free vector system. Finally, the transcriptionally silent vector LTRs may allow improved engineering of the transcriptional control of vector transgenes. These properties suggest that Bel-independent vectors will prove to be safe and effective for gene therapy applications.
ACKNOWLEDGMENTS
The authors thank Naoki Inoue and Elizabeth Rutledge for helpful suggestions, and Roli Hirata and John Weller for technical assistance. This work was supported by grants from the National Institutes of Health, the March of Dimes Birth Defects Foundation, and the American Society of Hematology. Grant Trobridge is the recipient of a Jesse Davidson Postdoctoral Fellowship from the Medical Research Council of Canada.
REFERENCES
- ACHONG BG, MANSELL PW, EPSTEIN MA, CLIFFORD P. An unusual virus in cultures from a human nasopharyngeal carcinoma. J. Natl. Cancer Inst. 1971;46:299–307. [PubMed] [Google Scholar]
- BAUNACH G, MAURER B, HAHN H, KRANZ M, RETHWILM A. Functional analysis of human foamy virus accessory reading frames. J. Virol. 1993;67:5411–5418. doi: 10.1128/jvi.67.9.5411-5418.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BIENIASZ PD, ERLWEIN O, AGUZZI A, RETHWILM A, MCCLURE MO. Gene transfer using replication-defective human foamy virus vectors. Virology. 1997;235:65–72. doi: 10.1006/viro.1997.8658. [DOI] [PubMed] [Google Scholar]
- DUBRIDGE RB, TANG P, HSIA HC, LEONG PM, MILLER JH, CALOS MP. Analysis of mutation in human cells by using an Epstein–Barr virus shuttle system. Mol. Cell. Biol. 1987;7:379–387. doi: 10.1128/mcb.7.1.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENSSLE J, JORDAN I, MAUER B, RETHWILM A. Foamy virus reverse transcriptase is expressed independently from the Gag protein. Proc. Natl. Acad. Sci. U.S.A. 1996;93:4137–4141. doi: 10.1073/pnas.93.9.4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FIELDS-BERRY SC, HALLIDAY AL, CEPKO CL. A recombinant retrovirus encoding alkaline phosphatase confirms clonal boundary assignment in lineage analysis of murine retina. Proc. Natl. Acad. Sci. U.S.A. 1992;89:693–697. doi: 10.1073/pnas.89.2.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FISCHER N, HEINKELEIN M, LINDEMANN D, ENSSLE J, BAUM C, WERDER E, ZENTGRAF H, MULLER JG, RETHWILM A. Foamy virus particle formation. J. Virol. 1998;72:1610–1615. doi: 10.1128/jvi.72.2.1610-1615.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FLUGEL RM, RETHWILM A, MAURER B, DARAI G. Nucleotide sequence analysis of the env gene and its flanking regions of the human spumaretrovirus reveals two novel genes. EMBO J. 1987;6:2077–2084. doi: 10.1002/j.1460-2075.1987.tb02473.x. [Published erratum appears in EMBO J. 1990, 9, 3806] [DOI] [PMC free article] [PubMed] [Google Scholar]
- GROSS-BELLARD M, OUDET P, CHAMBON P. Isolation of high molecular weight DNA from mammalian cells. Eur. J. Biochem. 1973;36:32–38. doi: 10.1111/j.1432-1033.1973.tb02881.x. [DOI] [PubMed] [Google Scholar]
- HIRATA RK, MILLER AD, ANDREWS RG, RUSSELL DW. Transduction of hematopoietic cells by foamy virus vectors. Blood. 1996;88:3654–3661. [PubMed] [Google Scholar]
- JORDAN I, ENSSLE J, GUTTLER E, MAUER B, RETHWILM A. Expression of human foamy virus reverse transcriptase involves a spliced pol mRNA. Virology. 1996;224:314–319. doi: 10.1006/viro.1996.0534. [DOI] [PubMed] [Google Scholar]
- KANG Y, BLAIR WS, CULLEN BR. Identification and functional characterization of a high-affinity Bel-1 DNA binding site located in the human foamy virus internal promoter. J. Virol. 1998;72:504–511. doi: 10.1128/jvi.72.1.504-511.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KELLER A, PARTIN KM, LOCHELT M, BANNERT H, FLUGEL RM, CULLEN BR. Characterization of the transcriptional trans activator of human foamy retrovirus. J. Virol. 1991;65:2589–2594. doi: 10.1128/jvi.65.5.2589-2594.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LOCHELT M, FLUGEL RM. The human foamy virus pol gene is expressed as a Pro–Pol polyprotein and not as a Gag–Pol fusion protein. J. Virol. 1996;70:1033–1040. doi: 10.1128/jvi.70.2.1033-1040.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LOCHELT M, ZENTGRAF H, FLUGEL RM. Construction of an infectious DNA clone of the full-length human spumaretrovirus genome and mutagenesis of the bel 1 gene. Virology. 1991;184:43–54. doi: 10.1016/0042-6822(91)90820-2. [DOI] [PubMed] [Google Scholar]
- LOCHELT M, MURANYI W, FLUGEL RM. Human foamy virus genome possesses an internal, Bel-1-dependent and functional promoter. Proc. Natl. Acad. Sci. U.S.A. 1993a;90:7317–7321. doi: 10.1073/pnas.90.15.7317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LOCHELT M, ABOUD M, FLUGEL RM. Increase in the basal transcriptional activity of the human foamy virus internal promoter by the homologous long terminal repeat in cis. Nucleic Acids Res. 1993b;21:4226–4230. doi: 10.1093/nar/21.18.4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MACPHERSON I, STOKER M. Polyoma transformation of hamster cell clones—an investigation of genetic factors affecting cell competence. Virology. 1962;16:147–151. doi: 10.1016/0042-6822(62)90290-8. [DOI] [PubMed] [Google Scholar]
- MANIATIS T, FRITSCH E, SAMBROOK S. Molecular Cloning: A Laboratory Manual. 2nd Ed Cold Spring Harbor Laboratory Press; Plainview, NY: 1989. Analysis of genomic DNA by Southern hybridisation; pp. 9.31–9.37. [Google Scholar]
- MAURER B, BANNERT H, DARAI G, FLUGEL RM. Analysis of the primary structure of the long terminal repeat and the gag and pol genes of the human spumaretrovirus. J. Virol. 1988;62:1590–1597. doi: 10.1128/jvi.62.5.1590-1597.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOEBES A, ENSSLE J, BIENIASZ PD, HEINKELEIN M, LINDEMANN D, BOCK M, MCCLURE MO, RETHWILM A. Human foamy virus reverse transcription that occurs late in the viral replication cycle. J. Virol. 1997;71:7305–7311. doi: 10.1128/jvi.71.10.7305-7311.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NESTLER U, HEINKELEIN M, LUCKE M, MEIXENSBERGER J, SCHEURLEN W, KRETSCHMER A, RETHWILM A. Foamy virus vectors for suicide gene therapy. Gene Ther. 1997;4:1270–1277. doi: 10.1038/sj.gt.3300561. [DOI] [PubMed] [Google Scholar]
- ORY DS, NEUGEBOREN BA, MULLIGAN RC. A stable human-derived packaging cell line for production of high titer retrovirus/vesicular stomatitis virus G pseudotypes. Proc. Natl. Acad. Sci. U.S.A. 1996;93:11400–11406. doi: 10.1073/pnas.93.21.11400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PALMER TD, HOCK RA, OSBORNE WR, MILLER AD. Efficient retrovirus-mediated transfer and expression of a human adenosine deaminase gene in diploid skin fibroblasts from an adenosine deaminase-deficient human. Proc. Natl. Acad. Sci. U.S.A. 1987;84:1055–1059. doi: 10.1073/pnas.84.4.1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PEAR WS, NOLAN GP, SCOTT ML, BALTIMORE D. Production of high-titer helper-free retroviruses by transient transfection. Proc. Natl. Acad. Sci. U.S.A. 1993;90:8392–8396. doi: 10.1073/pnas.90.18.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RASHEED S, NELSON-REES WA, TOTH EM, ARNSTEIN P, GARDNER MB. Characterization of a newly derived human sarcoma cell line (HT-1080) Cancer. 1974;33:1027–1033. doi: 10.1002/1097-0142(197404)33:4<1027::aid-cncr2820330419>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- RETHWILM A, ERLWEIN O, BAUNACH G, MAURER B, TER MEULEN V. The transcriptional transactivator of human foamy virus maps to the bel 1 genomic region. Proc. Natl. Acad. Sci. U.S.A. 1991;88:941–945. doi: 10.1073/pnas.88.3.941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RUSSELL DW, MILLER AD. Foamy virus vectors. J. Virol. 1996;70:217–222. doi: 10.1128/jvi.70.1.217-222.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHMIDT M, RETHWILM A. Replicating foamy virus-based vectors directing high level expression of foreign genes. Virology. 1995;210:167–178. doi: 10.1006/viro.1995.1328. [DOI] [PubMed] [Google Scholar]
- SCHWEIZER M, TUREK R, HAHN H, SCHLIEPHAKE A, NETZER KO, EDER G, REINHARDT M, RETHWILM A, NEUMANN-HAEFELIN D. Markers of foamy virus infections in monkeys, apes, and accidentally infected humans: Appropriate testing fails to confirm suspected foamy virus prevalence in humans. AIDS Res. Hum. Retroviruses. 1995;11:161–170. doi: 10.1089/aid.1995.11.161. [DOI] [PubMed] [Google Scholar]
- SONEOKA Y, CANNON PM, RAMSDALE EE, GRIFFITHS JC, ROMANO G, KINGSMAN SM, KINGSMAN AJ. A transient three-plasmid expression system for the production of high titer retroviral vectors. Nucleic Acids Res. 1995;23:628–633. doi: 10.1093/nar/23.4.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WEI CM, GIBSON M, SPEAR PG, SCOLNICK EM. Construction and isolation of a transmissible retrovirus containing the src gene of Harvey murine sarcoma virus and the thymidine kinase gene of herpes simplex virus type 1. J. Virol. 1981;39:935–944. doi: 10.1128/jvi.39.3.935-944.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YU SF, LINIAL ML. Analysis of the role of the bel and bet open reading frames of human foamy virus by using a new quantitative assay. J. Virol. 1993;67:6618–6624. doi: 10.1128/jvi.67.11.6618-6624.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YU SF, BALDWIN DN, GWYNN SR, YENDAPALLI S, LINIAL ML. Human foamy virus replication: A pathway distinct from that of retroviruses and hepadnaviruses. Science. 1996;271:1579–1582. doi: 10.1126/science.271.5255.1579. [DOI] [PubMed] [Google Scholar]