

Abstract
We previously characterized the matrix 1 (M1)-binding domain of the influenza A virus NS2/nuclear export protein (NEP), reporting a critical role for the tryptophan (W78) residue that is surrounded by a cluster of glutamate residues in the C-terminal region that interacts with the M1 protein (Akarsu et al., 2003). To gain further insight into the functional role of this interaction, here we used reverse genetics to generate a series of A/WSN/33 (H1N1)-based NS2/NEP mutants for W78 or the C-terminal glutamate residues and assessed their effect on virus growth. We found that simultaneous mutations at three positions (E67S/E74S/E75S) of NS2/NEP were important for inhibition of influenza viral polymerase activity, although the W78S mutant and other glutamate mutants with single substitutions were not. In addition, double and triple substitutions in the NS2/NEP glutamine residues, which resulted in the addition of seven amino acids to the C-terminus of NS1 due to gene overlapping, resulted in virus attenuation in mice. Animal studies with this mutant suggest a potential benefit to incorporating these NS mutations into live vaccines.
Keywords: NS2/NEP, influenza A virus, reverse genetics, polymerase activity, virulence
1. Introduction
The influenza A virus undergoes transcription and replication entirely inside the nucleus. The newly synthesised viral ribonucleoproteins (vRNPs) are then exported for incorporation into virions at the plasma membrane (for review, see Nayak et al., 2004). The viral proteins NS1 and NS2, designated as non-structural (NS) proteins in early studies (Lamb & Choppin, l983), are both encoded by the smallest segment, segment 8 (Inglis et al., 1979; Lamb et al., 1980). Both of these proteins play major roles in viral replication, and could, therefore, be of interest as drug targets.
NS1 has multiple functions during virus infection (for review, see Krug et al., 2003; Hale et al., 2008), including inhibiting the early interferon-α/β-independent (IFN-α/β) antiviral response of cells, by blocking the posttranscriptional processing of cellular antiviral pre-mRNAs (Fortes et al., 1994; Kim et al., 2002). The C-terminal domain of NS1, named the effector domain, binds the 30 kDa subunit of the cleavage and polyadenylation specificity factor (CPSF) (Nemeroff et al., 1998) and the poly(A)-binding protein II (PABII) via residues 223–237 (Chen et al., 1999). These binding events prevent processing of the 3’ ends of cellular pre-mRNAs and thus their nuclear export. The C-terminal domain also directly targets the export machinery and the nuclear pore complex (Satterly et al., 2007). NS1 also blocks the cellular antiviral response mediated by protein kinase R (PKR) (Bargmann et al., 2000; Hatada et al., 1999; Lu et al., 1995) by directly binding to this protein (Tan et al., 1998). It also enhances the translation of viral mRNAs (de la Luna et al., 1995; Enami et al., 1994; Park & Katze, 1995), and shuts off cellular protein synthesis, by interacting with the eukaryotic translational factor 4GI (eIF4GI) (Argon et al., 2000; Burgui et al., 2003) and possibly with the host mRNA binding protein guanine-rich sequence factor 1 (GRSF-1) (Park et al., 1999).
By contrast, the NS2 protein is less well characterised. Although initially designated as a non-structural protein (Lamb et al., 1980), NS2 has since been found in purified viral particles (Richardson & Akkina, 1991). It also accumulates preferentially in the nuclei of infected eukaryotic cells (Greenspan et al., 1985; Smith et al., 1987). Some studies have suggested a role for NS2 in regulating viral RNA replication (Bullido et al., 2001; Odagiri & Tobita, 1990; Odagiri et al., 1994; Perez et al., 2010; Robb et al., 2009; Varble et al., 2010), thereby providing a possible explanation for its nuclear accumulation. The function of NS2 during the viral life cycle became more apparent when O’Neill and colleagues (O'Neill et al., 1998) showed that NS2 was the adaptor between the cellular nuclear export machinery Crm1 and the newly amplified viral genomic segments (vRNPs). NS2 was then renamed nuclear export protein (NEP). The central role of NS2/NEP in vRNP nuclear export through Crm1 was later confirmed by in vivo (Iwatsuki-Horimoto et al., 2004; Neumann et al., 2000) and in vitro studies (Akarsu et al., 2003). Conflicting data, however, have also been published, regarding whether or not the viral matrix protein M1 is sufficient for the nuclear export of vRNPs in the absence of NS2/NEP (Bui et al., 2000), and whether the viral nucleoprotein NP interacts directly with Crm1 (Elton et al., 2001). Despite these contradictory studies, there is good evidence showing the interaction of NS2/NEP with Crm1 (Akarsu et al., 2003; Neumann et al., 2000; O'Neill et al., 1998) and with M1 (Akarsu et al., 2003; Yasuda et al., 1993) and of M1 with the vRNPs (Baudin et al.,2001; Bui et al., 2000; Noton et al., 2007; Ye et al., 1999) suggest the following model for the nuclear export of newly synthesized vRNPs: the C-terminal region of M1 interacts with NP, while its N-terminal nuclear localization signal (NLS) binds to the C-terminal region of NS2/NEP. In turn, the N-terminal region of NS2/NEP recognizes Crm1 and permits the nuclear export of the vRNPs (for review, see Boulo et al., 2007).
We previously solved the three-dimensional structure of the M1-binding domain of NS2/NEP and found that a tryptophan residue (W78) surrounded by a cluster of glutamate residues (E67, E74, E75, E81 and E82) form a negatively charged patch that is a major M1-binding epitope for this protein (Akarsu et al., 2003). All of these residues are highly conserved among the NS2 proteins of the influenza A strains, and residues E67, E74, and E75 are also found in influenza B viruses. Pull-down experiments showed that the W78 of NS2/NEP is a critical binding center for the basic nuclear localization signal (NLS) motif 101RKLKR105 in the N-terminal fragment of M1 (Arzt et al., 2001; Sha & Luo, 1997) via its support of electrostatic interactions with the surrounding acidic glutamate residues.
To further characterize NS2/NEP, here we generated a series of NS2/NEP mutants for W78 or the glutamate residues by using reverse genetics and looked for phenotypic changes in virus replication. Because of the overlap of the E67 and E74 residues of NS2/NEP with the C-terminal region of NS1, mutations at the glutamate residues also cause NS1 to gain mutations or insertions of additional residues at its C-terminus. The effect, if any, of these alterations in NS1 are also discussed, as is the role of NS2/NEP in influenza virus morphogenesis and pathogenesis.
2. Materials and methods
2.1. Cells
293T human embryonic kidney (HEK) cells and HEK293 cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum. Madin-Darby canine kidney (MDCK) cells were maintained in minimal essential medium (MEM) containing 5% newborn calf serum. The 293T cell line is a derivative of the HEK293 cell line into which the gene for the simian virus 40 T antigen was inserted. All cells were maintained at 37°C in 5% CO2.
2.2. Plasmid construction
Genes of the A/WSN/33 (H1N1) virus were cloned into the pPolI vector, as described previously (Neumann et al., 1999). Briefly, vRNAs were reverse-transcribed, the corresponding cDNAs were amplified with a BsmBI site at their 5’ end and cloned into a pPolI plasmid containing the human RNA polymerase I promoter and the mouse RNA polymerase I terminator separated by BsmBI sites. The NS2/NEP mutant genes used in the experiments were constructed by PCR using the following primers (5’-gagagacgtctcgaaataagaagcttgattgaagaagtgagacacaaactgaagataacagag-3’) and (5’-gagagacgtctctatttcttcaaacttctgacctaattgttcccgc-3’) for mut-W78, (5’-gagagacgtctcctgacctaattgcgaccgccattttccgtttctgttttggagtgagtgg-3’) and (5’-gagagacgtctcggtcagaagttttcggaaataagatggttgattgaagaagtgagacacagactg-3’) for mut-E67/E74/tail, and (5’-gagagacgtctcctgacctaattgcgaccgccattttccgtttctgttttggagtgagtgg-3’) and (5’-gagagacgtctcggtcagaagttttcgtcgataagatggttgattgaagaagtgagacacagactg-3’) for mut-E67/E74/E75/tail. These primers have a BsmB1 site at their 5’ ends. The PCR products were isolated by use of a reaction clean-up kit (Qiagen), digested with BsmBI and DpnI (Biolabs) and then transformed into the Escherichia coli strain DH5 for amplification. All constructs were subsequently sequenced to verify the mutations.
To construct pCAGGS plasmids for the expression of NS2/NEP wild-type and mutants E67S/E74S and E67S/E74S/E75S, MDCK cells were infected with recombinant viruses and harvested at 37°C for 5 h. RNA was extracted and reverse transcribed with the NS2/NEP mRNA specific primer (5’-ttttttttttttttttattattaaataagc-3’). Corresponding cDNAs were amplified by PCR with primers NS_EcoRI_F (5’-cacacagaattcatggatccaaacactgtgtcaag-3’) and NS_SalI_R (5’-cacacagtcgacttaaataagctgaaacgagaaagttcttatc-3’). PCR products were separated by agarose gel electrophoresis. Bands corresponding to NS2/NEP were extracted and purified by use of a gel extraction kit (QIAGEN), digested with EcoRI and SalI (NEB) and then cloned into pCAGGS/MCS (controlled by the chicken β-actin promoter) (Kobasa et al., 1997; Niwa et al., 1991).
2.3. Plasmid-based reverse genetics
In all experiments, A/WSN/33 genes were used to produce the viruses, except for the M gene, which was derived from the A/Puerto Rico/8/34 (PR8; H1N1) strain, as described by Neumann et al. (1999). Briefly, recombinant viruses were generated using eight plasmids expressing all viral RNAs, and four plasmids expressing the viral nucleoprotein (NP) and the three subunits of the viral polymerase PB1, PB2 and PA. Transfectant viruses were collected approximately 48 hours post-infection (h.p.i.) and stored as viral stocks for subsequent use. Virus titers were determined by 10-fold serial dilutions of supernatant by a standard plaque assay on MDCK cells in triplicate for each dilution and are given in plaque forming units (PFU).
2.4. Immunofluorescence staining
MDCK cells were infected with recombinant viruses at a multiplicity of infection (MOI) of 5 PFU/cell. At 7 h.p.i., cells were fixed with 4% paraformaldehyde and permeabilized with 0.5% Triton X-100. The cells were then incubated with the primary antibodies specific to the viral proteins. The anti-NS2 antibody was a rabbit polyclonal antibody (laboratory clone R5023) raised against recombinant NS2; the anti-NP antibody was a mouse monoclonal antibody (laboratory clone 68D2) raised against viral NP; the anti-M1 antibody was a mouse monoclonal antibody (laboratory clone 174/4) raised against viral M1; the anti-NS1 antibody was a mouse monoclonal antibody (laboratory clone P601) raised against the recombinant protein. After 3 washes with phosphate-buffered saline (PBS), the cells were incubated with a goat polyclonal secondary antibody coupled with Alexa488 to the anti-mouse and Alexa594 to the anti-rabbit IgGs (Molecular Probes), and washed for a further 3 times. The sub-cellular localizations of the different viral proteins were visualized with an epifluorescent UV microscope (Nikon).
2.5. Replicative properties of mutant viruses
MDCK cells in duplicate wells of 24-well plates were infected with virus at a MOI of 0.01 PFU/cell, overlaid with MEM containing 0.3% BSA, and incubated at 37°C. At selected times, supernatants were measured for infectious virus in plaque assay with MDCK cells.
2.6. Viral pathogenicity in mice
Four-week-old female BALB/c mice (n=4/group) were infected intranasally with 50 μl of viral suspension containing 105 PFU of a recombinant virus in sterile 0.9 % sodium chloride. Animals were monitored daily for survival or changes in body weight over the next 2 weeks.
2.7. Electron microscopy
For thin-section electron microscopy, MDCK cells were infected with virus at an MOI of 5 PFU/cell and incubated in MEM with TPCK-trypsin (1 μg/ml) at 37°C. At 12 h.p.i., the MDCK cells were washed with PBS, prefixed with 2.5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.4) for 1 h at 4°C, and then fixed with 2% osmium tetroxide in the same buffer for 1 h at 4°C. Specimens were subsequently stained en bloc with 1% aqueous uranyl acetate for 30 min at 4°C and processed as previously described (Noda et al., 2006). For negative staining, the culture medium from the MDCK cells infected with wild-type or mutant viruses was collected at 24 h.p.i. onto a Formvar-coated copper grid, stained with 2% phosphotungstic acid solution, and examined with an Hitachi H-7500 electron microscope at 80 kV.
2.8. Replicon assay
HEK293 cells were transfected with pCAGGS plasmids for the expression of the viral proteins PA, PB1, PB2, and NP and pPolI/NP(0)Fluc(0), which encodes vRNA possessing a reporter firefly luciferase gene flanked by 3' and 5' noncoding regions. Either pCAGGS empty vector or pCAGGS plasmids encoding the M1 and/or NS2/NEP protein (wild-type or mutated NS2/NEP) was co-transfected into the cells. As an internal control for the dual-luciferase assay, pGL4.74[hRluc/TK] (Promega, Madison, WI) was used. After transfection, the cells were incubated at 37°C for 24 h, and then luciferase activity was measured on a Glomax microplate luminometer (Promega) according to the manufacturer’s instructions.
2.9. Western blot analysis
The expression levels of influenza viral proteins were measured by western blotting. Cells were lysed in Tris-Glycine SDS sample buffer (Invitrogen, Carlsbad, CA, USA) with 100mM dithiothreitol (DTT), and proteins resolved on 4%–20% tris-glycine gels (BIORAD, Richmond, CA, USA) before being transferred to Immobilon-P transfer membranes (Millipore Corporation, MA, USA). The membranes were blocked in a commercial reagent (Blocking One; Nakarai tesque, Kyoto, Japan) and then incubated with appropriately diluted primary antibodies (clone 65/4 served as an anti-PA monoclonal antibody; clone136/1 as an anti-PB1 monoclonal antibody; clone 18/1 as an anti-PB2 monoclonal antibody; clone R5023 as an anti-NS2/NEP polyclonal antibody; clone 347/4 as an anti-NP monoclonal antibody; and clone 174/4 as an anti-M1 monoclonal antibody) for 2 hours at room temperature or 4°C overnight. Horseradish peroxidase (HRP)-conjugated anti-mouse IgG (GE Healthcare UK Limited, Little Chalfont, UK) or anti-rabbit IgG (CHEMICON, Temecula, CA, USA) and ECL Plus (GE Healthcare, Tokyo, Japan) were used to detect antibody-antigen complexes. In some assays, an immunodetection kit (ABC kit; Vector Laboratories, Burlingame, CA, USA) was used instead of the HRP-conjugated secondary antibody.
2.10. Vaccine studies
Four-week-old female BALB/c mice (n=12) were infected intranasally with the mutant virus (mut-E67/E74/tail); control mice (n=11) were inoculated with PBS. At day 25 post-vaccination (challenge day), all of the mice were similarly infected with 10 median mouse lethal doses (MLD50) of wild-type virus A/WSN/33. Survival was recorded daily for the next 14 days. On days 3 and 6 post-challenge, 3 mice were euthanized and their lungs were harvested and titrated for the presence of virus.
3. Results
3.1. Generation of NS mutants
Based on the crystal structure of the C-terminal M1-binding domain of NS2/NEP (Akarsu et al., 2003), we used reverse genetics to generate a W78S mutant as well as a series of glutamate mutants including single (E75S), double (E67S/E74S), or triple (E67S/E74S/E75S) mutations to look for phenotypic changes in virus replication (Fig. 1). Of note, due to gene overlapping, the NS2/NEP E67S mutation caused G224V/T225A mutations in NS1 and the NS2/NEP E74S mutation suppressed the stop codon of NS1, resulting in the addition of seven residues (termed the “tail” here, residues 231-237) to the C-terminus of NS1 (Fig. 1). We designated each mutant virus as mut-W78, -E75, -E67/E74/tail, or -E67/E74/E75/tail, respectively.
Fig. 1.

NS mutants used in this study. (A) The structure of NS2 reveals an amphipatic helical hairpin that dimerizes as a four-helix bundle. W78 is surrounded by a patch of negative charges (E67, E74, E75, E81, and E82) and appears to be involved in the interaction between NS2/NEP and the NLS of M1 (1). (B) Point mutants created in NS2 are located in the M1-binding domain. The C-terminal sequence of NS1 is also shown. Mutated residues are shown in red.
3.2 Intracellular localization of the NS mutant proteins
We first tested the expression and stability of each mutant by using an immunofluorescence assay. In mut-W78- (Fig. 2) and mut-E75- (data not shown) infected cells, NS1 appeared mostly in the cytoplasm in a distribution pattern similar to that seen with wild-type virus-infected cells, attesting to the expression of intact NS1 protein in these two viruses. NS2/NEP of mut-W78 was present in small amounts in the cytoplasm but was more concentrated in the nucleus, where it was strictly excluded from the nucleolus, as was that of the wild-type virus. Similarly, the localization of mut-E67/E74/tail NS1 emulated that of the wild-type virus, indicating that the additional tail sequence did not affect NS1 expression (Fig. 2). By contrast, the subcellular distribution of mut-E67/E74/E75/tail NS1 shifted towards the nucleoplasm and away from the cytoplasm, indicating that the sequence difference between the additional NS1 tails of mut-E67/E74/tail and mut-E67/E74/E75/tail led to different intracellular localization of these NS1 mutants. NS2/NEP expression and localization of all of the mutants was equivalent to that of the wild-type virus, except for mut-E67/E74/E75/tail, whose nuclear NS2/NEP expression was greater than that of the wild-type virus.
Fig. 2.
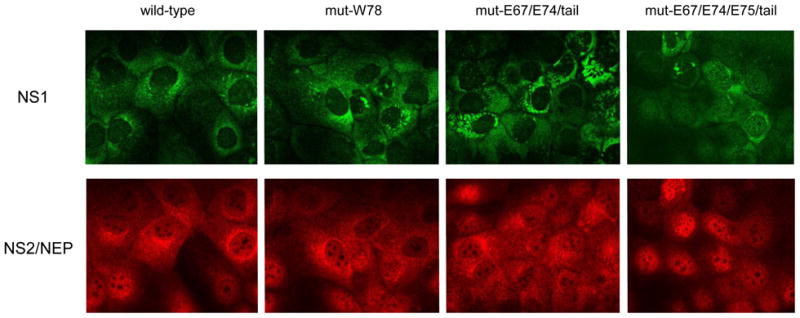
Subcellular localization of the NS2/NEP and NS1 proteins of NS mutants. Virus-infected MDCK cells were fixed at 9 hours post-infection and the viral proteins were visualised by indirect immunofluorescence. NS2 was visualised by staining with a polyclonal primary rabbit and an Alexa594-conjugated secondary antibody. NS1 was stained with a mouse monoclonal primary and an Alexa488-conjugated secondary antibody.
3.3. Growth properties of NS mutants
We compared the growth properties of the mutants with that of wild-type virus in MDCK cells. Each virus was infected at an MOI of 0.01, and growth kinetics were monitored. We found no significant differences between the growth rates of the mutants and the wild-type virus (Fig. 3).
Fig. 3.

Growth curves of NS2/NEP mutant virus in MDCK cells. Growth curves were determined by plaque titration on 12-well plaques, with triplicates for each dilution, followed by coloration with crystal violet to count the formed plaques.
3.4. Virulence in mice of NS mutants
We next tested the pathogenicity in mice of the mutants as an indicator of in vivo virus growth. We found that mut-E67/E74/tail and mut-E67/E74/E75/tail caused no weight loss in mice after intranasal infection with 105 PFU, unlike the wild-type virus, which caused 50% of the mice to die by day 9 post-inoculation, indicating an attenuated phenotype for these mutants in this species (Fig. 4). By contrast, mut-W78 induced transient weight loss in mice but no deaths, indicating that this mutant also exhibited an attenuated phenotype, albeit to a lesser extent than the other mutants.
Fig. 4.

Virulence of NS mutants in mice. Viruses were inoculated intranasally at 105 PFU per mouse (n=4/group). Body weight was measured each day after inoculation. Wild-type virus killed 2 mice at day 9 (50% lethality), whereas none of the mutants killed any mice. Changes in body weight are shown.
3.5. Morphology of NS mutants
The wild-type virus formed mainly spherical virions of 80–120 nm in diameter, as shown by negative staining (Fig. 5). Mut-W78 and mut-E67/E74/tail showed a similar morphology to that of the wild-type, whereas, the morphology of mut-E67/E74/E75/tail changed dramatically with a high degree of pleiomorphism. Moreover, compared to the wild-type and other mutants, thin sections of mut-E67/E74/E75/tail-infected cells showed elongated virus particles and a large population of “empty” particles that appeared to lack any ribonucleoprotein complexes (Fig. 5). Since virion shape and viral genome incorporation of the double glutamate mutant (mut-E67/E74/tail) were similar to those of the wild-type, the seven amino acid extension at the NS1 C-terminus is unlikely to be responsible for the changes in these features in the glutamate triple mutant (mut-E67/E74/E75/tail). Therefore, the cluster of glutamate residues in the M1-binding domain of NS2/NEP appears to plays a role in spherical virion formation.
Fig. 5.

Electron microscopy of NS mutants. Viruses in the supernatant of infected MDCK cells were observed after negative staining. For thin-section observations, virus-infected cells were fixed at 12 hours post-infection and then processed for testing. Bars; 200nm.
3.6. Evaluation of influenza viral polymerase activity
M1 and NS2 are known inhibitors of influenza polymerase (Bullido et al., 2001; Robb et al, 2009). Therefore, we performed a luciferase assay to evaluate the influenza viral polymerase activity of our mutants. Inhibition was observed with NS2/NEP alone, but this inhibition was enhanced when M1 was included in the assay (Fig. 6A). This finding indicates that M1 and NS2/NEP cooperate to inhibit viral polymerase activity. Double (E67S/E74S) mutations at the cluster of NS2/NEP glutamates reduced NS2/NEP the ability to inhibit the polymerase activity. An addition mutation in this glutamate cluster (triple mutant; E67S/E74S/E75S) further reduced this activity. In the presence of M1, further inhibition was observed with these mutant NS2/NEPs. Western blot analyses of these mutants demonstrated that the mutations did not alter NS2/NES stability (Fig. 6B). These results indicate that residues E67/E74/E75 are important for the interaction of NS2/NEP with M1 and for the inhibition of influenza viral polymerase activity.
Fig. 6.

Comparison of the effect of NS mutations on viral polymerase activity. Influenza polymerase activity was measured as reporter luciferase activity. Plasmids for the expression of mutated NS2/NEP without M1 (dark gray bars) or with M1 (light gray bars) were transfected together with protein expression plasmids for the viral polymerase subunits and pPolI/NP(0)Fluc(0). At 24 h post-transfection, cells were subjected to the dual-luciferase assay (A) and Western blot analysis (B). Error bars indicate the standard error of the mean of three independent experiments.
3.7. Potential of an NS mutant as a live vaccine
For live vaccines, viruses must replicate to high titers and be attenuated. Mut-E67/E74 appears to fulfill these criteria. We therefore tested the immunogenicity of this virus in a mouse model. We intranasally immunized mice with mut-E67/E74 and 25 days after immunization, challenged them with 10 LD50 of wild-type WSN virus. All immunized mice survived lethal WSN challenge and no virus was recovered from the lungs of the challenged mice, unlike the controls (Table 1). These results demonstrate that this NS mutant has potential as a live vaccine.
Table 1.
Protective immunity of an NS mutant to lethal challenge with a virulent virusX
| Virus | Virus titer in lung (log10PFU/g) |
Lethality (dead no./total no.) | |
|---|---|---|---|
| Day 3 | Day 6 | ||
| mut-E67/E74/tail | <2.5, <2.5, <2.5 | <2.5, <2.5, <2.5 | 0/11 (0%) |
| mock (PBS) | 7.18, 7.25, 7.36 | 3.70, 4.81, 5.41 | 11/11 (100%) |
Mice were intranasally immunized with mut-E67/E74/tail (105 PFU/mouse; n=12) or Mock (PBS; n=11). Twenty-five days after immunization, they were challenged with 10 LD50 of wild-type WSN virus.
Virus titers in lungs were determined from 3 mice per group; individual titers are shown (n=3). Detection limit is 102.5 PFU/g.
Lethality was determined at 14 days post-challenge with wild-type WSN virus (10MLD50).
4. Discussion
We previously identified an important role for a tryptophan (W78) residue surrounded by a cluster of glutamate residues in the C-terminal region of the NS2/NEP protein of influenza A virus for binding to the N-terminal region of M1 (Akarsu et al., 2003). In this study, we generated a series of NS2/NEP mutants for W78 and three glutamate residues (E67, E74, and E75) conserved among influenza A and B viruses and assessed the effect of these mutations on the interaction between M1 and NS2/NEP in virus replication in vitro and in vivo. We found that these residues, in concert, are important for efficient virus growth in mice, but not in cell culture, an important attribute for attenuating mutations in live vaccines.
Amino acid substitution E67S or E74S in NS2/NEP was accompanied by G224V/T225A mutations or disruption of the stop codon by adding seven amino acid residues (231FGNKMVD237) to NS1, due to gene overlapping. It is likely that the additional C-terminal amino acids of NS1 alter its function, since this region contains the PABII-binding domain (Chen et al., 1999) and a possible PDZ element (Jackson et al., 2008; Obenauer et al., 2006), although several natural isolates, such as A/Udorn/72 (H3N2), possess extended NS1s (237 amino compared to 230 in WSN NS1), which are the same size as those of the mutants containing E74S. Here, we showed that the mut-E67/E74/tail possessed similar virion morphology and growth kinetics to those of the wild-type virus in MDCK cells, indicating that NS1 mutants containing an artificially extended tail do not affect virus replication in this cell line. However, we found that the intracellular localization of mut-E67/E74/E75/tail NS1 was different from that of mut-E67/E74/tail NS1, suggesting that the sequence difference between these NS1 tails could alter the properties of NS1 in cells. A recent study indicated that a C-terminal extended tail containing two arginines at positions 231 and 232 of NS1 (e.g., Udorn strain) contributes to the formation of a second nuclear localization signal (NLS2) which also functions as a nucleolar localization signal (NoLS) (Melen et al., 2007). The total charge at this extended region of NS1 differs between mut-E67/E74/E75/tail (231FVDKMVD237) and mut-E67/E74/tail (231FGNKMVD237), raising the possibility that the different localization observed for these two NS1s may be explained by this property, without any significant difference in overall growth of the viruses in MDCK cells. But, we cannot rule out the effects of NS1 extension on the viral properties of mice.
Previous studies have shown that the two NS1 lengths—230 amino acids (e.g., the WSN strain) and 237 amino acids (e.g., the Udorn strain)—were evident among human influenza viruses, whereas the NS1s of avian-origin viruses are consistently 230 amino acids (with the exception of A/turkey/Oregon/71 (H7N3) and A/Vietnam/1203/2004 (H5N1)), suggesting the possiblity of host-specific selection pressure (Suarez & Perdue, 1998). This idea may be supported by our results that mutants with NS1s containing extended tails were attenuated in mice, although the precise molecular mechanism for this attenuation is unknown due to the presence of the concomitant NS2/NEP mutations.
NS2/NEP regulates RNA synthesis (Bullido et al., 2001; Odagiri & Tobita, 1990; Odagiri et al., 1994; Perez et al., 2010; Robb et al., 2009; Varble et al., 2010), while M1 inhibits the transcriptional activity of RNPs (5, Elster et al., 1997; Perez & Donis, 1998; Watanabe et al., 1996; Ye et al., 1989; Zvonarjev & Ghendon, 1980). However the mechanisms by which these viral proteins affect RNA synthesis remained unknown. Previously, we demonstrated the interaction of NS2/NEP with M1 at the structural level (Akarsu et al., 2003). In this study, we showed that M1 and NS2/NEP cooperate to inhibit the viral polymerase activity, and that the glutamate residues at 67, 74, and 75 of NS2/NEP are important for both the M1 interaction and the inhibitory effect on the viral polymerase. Robb et al. (2009) found that a point mutation at W78 of NS2/NEP does not affect the ability of NS2/NEP to regulate viral RNA levels. Our analyses are consistent with theirs in that a single mutation at W78 showed no effect in vitro. Previously, we found that this tryptophan residue is surrounded by a ring of glutamate residues (see Fig. 1), and that the interaction between NS2/NEP and M1 probably has a significant electrostatic component (Akarsu et al., 2003). Therefore, the entire charge of this domain is important for MS2/NEP-M1 interaction and function.
We also found that simultaneous mutations at three glutamate residues (mut-E67/E74/E75/tail) of NS2/NEP resulted in a change in virion shape, showing pleiomorphism as well as a decrease in the ribonucleoprotein complex content inside the virus particles; this was not apparent with mut-W78 and mut-E67/E74/tail. These results establish, for the first time, that NS2/NEP influences viral particle shape and is involved in the assembly/incorporation of genomic segments. Such morphological changes look similar to those that occur in viruses with mutations in the NLS of M1 (Burleigh et al., 2005) and those with mutations in the cytoplasmic tails of their HA, NA (Jin et al., 1997), or M2 (Iwatsuki-Horimoto et al., 2006). It has been shown that i) NS2/NEP is present in a viral particle (Richardson & Akkina, 1991), that ii) NS2/NEP interacts directly with the NLS motif of M1 (Akarsu et al., 2003), and that iii) M1 binds not only to the lipids (Baudin et al., 2001; Ruigrok & Baudin, 1995), thus possibly covering the inside surface of the viral envelope, but also to the viral glycoproteins HA and NA (Ali et al., 2000; Enami & Enami, 1996) and the vRNPs (Noton et al., 2007; Watanabe et al., 1996; Ye et al., 1999) through its C-terminal region (Akarsu et al., 2003; Baudin et al., 2001). Our observations, together with these latest findings, lead us to the hypothesis for virion assembly that the complex of nuclear M1 (ncM1) and NS2/NEP with vRNPs is translocated to the cytoplasm and goes to the plasma membrane covered by cytoplasmic M1 (cyM1), which interacts with the HA, NA, and M2 cytoplasmic tails, resulting in binding to cyM1 via an ncM1/cyM1 interaction by M1 self-polymerization (Baudin et al., 2001). Thus, any decreased interaction between ncM1 and NS2/NEP as a result of a mutation(s) would alter virion morphology.
Here, we found that mut-W78 showed a slightly attenuated phenotype compared to the wild-type virus, indicating some effect of the W78 of NS2/NEP in viral replication in vivo and in vitro. By contrast, the NS2/NEP mutants with double and triple glutamate substitutions showed a strikingly attenuated phenotype in mice, possibly suggesting weaker binding of these NS2/NEPs mutants to M1. In addition, as discussed above, we cannot rule out the possibility that mutations in NS1 contribute to the attenuated phenotype in mice of these mutants; the extended tail of NS1 with mut-E67/E74/tail and mut-E67/E74/E75/tail may mask the C-terminal cis-acting motifs, such as the PABII-binding and PDZ domains, that may indirectly affect viral polymerase activity in mice but not in cell culture.
Our study demonstrates that mutations introduced into mut-E67/E74/tail can be used to attenuate influenza viruses for use as live vaccines. Since mut-E67/E74/tail replicated as well as the wild-type virus in cell culture, it and similar viruses may serve as cell culture-based live influenza vaccines, negating issues of antigenic alteration caused by propagation of seed viruses in embryonated chicken eggs (Robertson, 1993). NS mutants should be considered as an option for the development of live vaccines against influenza.
Acknowledgments
We thank S. Watson for editing the manuscript. This work was supported, in part, by the ERATO (Exploratory Research for Advanced Technology) grant from the Japan Science and Technology Agency and by Grants-in-Aid for Specially Promoted Research and for Scientific Research B from the Ministry of Education, Culture, Sports, Science, and Technology of Japan, and by National Institute of Allergy and Infectious Diseases Public Health Service research grants, USA. H.A. was supported by a JSPS (Japan Society for Promotion of Science) Fellowship for Foreign Researchers (Short-term).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akarsu H, Burmeister WP, Petosa C, Petit I, Muller CW, Ruigrok RW, Baudin F. Crystal structure of the M1 protein-binding domain of the influenza A virus nuclear export protein (NEP/NS2) EMBO J. 2003;22:4646–4655. doi: 10.1093/emboj/cdg449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali A, Avalos RT, Ponimaskin E, Nayak DP. Influenza virus assembly: effect of influenza virus glycoproteins on the membrane association of M1 protein. J Virol. 2000;74:8709–8719. doi: 10.1128/jvi.74.18.8709-8719.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aragon T, de la Luna S, Novoa I, Carrasco L, Ortin J, Nieto A. Eukaryotic translation initiation factor 4GI is a cellular target for NS1 protein, a translational activator of influenza virus. Mol Cell Biol. 2000;20:6259–6268. doi: 10.1128/mcb.20.17.6259-6268.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arzt S, Baudin F, Barge A, Timmins P, Burmeister WP, Ruigrok RW. Combined results from solution studies on intact influenza virus M1 protein and from a new crystal form of its N-terminal domain show that M1 is an elongated monomer. Virology. 2001;279:439–446. doi: 10.1006/viro.2000.0727. [DOI] [PubMed] [Google Scholar]
- Baudin F, Petit I, Weissenhorn W, Ruigrok RW. In vitro dissection of the membrane and RNP binding activities of influenza virus M1 protein. Virology. 2001;281:102–108. doi: 10.1006/viro.2000.0804. [DOI] [PubMed] [Google Scholar]
- Bergmann M, Garcia-Sastre A, Carnero E, Pehamberger H, Wolff K, Palese P, Muster T. Influenza virus NS1 protein counteracts PKR-mediated inhibition of replication. J Virol. 2000;74:6203–6206. doi: 10.1128/jvi.74.13.6203-6206.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulo S, Akarsu H, Ruigrok RW, Baudin F. Nuclear traffic of influenza virus proteins and ribonucleoprotein complexes. Virus Res. 2007;124:12–21. doi: 10.1016/j.virusres.2006.09.013. [DOI] [PubMed] [Google Scholar]
- Bui M, Wills EG, Helenius A, Whittaker GR. Role of the influenza virus M1 protein in nuclear export of viral ribonucleoproteins. J Virol. 2000;74:1781–1786. doi: 10.1128/jvi.74.4.1781-1786.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullido R, Gomez-Puertas P, Saiz MJ, Portela A. Influenza A virus NEP (NS2 protein) downregulates RNA synthesis of model template RNAs. J Virol. 2001;75:4912–4917. doi: 10.1128/JVI.75.10.4912-4917.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgui I, Aragon T, Ortin J, Nieto A. PABP1 and eIF4GI associate with influenza virus NS1 protein in viral mRNA translation initiation complexes. J Gen Virol. 2003;84:3263–3274. doi: 10.1099/vir.0.19487-0. [DOI] [PubMed] [Google Scholar]
- Burleigh LM, Calder LJ, Skehel JJ, Steinhauer DA. Influenza a viruses with mutations in the m1 helix six domain display a wide variety of morphological phenotypes. J Virol. 2005;79:1262–1270. doi: 10.1128/JVI.79.2.1262-1270.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Li Y, Krug RM. Influenza A virus NS1 protein targets poly(A)-binding protein II of the cellular 3'-end processing machinery. EMBO J. 1999;18:2273–2283. doi: 10.1093/emboj/18.8.2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Luna S, Fortes P, Beloso A, Ortin J. Influenza virus NS1 protein enhances the rate of translation initiation of viral mRNAs. J Virol. 1995;69:2427–2433. doi: 10.1128/jvi.69.4.2427-2433.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elster C, Larsen K, Gagnon J, Ruigrok RWH, Baudin F. Influenza virus M1 protein binds to RNA through its nuclear localization signal. J Gen Virol. 1997;78:1589–1596. doi: 10.1099/0022-1317-78-7-1589. [DOI] [PubMed] [Google Scholar]
- Elton D, Simpson-Holley M, Archer K, Medcalf L, Hallam R, McCauley J, Digard P. Interaction of the influenza virus nucleoprotein with the cellular CRM1-mediated nuclear export pathway. J Virol. 2001;75:408–419. doi: 10.1128/JVI.75.1.408-419.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enami K, Sato TA, Nakada S, Enami M. Influenza virus NS1 protein stimulates translation of the M1 protein. J Virol. 1994;68:1432–1437. doi: 10.1128/jvi.68.3.1432-1437.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enami M, Enami K. Influenza virus hemagglutinin and neuraminidase glycoproteins stimulate the membrane association of the matrix protein. J Virol. 1996;70:6653–6657. doi: 10.1128/jvi.70.10.6653-6657.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortes P, Beloso A, Ortin J. Influenza virus NS1 protein inhibits pre-mRNA splicing and blocks mRNA nucleocytoplasmic transport. EMBO J. 1994;13:704–712. doi: 10.1002/j.1460-2075.1994.tb06310.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenspan D, Krystal M, Nakada S, Arnheiter H, Lyles DS, Palese P. Expression of influenza virus NS2 nonstructural protein in bacteria and localization of NS2 in infected eucaryotic cells. J Virol. 1985;54:833–843. doi: 10.1128/jvi.54.3.833-843.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale BG, Randall RE, Ortín J, Jackson D. The multifunctional NS1 protein of influenza A viruses. J Gen Virol. 2008;89:2359–76. doi: 10.1099/vir.0.2008/004606-0. [DOI] [PubMed] [Google Scholar]
- Hatada E, Saito S, Fukuda R. Mutant influenza viruses with a defective NS1 protein cannot block the activation of PKR in infected cells. J Virol. 1999;73:2425–2433. doi: 10.1128/jvi.73.3.2425-2433.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inglis SC, Barrett T, Brown CM, Almond JW. The smallest genome RNA segment of influenza virus contains two genes that may overlap. Proc Natl Acad Sci USA. 1979;76:3790–3794. doi: 10.1073/pnas.76.8.3790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwatsuki-Horimoto K, Horimoto T, Fujii Y, Kawaoka Y. Generation of influenza A virus NS2 (NEP) mutants with an altered nuclear export signal sequence. J Virol. 2004;78:10149–10155. doi: 10.1128/JVI.78.18.10149-10155.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwatsuki-Horimoto K, Horimoto T, Noda T, Kiso M, Maeda J, Watanabe S, Muramoto Y, Fujii K, Kawaoka Y. The cytoplasmic tail of the influenza A virus M2 protein plays a role in viral assembly. J Virol. 2006;80:5233–5240. doi: 10.1128/JVI.00049-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson D, Hossain MJ, Hickman D, Perez DR, Lamb RA. A new influenza virus virulence determinant: the NS1 protein four C-terminal residues modulate pathogenicity. Proc Natl Acad Sc USA. 2008;105:4381–4386. doi: 10.1073/pnas.0800482105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin H, Leser GP, Zhang J, Lamb RA. Influenza virus hemagglutinin and neuraminidase cytoplasmic tails control particle shape. EMBO J. 1997;16:1236–1247. doi: 10.1093/emboj/16.6.1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MJ, Latham AG, Krug RM. Human influenza viruses activate an interferon-independent transcription of cellular antiviral genes: outcome with influenza A virus is unique. Proc Natl Acad Sci USA. 2002;99:10096–10101. doi: 10.1073/pnas.152327499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobasa D, Rogeres ME, Wells K, Kawaoka Y. Neuraminidase hemadsorption activity, conserved in avian influenza A viruses, does not influence viral replication in ducks. J Virol. 1997;71:6706–6713. doi: 10.1128/jvi.71.9.6706-6713.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug RM, Yuan W, Noah DL, Latham AG. Intracellular warfare between human influenza viruses and human cells: the roles of the viral NS1 protein. Virology. 2003;309:181–189. doi: 10.1016/s0042-6822(03)00119-3. [DOI] [PubMed] [Google Scholar]
- Lamb RA, Choppin PW. The gene structure and replication of influenza virus. Annu Rev Biochem. 1983;52:467–506. doi: 10.1146/annurev.bi.52.070183.002343. [DOI] [PubMed] [Google Scholar]
- Lamb RA, Choppin PW, Chanock RM, Lai CJ. Mapping of the two overlapping genes for polypeptides NS1 and NS2 on RNA segment 8 of influenza virus genome. Proc Natl Acad Sci USA. 1980;77:1857–1861. doi: 10.1073/pnas.77.4.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Wambach M, Katze MG, Krug RM. Binding of the influenza virus NS1 protein to double-stranded RNA inhibits the activation of the protein kinase that phosphorylates the elF-2 translation initiation factor. Virology. 1995;214:222–228. doi: 10.1006/viro.1995.9937. [DOI] [PubMed] [Google Scholar]
- Melen K, Kinnunen L, Fagerlund R, Ikonen N, Twu KY, Krug RM, Julkunen I. Nuclear and nucleolar targeting of influenza A virus NS1 protein: striking differences between different virus subtypes. J Virol. 2007;81:5995–6006. doi: 10.1128/JVI.01714-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak DP, Hui EK, Barman S. Assembly and budding of influenza virus. Virus Res. 2004;106:147–165. doi: 10.1016/j.virusres.2004.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemeroff ME, Barabino SM, Li Y, Keller W, Krug RM. Influenza virus NS1 protein interacts with the cellular 30 kDa subunit of CPSF and inhibits 3'end formation of cellular pre-mRNAs. Mol Cell. 1998;1:991–1000. doi: 10.1016/s1097-2765(00)80099-4. [DOI] [PubMed] [Google Scholar]
- Neumann G, Hughes MT, Kawaoka Y. Influenza A virus NS2 protein mediates vRNP nuclear export through NES-independent interaction with hCRM1. EMBO J. 2000;19:6751–6758. doi: 10.1093/emboj/19.24.6751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann G, Watanabe T, Ito H, Watanabe S, Goto H, Gao P, Hughes M, Perez DR, Donis R, Hoffmann E, Hobom G, Kawaoka Y. Generation of influenza A viruses entirely from cloned cDNAs. Proc Natl Acad Sci USA. 1999;96:9345–50. doi: 10.1073/pnas.96.16.9345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–199. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- Noda T, Sagara H, Yen A, Takada A, Kida H, Cheng RH, Kawaoka Y. Architecture of ribonucleoprotein complexes in influenza A virus particles. Nature. 2006;439:490–492. doi: 10.1038/nature04378. [DOI] [PubMed] [Google Scholar]
- Noton SL, Medcalf E, Fisher D, Mullin AE, Elton D, Digard P. Identification of the domains of the influenza A virus M1 matrix protein required for NP binding, oligomerization and incoporation into virions. J Gen Virol. 2007;88:2280–2290. doi: 10.1099/vir.0.82809-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obenauer JC, Denson J, Mehta PK, Su X, Mukatira S, Finkelstein DB, Xu X, Wang J, Ma J, Fan Y, Rakestraw KW, Webster RG, Hoffmann E, Krauss S, Zheng J, Zhang Z, Naeve W. Large-scale sequence analysis of avian influenza isolates. Science. 2006;311:1576–1580. doi: 10.1126/science.1121586. [DOI] [PubMed] [Google Scholar]
- Odagiri T, Tobita K. Mutation in NS2, a nonstructural protein of influenza A virus, extragenically causes aberrant replication and expression of the PA gene and leads to generation of defective interfering particles. Proc Natl Acad Sci USA. 1990;87:5988–5992. doi: 10.1073/pnas.87.15.5988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odagiri T, Tominaga K, Tobita K, Ohta S. An amino acid change in the non-structural NS2 protein of an influenza A virus mutant is responsible for the generation of defective interfering (DI) particles by amplifying DI RNAs and suppressing complementary RNA synthesis. J Gen Virol. 1994;75 (Pt 1):43–53. doi: 10.1099/0022-1317-75-1-43. [DOI] [PubMed] [Google Scholar]
- O'Neill RE, Talon J, Palese P. The influenza virus NEP (NS2 protein) mediates the nuclear export of viral ribonucleoproteins. EMBO J. 1998;17:288–296. doi: 10.1093/emboj/17.1.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park YW, Katze MG. Translational control by influenza virus. Identification of cis-acting sequences and trans-acting factors which may regulate selective viral mRNA translation. J Biol Chem. 1995;270:28433–28439. doi: 10.1074/jbc.270.47.28433. [DOI] [PubMed] [Google Scholar]
- Park YW, Wilusz J, Katze MG. Regulation of eukaryotic protein synthesis: selective influenza viral mRNA translation is mediated by the cellular RNA-binding protein GRSF-1. Proc Natl Acad Sci USA. 1999;96:6694–6699. doi: 10.1073/pnas.96.12.6694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez DR, Donis RO. The matrix 1 protein of influenza A virus inhibits the transcriptase activity of a model influenza reporter genome in vivo. Virology. 1998;249:52–61. doi: 10.1006/viro.1998.9318. [DOI] [PubMed] [Google Scholar]
- Perez JT, Varble A, Sachidanandam R, Zlatev I, Manoharan M, García-Sastre A, Tenoever BR. Influenza A virus-generated small RNAs regulate the switch from transcription to replication. Proc Natl Acad Sci U S A. 2010;107:11525–11530. doi: 10.1073/pnas.1001984107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson JC, Akkina RK. NS2 protein of influenza virus is found in purified virus and phosphorylated in infected cells. Arch Virol. 1991;116:69–80. doi: 10.1007/BF01319232. [DOI] [PubMed] [Google Scholar]
- Robertson JS. Clinical influenza virus and the embryonated hen’s eggs. Rev Med Virol. 1993;3:97–108. [Google Scholar]
- Robb NC, Smith M, Vreede FT, Fodor E. NS2/NEP protein regulates transcription and replication of the influenza virus RNA genome. J Gen Virol. 2009;90:1398–1407. doi: 10.1099/vir.0.009639-0. [DOI] [PubMed] [Google Scholar]
- Ruigrok RW, Baudin F. Structure of influenza virus ribonucleoprotein particles. II. Purified RNA-free influenza virus ribonucleoprotein forms structures that are indistinguishable from the intact influenza virus ribonucleoprotein particles. J Gen Virol. 1995;76 (Pt 4):1009–1014. doi: 10.1099/0022-1317-76-4-1009. [DOI] [PubMed] [Google Scholar]
- Satterly N, Tsai PL, van Deursen J, Nussenzveig DR, Wang Y, Faria PA, Levay A, Levy DE, Fontoura BM. Influenza virus targets the mRNA export machinery and the nuclear pore complex. Proc Natl Acad Sci USA. 2007;104:1853–1858. doi: 10.1073/pnas.0610977104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha B, Luo M. Structure of a bifunctional membrane-RNA binding protein, influenza virus matrix protein M1. Nat Struct Biol. 1997;4:239–244. doi: 10.1038/nsb0397-239. [DOI] [PubMed] [Google Scholar]
- Smith GL, Levin JZ, Palese P, Moss B. Synthesis and cellular location of the ten influenza polypeptides individually expressed by recombinant vaccinia viruses. Virology. 1987;160:336–345. doi: 10.1016/0042-6822(87)90004-3. [DOI] [PubMed] [Google Scholar]
- Suarez DL, Perdue ML. Multiple alignment comparison of the non- structural genes of influenza A viruses. Virus Res. 1998;54:59–69. doi: 10.1016/s0168-1702(98)00011-2. [DOI] [PubMed] [Google Scholar]
- Tan SL, Katze MG. Biochemical and genetic evidence for complex formation between the influenza A virus NS1 protein and the interferon-induced PKR protein kinase. J Interferon Cytokine Res. 1998;18:757–766. doi: 10.1089/jir.1998.18.757. [DOI] [PubMed] [Google Scholar]
- Varble A, Chua MA, Perez JT, Manicassamy B, García-Sastre A, Tenoever BR. Engineered RNA viral synthesis of microRNAs. Proc Natl Acad Sci U S A. 2010:11519–11524. doi: 10.1073/pnas.1003115107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K, Handa H, Mizumoto K, Nagata K. Mechanism for inhibition of influenza virus RNA polymerase activity by matrix protein. J Virol. 1996;70:241–247. doi: 10.1128/jvi.70.1.241-247.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda J, Nakada S, Kato A, Toyoda T, Ishihama A. Molecular assembly of influenza virus: association of the NS2 protein with virion matrix. Virology. 1993;196:249–255. doi: 10.1006/viro.1993.1473. [DOI] [PubMed] [Google Scholar]
- Ye Z, Baylor NW, Wagner RR. Transcription-inhibition and RNA-binding domains of influenza A virus matrix protein mapped with anti-idiotype antibodies and systhetic peptides. J Virol. 1989;63:3586–3594. doi: 10.1128/jvi.63.9.3586-3594.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Z, Liu T, Offringa DP, McInnis J, Levandowski RA. Association of influenza virus matrix protein with ribonucleoproteins. J Virol. 1999;73:7467–7473. doi: 10.1128/jvi.73.9.7467-7473.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zvonarjev AY, Ghendon YZ. Influence of membrane (M) protein on influenza A virus virion transcriptase activity in vitro and its susceptibility to rimantadine. J Virol. 1980;33:583–586. doi: 10.1128/jvi.33.2.583-586.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]