

Abstract
The non-heme iron dioxygenase PtlH from the soil organism Streptomyces avermitilis is a member of the iron(II)/α-ketoglutarate–dependent dioxygenase superfamily and catalyzes an essential reaction in the biosynthesis of the sesquiterpenoid antibiotic pentalenolactone. To investigate the structural basis for substrate recognition and catalysis, we have determined the X-ray crystal structure of PtlH in several complexes with the cofactors iron, α-ketoglutarate, and the non-reactive enantiomer of the substrate, ent-1-deoxypentalenic acid, in four different crystal forms to up to 1.31 Å resolution. The overall structure of PtlH forms a double-stranded barrel helix fold and the cofactor-binding site for iron and α-keto-glutarate is similar to other double-stranded barrel helix fold enzymes. Additional secondary structure elements that contribute to the substrate-binding site in PtlH are not conserved in other double-stranded barrel helix fold enzymes. Binding of the substrate enantiomer induces a reorganization of the monoclinic crystal lattice leading to a disorder-order transition of a C-terminal α–helix. The newly formed helix blocks the major access to the active site and effectively traps the bound substrate. Kinetic analysis of wild type and site-directed mutant proteins confirms a critical function of two arginine residues in substrate binding, while simulated docking of the enzymatic reaction product reveals the likely orientation of bound substrate.
Streptomyces avermitilis is a gram-positive soil-dwelling organism that is used industrially for the production of the widely used anthelminthic avermectins. Recently, a S. avermitilis gene cluster for the biosynthesis of the sesquiterpenoid antibiotic pentalenolactone (Fig. 1a) has been characterized (1). Pentalenolactone was first identified from Streptomyces roseogriseus (2) and is also produced by several Streptomyces strains (3–5). The antibiotic is active against gram-positive and gram-negative bacteria as well as fungi and protozoa (6). In addition, it was shown to inhibit viral replication of HSV-1 and HSV-2 (7) and cell proliferation of rat vascular smooth muscle cells (8). Pentalenolactone inhibits glycolysis (9) by selectively inhibiting glyceraldehyde-3-phosphate dehydrogenase (10). The antibiotic was shown to irreversibly inactivate GAPDH by alkylation of the active site cysteine residue (11). An inducible, pentalenolactone-insensitive GAPDH isozyme that is part of the biosynthetic gene cluster confers self-resistance against the antibiotic in S. avermitilis and other Streptomyces strains (1,12–14).
Figure 1.
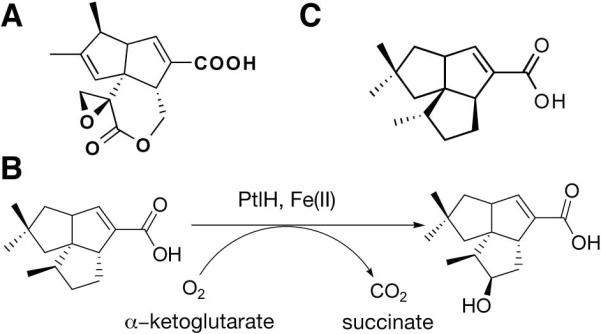
The PtlH reaction
(A) The sesquiterpenoid antibiotic pentalenolactone produced by S. avermitilis. (B) The enzymatic reaction catalyzed by PtlH. (C) The unreactive substrate homolog ent-1-deoxypentalenic acid.
We recently reported the molecular cloning and biochemical characterization of PtlH, an essential non-heme iron-dependent dioxygenase in the pentalenolactone biosynthesis pathway (15). The enzyme catalyzes the Fe(II)- and α-ketoglutarate-dependent hydroxylation of 1-deoxypentalenic acid to 11β-hydroxy-1-deoxypentalenic acid (Fig. 1b). Non-heme iron-dependent dioxygenases form a large family of structurally related enzymes that catalyze an unusual variety of chemical reactions. While the most common reaction is the hydroxylation of unreactive C-H bonds, other reactions include oxidative cyclization, ring fragmentation, desaturation, C-C bond cleavage, and epimerization (16,17). Some family members can modify several substrates or catalyze several reactions. For instance, the alkysulfatase AtsK desulfates a variety of linear and branched chain sulfate esters (18), whereas clavaminate synthase catalyzes three different reactions (hydroxylation, oxidative ring closure and desaturation) in the biosynthesis of the β-lactamase inhibitor clavulanic acid (19). A recent structure determination describing the SyrB2 halogenase underscores the large diversity of chemical reactions catalyzed by these enzymes (20).
Isotope labeling, kinetic, spectroscopic, theoretical, and crystallographic studies support a common mechanism of catalysis by non-heme Fe(II)/α-ketoglutarate-dependent dioxygenases in which binding of molecular oxygen to the Fe(II) is followed by oxidative decarboxylation of the α-ketoglutarate to generate carbon dioxide, succinate, and a key ferryl–oxygen species (G) (Fig. 2) (21–25). This highly reactive [Fe(IV)=O] is then directly responsible for oxidation of the bound organic substrate.
Figure 2.

Mechanism of oxidation by Fe(II)/a-ketoglutarate-dependent dioxygenases. Intermediate G is the highly reactive ferryl-oxo complex.
The first crystal structure for a non-heme iron and α-ketoglutarate-dependent oxygenase was reported for deacetoxycephalosporin C synthase in 1997 and several enzyme structures have been determined subsequently. The structural studies revealed a common double-stranded β-helix fold (DSBH), also termed the jelly-roll fold or double-greek key motif, which typically consists of two four-stranded β-sheets that form a β-sandwich structure. Additional secondary structure elements such as additional β-strands extending one of the core β-sheets and α-helical segments in the N- and C-terminal regions are observed in most family members. In all DSBH enzymes (and other non-heme oxygenases), the Fe(II) cofactor is bound by two histidines and one aspartate or glutamate residue in a common structural motif described as the 2-His-1-carboxylate facial triad (26). In addition, the position of these iron-coordinating residues is conserved as an HXD/E…H sequence motif (27,28), where the first two residues are located on the second β-strand and the second histidine residue is located on the seventh β-strand of the core DSBH fold. The available structural information for DSBH enzymes has recently been reviewed (17,21).
Here, we describe crystal structures of PtlH in complex with co-substrates iron and α-ketoglutarate and the non-reactive substrate enantiomer ent-1-deoxypentalenic acid. The structure reveals a typical DSBH core fold with additional structural elements unique to PtlH. The iron and α-ketoglutarate co-substrates are bound in a similar geometry to that in other DSBH enzymes. The active site cavity is open to the solvent and lined with mostly hydrophobic residues as well as two arginine residues that are strategically placed to bind the substrate carboxylate. A fortuitous rearrangement of the crystal packing during substrate soaking experiments trapped the enzyme in a closed active site conformation and reveals a dynamic disorder-order transition of a C-terminal α-helix to close the active site cavity during catalysis. Finally, docking calculations with the product 11β-hydroxy-1-deoxypentalenic acid suggest the actual orientation of the natural enantiomer of the substrate.
Experimental Procedures
Protein expression, purification and crystallization
Full length PtlH from S. avermitilis (Genbank NP_824167, 311 amino acids) was amplified by PCR from DNA of S. avermitilis cosmid clone CL_216_D07 and cloned into the pET28e vector (a variant of pET28a [Novagen] in which the XbaI site has been replaced by an EcoRI site) as described previously (15). After this protein failed to crystallize, however, reanalysis of the DNA sequence led to reassignment of the translational start site corresponding to a shorter encoded protein of 285 amino acids (residues 27 to 311 of the original genbank file, Genbank BAC70702, http://avermitilis.ls.kitasato-u.ac.jp). We therefore amplified the 858-bp ptlh gene by PCR from cosmid CL_216_D07 using the forward primer (5′-CGCG TCTCGCTCATATGACGAACGTGCTGGGGA CTAC-3′) and the same reverse primer as that previously used for the unnatural 311-aa version of PtlH (5′-GGCCGGAAGCTTACTAGTCAAT TGTCATTCCACGTCGGTGGGGGTA-3′) and subcloned the DNA for the shorter protein sequence into the pET28e vector. The resultant recombinant protein was over-expressed in E. coli BL21(DE3) (Invitrogen) at 20 °C using 0.4 mM IPTG. Bacterial cells were lysed by ultrasonification on ice. The soluble protein was bound to nickel-agarose affinity resin (Qiagen), washed with a buffer containing 20 mM Tris (pH 8.5), 250 mM NaCl, and 10 mM imidazole. His6-tagged protein was then eluted with a buffer containing 20 mM Tris (pH 8.5), 250 mM NaCl, and 150 mM imidazole. The protein was further purified with anion exchange chromatography at pH 8.5, using a linear gradient of 10 mM to 1 M NaCl concentration, and size exclusion chromatography at pH 8.5 and 200 mM NaCl. The purified protein was concentrated to 25 mg/ml in a buffer containing 10 mM Tris (pH 8.5), 20 mM NaCl, and 7% glycerol. The sample was flash-frozen in liquid nitrogen and stored at −80 °C. The N-terminal histidine-tag was removed by thrombin digestion before crystallization.
For the production of selenomethionyl proteins, the expression construct was transformed into B834(DE3) cells (Novagen). The bacterial growth was carried out in defined LeMaster media (29), and the protein was purified using the same protocol as for the wild-type protein.
Crystals of PtlH were obtained at 4°C with the sitting drop vapor diffusion method. The reservoir solution contained 100 mM TRIS pH 8.5, 200 mM MgCl2 and 20% PEG 3350 (w/v). 0.8 μl of protein solution (13 mg/ml, containing 2 mM α-keto- glutarate and 20 mM DTT) were mixed with 0.8 μl of reservoir solution. Initial small crystals grew within 24 hours and larger crystals were grown by microseeding techniques. Crystals grew to maximum dimensions of 0.45 × 0.12 × 0.12 mm. The crystals were cryo-protected by rapid soaking in a solution containing mother liquor with the addition of 20% (v/v) ethylene glycol and flash frozen in liquid nitrogen. The PtlH/α-ketoglutarate/ent-1PL complex (crystal form I) was obtained by adding 2 μl of a solution of 8 mM racemic (±)-1-deoxypentalenic acid and 22% ethylene glycol in mother liquor to the drop with crystals and soaking for 1 hour. Crystal forms III and IV were obtained from repeating the crystallization experiments with the same conditions using 100 mM Hepes at a pH of 7.5. Crystal form IV was additionally soaked for 15 min as described for crystal form I above.
Data Collection
X-ray diffraction data were collected on an ADSC CCD detector at the X4A beamline of the National Synchrotron Light Source in Brookhaven. For the structure determination, a selenomethionyl single-wavelength anomalous diffraction (SAD) data set to 1.35 Å resolution was collected at a wavelength of 0.9790 Å at 100K. The diffraction images were processed and scaled with the HKL package (30). The crystals belong to the space group P21, with cell dimensions of a = 43.5 Å, b = 70.1 Å, c = 48.7 Å, and β = 92.5°. There is one molecule in the asymmetric unit, giving a Vm of 2.3 Å3/Dalton. Diffraction data for the substrate-free crystals (crystal form II), which had been used for the substrate soaking experiments, were collected at a wavelength of 0.979 Å to a resolution of 1.31 Å on the X4A beamline. Crystal belong to space group P21 with cell dimensions a = 42.8 Å, b = 70.9 Å, c = 48.8 Å, and β = 98.2°. Diffraction data for the substrate-free enzyme in the third monoclinic space group (crystal form III) were collected at a wavelength of 1.54 Å on a Rigaku 007 rotating-anode generator equipped with an R-axis IV++ detector in space group P21 to 1.7 Å resolution. The cell dimensions for this crystal form are a = 45.8 Å, b = 70.6 Å, c = 48.0 Å, and β = 113.2°. Diffraction data for the second analogue-bound form (crystal form IV) were collected to 1.65 Å resolution in space group P21212 at 1.54 Å wavelength on the rotating-anode generator. The cell dimensions for this crystal form are a = 70.0 Å, b = 89.4 Å and c = 48.8 Å. The data processing statistics are summarized in Table 1.
Table 1.
| Crystal Form | I (anomalous) | II (anomalous) | III | IV |
|---|---|---|---|---|
| Data collection | ||||
| Wavelength | 0.979 Å | 0.979 Å | 1.54 Å | 1.54 Å |
| Cofactors/Substrate | Fe/α-KG/ent-1PL | Fe/α-KG | Fe/OGA | Fe/OGA/ent-1PL |
| Space Group | P21 | P21 | P21 | P21212 |
| Cell Dimensions (Å) | 43.5 × 70.1 × 48.7, β = 92.5° | 42.8 × 70.9 × 48.8, β = 98.2 | 45.8 × 70.6 × 48.0, β = 113.2° | 70.0 × 89.4 × 48.8 |
| Resolution range (Å) | 30−1.35 | 30−1.31 | 30−1.7 | 30−1.65 |
| Number of observations | 399,254 | 167,268 | 77,055 | 81,386 |
| Rmerge (%)a | 5.2 (44.6) | 5.8 (35.4) | 7.6 (52.4) | 3.1 (21.9) |
| I/σ(I) | 21.9 (2.8) | 11.0 (1.7) | 12.3 (1.8) | 25.0 (3.9) |
| Redundancy | 3.2 (3.1) | 1.5 (1.4) | 2.5 (2.4) | 2.2 (2.0) |
| Refinement | ||||
| Number of reflections | 60,549 | 61,369 | 28,959 | 34,334 |
| Completeness (%) | 99.5 (99.5) | 93.4 (97.2) | 98.3 (96.6) | 96.3 (95.0) |
| R factor (%)b | 17.2 (21.6) | 17.8 (24.4) | 17.4 (30.1)) | 18.2 (27.8) |
| Free R factor (%) | 19.6 (24.9) | 20.2 (26.2) | 21.7 (37.9) | 20.9 (34.3) |
| rmsd in bond lengths (Å) | 0.008 | 0.007 | 0.010 | 0.009 |
| rmsd in bond angles (°) | 1.2 | 1.2 | 1.2 | 1.2 |
| Average B for protein atoms (Å2) | 9.8 | 8.6 | 14.8 | 16.9 |
| Average B for ligand atoms (Å2) | 8.1 | 7.0 | 11.7 | 25.9 |
| Average B for solvent atoms (Å2) | 36.2 | 32.2 | 31.4 | 38.4 |
| Ramachandran Plot most favored region (%) | 92.4 | 91.7 | 92.1 | 90.6 |
| Allowed (%) | 7.6 | 8.3 | 7.9 | 9.4 |
. The numbers in parenthesis are for the highest resolution shell.
Structure Determination and Refinement
The locations of selenium atoms were determined with the program Solve (31) based on the anomalous differences in the SAD data set. The resulting experimental electron density map was of excellent quality. The initial atomic model was built with the program Resolve (32) after density modification. Coordinates for the substrate homolog were obtained from the PRODRG server (33). The protein model was further rebuilt manually with the program O (34). Crystallographic refinement was performed with the program Refmac (35) from the CCP4 program suite (36). The structures of the other crystal forms were determined by molecular replacement with the program COMO (37). The structure refinement statistics are summarized in Table 1. All molecular figures were produced with the program PyMOL (38).
Preparation of 1-deoxypentalenic acid
Racemic 1-deoxypentalenic acid was synthesized from α-humulene as described previously (1). Briefly, α-humulene was converted to (±)-pentalenene in three chemical steps, followed by oxidation of (±)-pentalenene to (±)-pentalen-13-ol and (±)-pentalen-13-al. (±)-Pentalen-13-al was converted to the methyl ester of (±)-1-deoxy-pentalenic acid. Finally, (±)-1-deoxypentalenic acid was obtained by hydrolyzing the methyl ester in the solution of KOH/MeOH/H2O.
Preparation of PtlH mutants R117Q and R188Q
Site-directed mutagenesis was carried out with the Quikchange II XL kit (Stratagene) using plasmid DNA pET28e/PtlH as template and the two sets of primer pairs – R117Q: 5′-CTCCACGACCATC CAGTACTACCCGTCGC-3′ (sense, Gln codon in bold), 5′-GCGACGGGTAGTACTGGATGGT CGTGGAG-3′ (antisense); R188Q: 5′-CG AGTC CTTCCGCCAGTTCGGCCACCCG-3′ (sense, Gln codon in bold), 5′-CGGGTGGCCGAACTG GCGGAAGGACTCG -3′ (antisense). The complete sequences of both mutants were confirmed by automated dideoxy sequencing. Both mutants were expressed in E. coli BL21(DE3) under the same conditions used for the wild-type protein and the resultant N-terminal His6-tag proteins were purified to homogeneity by Ni2+-NTA affinity chromatography.
Steady-State kinetics of PtlH
The steady-state kinetic parameters were determined in a manner to that previously described with small modifications (15). To determine the linearity of the enzyme reaction, incubations were performed at 23 °C with mixtures containing MES (95 mM), PtlH (0.018 μM), α-ketoglutarate (2.67 mM), L-ascorbate (2.67 mM), FeSO4 (1.33 mM), catalase (0.95 mg/mL) and DTT (3 mM) at pH of 6.0. Reactions were initiated by adding a solution of (±)-1-deoxy pentalenic acid in DMSO to a concentration of 300 μM (final DMSO concentration 2%). The reactions were quenched with HCl after incubation for 5 min, 10 min, 15 min, 20 min and 40 min. The quenched mixtures were extracted with ether, converted to the methyl ester with trimethylsiliyldiazomethane (TMS-CHN2), and subjected to GC-MS analysis. A plot of the amount of product versus time showed a good linear correlation within 10 min under these conditions (R2 = 0.98). Kinetic assays were therefore carried out for 10 min at 23 °C. Reactions were initiated by adding a solution of (±)-1-deoxypentalenic acid in DMSO to a concentration of 25–250 μM (final DMSO concentration 2%). The reactions were quenched with HCl at 10 min, and the mixtures were extracted with ether and treated with TMS-CHN2. Each sample was adjusted to the same volume (150 μL) with methanol before analysis by GC-MS. A 3 μL sample was injected and the abundance of product peak from each reaction was determined. The GC-MS peak abundance was calibrated by injecting various known amounts of the pure methyl ester of the enzymatic product. Fitting of the initial velocities to the Michaelis-Menten equation gave kcat = 3.0 ± 0.3 s−1 and a Km = 41 ± 13 μM, essentially the same kcat as that of the previously analyzed recombinant PtlH protein carrying the supernumerary 26-aa N-terminal peptide (4.2 ± 0.6 s−1), but with a substantially lower Km compared to the previously reported value of 570 ± 190 μM. Standard deviations in the steady-state kinetic parameters represent the calculated statistical errors in the non-linear, least squares regression analysis. GC-MS experiments were carried out on a Hewlett-Packard GCD Series II instrument with a DB5 (30 m × 0.25 mm) column. Kinetic measurements on the R117Q and R188Q mutants were performed at a single concentration of 0.4 mM (±)-1-deoxypentalenic acid, 10× the Km for the wild-type enzyme.
Atomic Coordinates
The atomic coordinates and structure factors have been deposited in the Protein Data Bank with accession codes 2RDN, 2RDQ, 2RDR and 2RDS for crystal forms I to IV, respectively.
Results and Discussion
Overall Structure
We have determined the structure of PtlH from S. avermitilis in four crystal forms at a resolution of up to 1.31 Å (Table 1). Crystal form I was obtained at pH 8.5 in space group P21 as a complex with Fe(II), α-ketoglutarate and the non-reactive substrate enantiomer ent-1-deoxypentalenic acid (ent-1PL, Fig. 1c) after soaking crystal form II with a racemic substrate mixture for one hour. Crystal form III crystallized in space group P21 as a complex with iron and the α-ketoglutarate-analogue inhibitor N-oxalylglycine (OGA) upon lowering the pH of the precipitant to 7.5. Crystal form IV crystallized at pH 7.5 in space group P21212 as a complex with iron, OGA and contained a partially occupied molecule of ent-1PL after soaking for 15 minutes. All crystal forms required the presence of either α-ketoglutarate or oxalylglycine and no crystals could be obtained with succinate.
The structure was solved by single anomalous dispersion methods of selenomethionyl-substituted protein using a 1.35 Å resolution data set collected for crystal form I. The crystallographic R/Rfree factors are 17.2/19.6, 17.8/20.2, 17.4/21.7, and 18.2/20.9 for the four crystal forms, respectively. There is one molecule in the asymmetric unit in each crystal form and the majority of the residues (92.4, 91.7, 92.1, 90.6) are in the most favored region of the Ramachandran plot. The purified recombinant protein was yellow and enzymatically active without addition of Fe(II), indicating that bound Fe(II) was retained throughout the protein purification procedure. Crystals were grown without Fe(II) and a fluorescence scan at the iron peak wavelength at beamline X4A of the National Synchrotron Light Source confirmed the presence of Fe(II). We cannot exclude, however, the possibility that the metal binding site is partially occupied by magnesium ions from the crystallization buffer, as subsequent experiments showed that magnesium can be bound in the metal binding site. In crystal forms I and II, two solvent magnesium ions coordinate to the protein in an octahedral environment. One of these magnesium ions (Mg4) mediates a crystal contact between Asp60 and His206 from a neighboring molecule. The magnesium ion coordinates to five solvent water molecules that form hydrogen bonds to Asp60 whereas His206 interacts directly with the magnesium ion to complete the octahedral coordination sphere. The bridging contact to His206 is lost in the two other crystal forms at pH 7.5 (Mg4 remains coordinated to Asp60 in crystal form III, but is not observed in crystal form IV).
In an attempt to produce crystals with the natural enantiomer of the substrate bound in the active site, we purified metal-free protein by including 50 mM of EDTA at every step of the purification protocol. The protein sample obtained was colorless and enzymatically inactive without addition of Fe(II). Crystals did not show an anomalous fluorescence peak at the characteristic iron peak wavelength. However, several independent data sets showed a strong electron density peak at the metal binding site, which likely corresponds to magnesium(II) (the crystallization conditions require the presence of 200 mM Mg(II)). Attempts to trap the substrate with these crystals were unsuccessful, while soaking experiments using oxalylyglycine-bound crystals resulted in partial binding of the inactive substrate enantiomer, ent-1PL (crystal form IV).
As predicted from sequence homology and the enzymatic reaction, the core region of PtlH forms a double-stranded β-helix (DSBH) fold (Fig. 3a). However, only seven of the eight β-strands of the DSBH fold are observed in the PtlH structure (a topology diagram is shown in Fig. 3b). The major antiparallel β-sheet consists of the strands β2, β10, β3, β8, and the additional strand β1. The minor β-sheet consists of only three strands, β9, β4 and β7. In PtlH, a loop segment between residues 133 and 138 (including the iron-binding His137) occupies the position of the fourth β-strand in other DSBH structures. Similar to other DSBH dioxygenases, the major β-sheet of the DSBH fold is stabilized by several α-helices. These helices are located in the N-terminal region of the PtlH sequence and include helices α1 and α2, which stabilize the additional strand β1 at the end of the β-sheet, and helices α3 – α6, which stabilize the back of the major β-sheet. Other additional secondary structure elements are either involved with iron binding (α7) or with the formation of the substrate-binding site (α9, α10, β5, β6).
Figure 3.

Structure of PtlH
(A) Stereo diagram showing a schematic representation of the structure of PtlH. The Fe(II) ion is shown as a light-green sphere, α-ketoglutarate is shown as sticks with carbon atoms in gray, and ent-1PL is shown as sticks with carbon atoms in blue. (B) Topology diagram of PtlH. The position of the β-strand not present in the PtlH DSBH fold is indicated with a gray arrow. (C) Final 2Fo-Fc electron density map (contoured at 1σ) of the active site region. Electron density for Fe(II), α-ketoglutarate, ent-1PL and the Fe(II)-coordinated solvent water molecule is shown in green, gray, blue and red, respectively.
The overall protein structure is similar in all four crystal forms with a root mean square deviation (rmsd) of 0.37 Å between form I and II, 0.44 Å between form I and III, and 0.37 Å between form I and IV. Significant structural differences are observed at the N- and C-termini and in the loop region between residues 56 to 58 following helix α3, where Gly57 moves by 4.3 Å between form I and III. Most importantly, the C-terminal region (residues 266 to 276) comprising helix α10 is only observed in crystal form I and is disordered in the other crystal forms (discussed below). Electron density for a loop region including the short helix α8 between residues 198 and 200 is weakly defined in all crystal forms indicating higher mobility of this protein segment.
In the PtlH structure, Fe(II) is bound by a canonical 2-His-1-Asp facial triad by His137, Asp139 and His226 (HXD…H sequence signature, Fig. 4a). α-Ketoglutarate coordinates to the metal in a bidentate fashion with its carboxylate oxygen O2 and the α-keto oxygen. The octahedral metal coordination sphere is completed by the solvent water molecule W1. The iron-coordinating Asp139 forms additional hydrogen bonds with Thr151 and Arg246, which also coordinates with the metal-bound carboxylate of α-ketoglutarate. The second carboxylate of the α-ketoglutarate co-substrate is bound to the protein by a salt bridge to Arg240 and additional hydrogen bonds to Ser228 and Trp153. The oxalylglycine inhibitor (observed in crystal form III and IV) binds in a highly similar fashion to the enzyme. The coordination of the carboxylate groups to the metal and to Arg240 remains undisturbed, but the molecule is slightly rotated with the largest positional shift being 0.4 Å for the nitrogen atom compared to C3 of α-ketoglutarate.
Figure 4.

The PtlH Active Site
(A) Stereo diagram of Fe(II)/α-ketoglutarate binding site. The Fe(II) ion is shown as a green sphere, α-ketoglutarate is shown as sticks with carbon atoms in gray and the iron-coordinated solvent water molecule is shown as a small red sphere. (B) Coordination of ent-1PL in the PtlH active site. ent-1PL is shown in sticks with blue carbon atoms and solvent water molecules are shown as small salmon-colored spheres. (C, D) Surface illustration of PtlH colored by electrostatic potential in the absence of helix α10 (c, crystal form II) and with helix α10 shown as cartoon representation to indicate the closure of the active site in the presence of this helix. The bound ent-1PL molecule is shown in blue sticks; bound α-ketoglutarate is shown in gray sticks.
Ent-1-deoxypentalenic acid binding
After soaking PtlH crystals with a 1:1 racemic substrate mixture, we observed the non-reactive enantiomer ent-1-deoxypentalenic acid bound in the active site (Fig. 4b). Soaking was performed for one hour and comparison with the substrate free crystals revealed that the crystal lattice had reorganized during the soaking experiment. While the cell dimensions remained similar, the β-angle of the monoclinic cell changed from 98° to 93° (crystal forms II and I). Soaking experiments were performed in the presence of oxygen and α-ketoglutarate and we believe that all substrate was converted to product during the prolonged time of the experiment before the non-reactive enantiomer was bound in a dead-end complex. However, shorter soaking times did not yield interpretable substrate density and it seems that the formation of crystal form I is required to trap the substrate in the active site.
Ent-1PL is bound in the active site by a salt bridge interaction formed between its carboxylate group and Arg117 and an interaction with Arg188 (Fig. 4b). The carboxylate O2 forms additional hydrogen bonds with the iron-coordinated solvent water W1 and the hydroxyl group of Y142 whereas the O1 oxygen interacts with the solvent water W175, which is part of an extended solvent network established between R117, R188, Trp262 and Ser270. The hydrophobic moiety of ent-1PL is located in a larger active site cavity that is lined with mostly hydrophobic residues including Thr134, His137, Tyr142, Ile273 and Val193. Substrate coordination by the two arginine residues serves as the major recognition mechanism in PtlH. Site-directed mutagenesis showed that the R117Q mutation essentially abolished enzymatic activity (>4700-fold less active at 10× the Km for wild-type PtlH) while the R188Q mutation also significantly reduced PtlH activity by a factor of 280. Due to the low activity of the mutants, no attempt was made to distinguish whether the effects of the mutations were primarily on kcat or Km.
The side chain of Tyr142 assumes an unusual coordination in the ent-1PL bound structure. This residue is observed in two distinct conformations in the absence of substrate or in the partially bound crystal form IV. In the presence of ent-1PL, Tyr142 is locked into one orientation towards the Fe(II) center and forms hydrogen bonds with the iron-coordinated solvent water W1 and the ent-1PL carboxylate group. An equivalent tyrosine residue was found in phytanoyl-CoA 2-hydroxylase (39), but a similar interaction with the Fe(II) center has not previously been observed. The observed conformational disorder suggests that Tyr142 might contribute to the stabilization of the solvent water coordination to the Fe(II) center in the absence of substrate so as to reduce uncoupled enzymatic turnover.
The observed order-disorder of the C-terminal helix α10 could provide another mechanism to enhance productive catalysis. Electron density for helix α10 between residues 267 and 276 was only observed for crystal form I. Within the PtlH molecule, the helical structure is only weakly stabilized by a hydrogen bond between Ser276 and Arg194 and hydrophobic contacts from Ile273 to Val193 and Tyr142. However, hydrogen bonds are established between a neighboring protein molecule and Lys271 and the reorganization of the crystal packing locks helix α10 into its ordered conformation. Formation of helix α10 effectively closes off the access for the substrate to the active site and could thus function as a `lid' to further stabilize the bound substrate during catalysis (Fig. 4c,d).
Modeling of substrate binding
To obtain further insights into substrate binding, we placed the substrate enantiomer manually in the active site with its carboxylate group in the same position as the inactive ent-1PL molecule. We examined many possible substrate orientations obtained from rotation of the carbon skeleton around the carboxylate group in combination with small translations. However, we could not identify a substrate orientation in which the substrate C11 atom was placed in a favorable position for catalysis of 11β-C–H hydroxylation. We therefore next performed docking calculations of the protein–product complex using the Dock6 program (http://dock. compbio.ucsf.edu). We used the closed active site cavity as observed in crystal form I while rotating the Tyr142 side chain away from the iron center in the orientation observed in the absence of ent-1PL. We also removed the iron-coordinated water W1 and replaced α-ketoglutarate with succinate (the docked position of the product did not differ when either cofactor was used for the calculations). We then performed a rigid-body docking calculation of the PtlH product, 11β-hydroxy-1-deoxypentalenic acid, keeping the orientation of protein side chains in the active site fixed. Dock6 placed the product, and by inference, the natural enantiomer of the substrate in an orientation different than that observed for ent-1PL, but fully consistent with the structural, stereochemical, and kinetic data (Fig. 5). Notably, the product hydroxyl group is placed in close proximity to the iron-coordinated solvent water molecule and the product carboxylate is close to both arginine residues. In the docked orientation, O1 of the product carboxylate is located at a distance of 3.3 Å to Arg188, and O2 is placed at a distance of 3.8 Å to NE1 and 3.9 Å to NE2 of Arg117. A small rotation around the product hydroxyl oxygen atom is sufficient to bring the carboxylate group into close hydrogen bonding distance to both arginine residues. The hydroxylated C11 atom in the product is located 3.9 Å from the iron atom, compared with a distance of 4.7 Å for the closest carbon (C12) – Fe(II) distance in the experimental ent-1PL structure. This suggests that while the non-reactive enantiomer of the substrate engages in the same hydrogen bonding interactions as the natural substrate, its orientation in the hydrophobic binding pocket places all carbon atoms too far from the iron center for productive catalysis to occur.
Figure 5.

Structural Model of Product Binding
(A) Orientation of ent-1PL in the PtlH active site (B) Model of the orientation of the reaction product, 11β-hydroxy-1-deoxypentalenic acid in the PtlH active site. The iron-coordinated water molecule (salmon) is not part of the model but is shown for reference.
Comparison with structurally related enzymes
A database search with the program Dali (40) identified phytanoyl-CoA 2-hydroxylase and the SyrB2 halogenase as the closest related protein structures. The PtlH sequence is 22.4% identical with the PAHX structure (295 residues, Pdb: 2A1X) and 169 Cα atoms can be aligned to a root mean square deviation of 1.7 Å. The comparison of the PtlH structure with PAHX shows that the core DSBH fold remains fairly conserved whereas most of the connecting loop regions and especially secondary structure motifs close to the substrate-binding site vary substantially (Fig. 6a). Not surprisingly, the positions of the iron-coordinating histidine and aspartate residues remain conserved as well as the identity of the α-ketoglutarate coordinating residues, although the coordination of the α-ketoglutarate 1-carboxylate is trans to His226 (PtlH numbering) in PAHX, in contrast to the coordination trans to His137 found in PtlH (Fig. 6b). Minor changes in side chain positions are sufficient to accommodate α-ketoglutarate in this coordination. The structural comparison between both enzymes clearly illustrates how comparatively small variations of additional secondary structure elements result in major changes in substrate specificity and how variation of only a few key residues in the active site is sufficient to accurately position a substrate molecule for hydroxylation.
Figure 6.

Comparison of PtlH with Phytanoyl-CoA 2-Hydroxylase
(A) Cartoon representation of a least-squares alignment of PtlH with phytanoyl-CoA 2-hydroxylase (Pdb Code: 2A1X). PtlH is colored in orange with the iron ion shown in green, α-ketoglutarate in gray sticks and ent-1PL in cyan sticks. Phytanoyl-CoA 2-hydroxylase and bound cofactors are shown in blue. (B) Comparison of the active site region after local least-squares alignment of Fe(II) and Fe(II)-coordinating histidine and aspartate residues. Color scheme as in (A).
In summary, the structure determination of the PtlH dioxygenase has revealed that a DSBH fold provides the structural framework for catalysis. The recognition of the Fe(II)- and α-ketoglutarate cofactors is conserved and is similar to other DSBH dioxygenases while the substrate-binding site is unique and specific to 1-deoxypentalenic acid binding and stereospecific hydroxylation to 11β-hydroxy-1-deoxypentalenic acid.
Acknowledgments
This work was supported in part by NIH grant GM30301 to D.E.C. We thank John Schwanof and Randy Abramowitz for access to the X4A beamline at the National Synchrotron Light Source and Hasan Demirci and Hua Li for help with data collection at the synchrotron.
1. The abbreviations used are
- DSBH
double-stranded barrel helix
- ent-1PL
ent-1-deoxypentalenic acid
- OGA
N-oxalylglycine
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
References
- 1.Tetzlaff CN, You Z, Cane DE, Takamatsu S, Omura S, Ikeda H. Biochemistry-Us. 2006;45(19):6179–6186. doi: 10.1021/bi060419n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koe BK, Sobin BA, Celmer WD. Antibiot. Annu. 1957:672–675. [PubMed] [Google Scholar]
- 3.Keller-Schierlein W, Lemke J, Nyfeler R, Zahner H. Arch. Mikrobiol. 1972;84:301–316. [PubMed] [Google Scholar]
- 4.Okazaki T, Enokita R, Torikata A, Inukai M, Takeuchi M, Takahashi S, Arai M. Ann. Rep. Sankyo Res. Lab. 1979;31:94–103. [Google Scholar]
- 5.Takahashi S, Takeuchi M, Arai M, Seto H, Otake N. J Antibiot. 1983;36(3):226–228. doi: 10.7164/antibiotics.36.226. [DOI] [PubMed] [Google Scholar]
- 6.English AR, McBride TJ, Lynch JE. Antibiot. Annu. 1957:682–687. [PubMed] [Google Scholar]
- 7.Nakagawa A, Tomoda H, Hao MV, Okano K, Iwai Y, Omura S. J Antibiot. 1985;38(8):1114–1115. doi: 10.7164/antibiotics.38.1114. [DOI] [PubMed] [Google Scholar]
- 8.Ikeda M, Fukuda A, Takagi M, Morita M, Shimada Y. Eur J Pharmacol. 2001;411(1–2):45–53. doi: 10.1016/s0014-2999(00)00894-3. [DOI] [PubMed] [Google Scholar]
- 9.Duszenko M, Balla H, Mecke D. Biochim Biophys Acta. 1982;714(2):344–350. doi: 10.1016/0304-4165(82)90343-9. [DOI] [PubMed] [Google Scholar]
- 10.Cane DE, Sohng JK. Arch Biochem Biophys. 1989;270(1):50–61. doi: 10.1016/0003-9861(89)90006-4. [DOI] [PubMed] [Google Scholar]
- 11.Cane DE, Sohng JK. Biochemistry-Us. 1994;33(21):6524–6530. doi: 10.1021/bi00187a020. [DOI] [PubMed] [Google Scholar]
- 12.Maurer KH, Mecke D. J Antibiot. 1986;39(2):266–271. doi: 10.7164/antibiotics.39.266. [DOI] [PubMed] [Google Scholar]
- 13.Maurer KH, Pfeiffer F, Zehender H, Mecke D. J Bacteriol. 1983;153(2):930–936. doi: 10.1128/jb.153.2.930-936.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frohlich KU, Kannwischer R, Rudiger M, Mecke D. Arch Microbiol. 1996;165(3):179–186. doi: 10.1007/BF01692859. [DOI] [PubMed] [Google Scholar]
- 15.You Z, Omura S, Ikeda H, Cane DE. J Am Chem Soc. 2006;128(20):6566–6567. doi: 10.1021/ja061469i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hewitson KS, Granatino N, Welford RWD, McDonough MA, Schofield CJ. Philosophical Transactions of the Royal Society a-Mathematical Physical and Engineering Sciences. 2005;363(1829):807–828. doi: 10.1098/rsta.2004.1540. [DOI] [PubMed] [Google Scholar]
- 17.Clifton IJ, McDonough MA, Ehrismann D, Kershaw NJ, Granatino N, Schofield CJ. Journal of Inorganic Biochemistry. 2006;100(4):644–669. doi: 10.1016/j.jinorgbio.2006.01.024. [DOI] [PubMed] [Google Scholar]
- 18.Muller I, Kahnert A, Pape T, Sheldrick GM, Meyer-Klaucke W, Dierks T, Kertesz M, Uson I. Biochemistry-Us. 2004;43(11):3075–3088. doi: 10.1021/bi035752v. [DOI] [PubMed] [Google Scholar]
- 19.Kershaw NJ, Caines MEC, Sleeman MC, Schofield CJ. Chemical Communications. 2005;(34):4251–4263. doi: 10.1039/b505964j. [DOI] [PubMed] [Google Scholar]
- 20.Blasiak LC, Vaillancourt FH, Walsh CT, Drennan CL. Nature. 2006;440(7082):368–371. doi: 10.1038/nature04544. [DOI] [PubMed] [Google Scholar]
- 21.Hausinger RP. Critical Reviews in Biochemistry and Molecular Biology. 2004;39(1):21–68. doi: 10.1080/10409230490440541. [DOI] [PubMed] [Google Scholar]
- 22.Hoffart LM, Barr EW, Guyer RB, Bollinger JM, Krebs C. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(40):14738–14743. doi: 10.1073/pnas.0604005103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Price JC, Barr EW, Glass TE, Krebs C, Bollinger JM. J Am Chem Soc. 2003;125(43):13008–13009. doi: 10.1021/ja037400h. [DOI] [PubMed] [Google Scholar]
- 24.Price JC, Barr EW, Tirupati B, Bollinger JM, Krebs C. Biochemistry-Us. 2003;42(24):7497–7508. doi: 10.1021/bi030011f. [DOI] [PubMed] [Google Scholar]
- 25.Borowski T, Bassan A, Siegbahn PEM. Chemistry-a European Journal. 2004;10(4):1031–1041. doi: 10.1002/chem.200305306. [DOI] [PubMed] [Google Scholar]
- 26.Koehntop KD, Emerson JP, Que L. Journal of Biological Inorganic Chemistry. 2005;10(2):87–93. doi: 10.1007/s00775-005-0624-x. [DOI] [PubMed] [Google Scholar]
- 27.Costas M, Mehn MP, Jensen MP, Que L. Chemical Reviews. 2004;104(2):939–986. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]
- 28.Hogan DA, Smith SR, Saari EA, McCracken J, Hausinger RP. Journal of Biological Chemistry. 2000;275(17):12400–12409. doi: 10.1074/jbc.275.17.12400. [DOI] [PubMed] [Google Scholar]
- 29.Hendrickson WA, Horton JR, Lemaster DM. EMBO J. 1990;9(5):1665–1672. doi: 10.1002/j.1460-2075.1990.tb08287.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Otwinowski Z, Minor W. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 31.Terwilliger TC, Berendzen J. Acta Crystallographica Section D-Biological Crystallography. 1999;55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Terwilliger TC. Acta Crystallographica Section D-Biological Crystallography. 2000;56:965–972. doi: 10.1107/S0907444900005072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schuttelkopf AW, van Aalten DMF. Acta Crystallographica Section D-Biological Crystallography. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 34.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Acta Crystallogr. A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 35.Murshudov GN, Vagin AA, Dodson EJ. Acta Crystallogr. D. Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 36.Bailey S. Acta Crystallogr. D. Biol. Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 37.Jogl G, Tao X, Xu YW, Tong L. Acta Crystallographica Section D-Biological Crystallography. 2001;57:1127–1134. doi: 10.1107/s0907444901006783. [DOI] [PubMed] [Google Scholar]
- 38.DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific; San Carlos, USA: 2002. [Google Scholar]
- 39.McDonough MA, Kavanagh KL, Butler D, Searls T, Oppermann U, Schofield CJ. Journal of Biological Chemistry. 2005;280(49):41101–41110. doi: 10.1074/jbc.M507528200. [DOI] [PubMed] [Google Scholar]
- 40.Holm L, Sander C. Journal of Molecular Biology. 1993;233(1):123–138. doi: 10.1006/jmbi.1993.1489. [DOI] [PubMed] [Google Scholar]