

Abstract
Understanding mechanisms of glaucomatous optic nerve damage is essential for developing effective therapies to augment conventional pressure-lowering treatments. This requires that we understand not only the physical forces in play, but the cellular responses that translate these forces into axonal injury. The former are best understood by using primate models, in which a well-developed lamina cribrosa, peripapillary sclera and blood supply are most like that of the human optic nerve head. However, determining cellular responses to elevated intraocular pressure (IOP) and relating their contribution to axonal injury requires cell biology techniques, using animals in numbers sufficient to perform reliable statistical analyses and draw meaningful conclusions. Over the years, models of chronically elevated IOP in laboratory rats and mice have proven increasingly useful for these purposes. While lacking a distinct collagenous lamina cribrosa, the rodent optic nerve head (ONH) possesses a cellular arrangement of astrocytes, or glial lamina, that ultrastructurally closely resembles that of the primate. Using these tools, major insights have been gained into ONH and the retinal cellular responses to elevated IOP that, in time, can be applied to the primate model and, ultimately, human glaucoma.
Keywords: glaucoma, intraocular pressure, animal models, pressure-induced optic nerve damage, optic nerve head
1. Introduction
Clinically, glaucomatous optic nerve damage is recognized by enlargement and deepening of the optic cup, accompanied by thinning of the neural rim. In a majority of these cases, the thinning appears regional, with a predilection for the superior and inferior portions of the optic nerve head (ONH), and progresses to result in expansion of the nerve tissue underneath the scleral rim, or undermining of the disc margin. This is accompanied by corresponding thinning and absence of the nerve fiber layer in these regions. It is the loss of axons in the nerve fiber layer that leads to loss of vision, with characteristic patterns of visual field loss that correspond to the above regional damage.
Although these characteristics are well known, the cellular mechanisms involved are poorly understood. Understanding these processes will lead to new methods of preventing chronic glaucomatous vision loss when conventional IOP-lowering treatments either fail to prevent progression or adequate target pressures cannot be achieved due to ineffectiveness or poor tolerance to medication, or failure of conventional filtering surgery. Because studies designed to understand the cellular mechanisms of this process are not appropriate in humans, development of appropriate animal models of this disease is critical to gaining this knowledge.
2. Anatomy of the rodent optic nerve head
The ONH is generally considered the primary site of injury in glaucoma. This is supported by histologic evaluation of the ONH in human glaucoma, and the observation that experimental elevation of intraocular pressure (IOP) produces obstruction of orthograde and retrograde axonal transport at the level of the ONH.(Quigley & Addicks 1980; Quigley 1981) Because this obstruction has been localized ultrastructurally to coincide with the collagenous beams of the lamina cribrosa, and because the pattern of axonal damage and subsequent visual field loss in human glaucoma corresponds best to its structure, the lamina has received significant attention in our thinking about mechanisms of glaucomatous optic nerve damage. It is therefore important to understand the anatomy of the rodent ONH if we are to fully appreciate what rodent models of glaucoma can teach us about human glaucoma.
The rodent ONH possesses several similarities to that of the primate eye, in addition to some important differences. (Morrison 1995) When viewed in a vertical longitudinal section, the neural component of the nerve head, which consists primarily of unmyelinated optic nerve fiber bundles, astrocytes and capillaries, is narrow at the level of the sclera, and then expands gradually to form the fully myelinated retrobulbar optic nerve, which begins well posterior to the sclera. (Figure 1)
Figure 1.
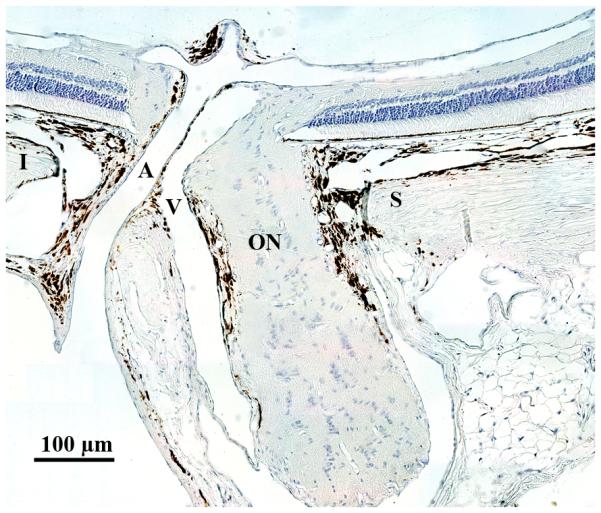

Anatomy of the rat ONH. (A) Vertical longitudinal section shows relationships of the unmyelinated optic nerve (ON) to the superior (S) and inferior (I) sclera and to the central retinal artery (A) and vein (V). The optic nerve becomes fully myelinated approximated 1 mm posterior to the sclera. Cross section (B) demonstrates these same relationships, although inferior sclera is beyond the central retinal artery, outside of the frame of this image.
Scattered patches of connective tissue are seen throughout the transition zone, many of which contain capillaries and may represent a rudimentary lamina, with a collagenous composition similar to that of the lamina cribrosa of the primate.(Morrison et al. 1989; Morrison 1995) Although the rodent ONH has a poorly developed collagenous lamina cribrosa,(Morrison 1995; May & Lutjen-Drecoll 2002; May 2003) it does possess astrocytes that are oriented transversely and arranged in columns that border the axon bundles, analogous to glial columns of the primate ONH. (Figure 2A) Ultrastructurally, these astrocytes extend their fine processes into the axonal bundles.(Figure 2B) Anatomic studies of transgenic mice that express green fluorescent protein in many, but not all astrocytes, indicate that their processes reach well into the ONH, often overlapping as they ensheath axonal bundles.(Sun et al. 2009) Because these extensions intimately contact axons throughout the ONH, including the bordering connective tissues, the astrocytes may provide a direct link between the axons, sclera and blood vessels of the ONH by which physical forces and the physiologic effects of changing perfusion from fluctuating IOP produce axonal injury.
Figure 2.


Cellular anatomy of the rat ONH. (A) Immunofluorescent staining of the anterior ONH with antibodies to GFAP showing arrangement of astrocytes (red) in glial columns, with attachment of their processes to the connective tissue borders of the optic nerve. Blue, DAPI- stained nuclei; S, superior sclera; V, retinal vein. (B) Transmission electron micrograph cross section of axon bundle in the rat ONH. Relatively dark astrocyte (A) processes are seen throughout the axon bundle to form intimate contact with nearly all of the lighter axons, cut in cross section.
Scanning electron microscopy of microvascular corrosion castings demonstrates that the blood supply to the rat ONH originates in the ophthalmic artery, which lies inferior to the optic nerve in the optic nerve sheath, and then trifurcates immediately posterior to the globe into the central retinal artery and two long posterior ciliary arteries.(Morrison et al. 1999; Sugiyama et al. 1999; Morrison et al. 2005) In the rat, the central retinal artery enters the eye inferior to, rather than in the center, of the neural ONH.(Figure 1) As with the primate,(Cioffi & Van Buskirk 1994) the nerve fiber layer of the ONH and retina is perfused by the central retinal artery, while the majority of blood supply to the deeper, “laminar” layers appears to be centripetal, from branches of the posterior ciliary arteries that produce an incomplete, “Zinn-Haller”-like circle.
At all levels of the rat ONH, venous blood drains into the central retinal vein, which lies between the neural optic nerve and central retinal artery. Originating from the major retinal veins, this vein also joins a broad sinus which is derived, in part, from the choroidal system.(Morrison et al. 1999; Eida et al. 2001) Posteriorly, the central retinal vein travels in the optic nerve sheath along with other large veins that are connected to the choroidal vasculature.
Similar studies of the mouse ONH demonstrate that the central retinal artery and vein also lie mostly inferior to the neural portion of the ONH.(May & Lutjen-Drecoll 2002) However, the unmyelinated portion of the mouse ONH appears to receive its capillary supply from scattered, posteriorly directed vessels that originate from the central retinal artery in the inner retina, rather than from posterior ciliary arteries. This fundamental difference in ONH vasculature may be important when comparing results between mouse and rat glaucoma models.
3. Animal models for studying glaucomatous optic neuropathy
Animal models developed for studying the cellular events of glaucomatous optic neuropathy can be broadly separated into those that are independent of IOP and those that rely on elevation of IOP. The former are generally based on specific hypotheses of the mechanism of glaucomatous retinal ganglion cell death. These have been thoroughly presented in several published reviews and will not be discussed here. (Lindsey & Weinreb 2005; Pang & Clark 2007; Johnson & Tomarev 2009) The latter all rely on elevation of IOP to reproduce a major risk factor for glaucoma and its effects on the visual system. These will be presented briefly as they are the primary sources for the data to be reviewed here. More detailed discussions of each can be found in the above-mentioned reviews, as well as others.(Morrison et al. 2005; Morrison et al. 2008; McKinnon et al. 2009)
3.1. “Pressure-dependent” models of glaucomatous optic neuropathy
Elevated IOP is a widely recognized and well-documented glaucoma risk factor.(Sommer 1989; Klein et al. 1992) In addition, several large multicenter trials have demonstrated that lowering IOP has a beneficial effect on either preventing or at least slowing the rate of progression of vision loss in patients with normal tension glaucoma, ocular hypertension, early glaucoma and advanced glaucoma.(Drance 1999; AGIS-Investigators 2000; Heijl et al. 2002; Kass et al. 2002) Thus, models of elevated IOP should provide useful insights into many, if not all, forms of glaucoma.
Rodent models of elevated IOP can either occur spontaneously or be produced experimentally. Spontaneous models of elevated IOP in mice have the advantage of producing a gradual, moderate pressure rise, similar to that which occurs in chronic, primary open angle glaucoma. These models, when combined with mouse genetics to manipulate specific cellular pathways, represent a powerful tool for understanding the importance of specific pathways in the production of optic nerve damage. The disadvantage of these models is that pressure elevation is generally bilateral, and, because it is not possible to perform continuous IOP monitoring, the exact level and duration of pressure elevation is unknown.
The DBA/2J mouse line, first described by John, and his colleagues, is the most well-characterized of the spontaneous rodent glaucoma models.(John et al. 1998; John 2005; Libby et al. 2005b) In this model, mutations of 2 genes results in iris stroma atrophy and pigment dispersion in the anterior segment and the trabecular meshwork, which results in reduced aqueous humor outflow. In most animals, IOP elevation occurs at about 7 to 8 months of age, although its extent varies among animals of the same age, and between the two eyes of a single animal.(Schlamp et al. 2006) The resulting IOP elevation produces loss of retinal ganglion cells (RGC's) as well as damage to the optic nerve.(John et al. 1998; Moon et al. 2005; Schlamp et al. 2006) Several other spontaneous mouse glaucoma models have also been described and are discussed in specific reviews. (Lindsey & Weinreb 2005; Pang & Clark 2007; Johnson & Tomarev 2009)
Other “pressure-dependent” rodent models rely on experimental production of elevated IOP. With this approach, the experimenter knows when the pressure-inducing insult is applied, making it easier to determine the timing of cellular responses to elevated IOP. Because the animals are, generally, otherwise normal, the effects of pressure alone can be better isolated, and since pressure elevation is unilateral, the fellow eye provides an excellent internal control. However, the exact onset of pressure elevation may still be uncertain, and the level of IOP attained can be hard to control.
The most relevant rodent models of experimental glaucomatous optic neuropathy are those that obstruct aqueous humor outflow. In rats, this can be accomplished by retrograde injections of hypertonic saline into the aqueous humor outflow pathways via episcleral veins(Morrison et al. 1997) or using external laser to the anterior chamber angle.(Ueda et al. 1998; WoldeMussie et al. 2001; Levkovitch-Verbin et al. 2002) (Figure 3) Although these two approaches have their individual strengths and weaknesses, both result in a combination of trabecular meshwork scarring and anterior chamber angle closure, and produce a similar extent and pattern of optic nerve damage.(Morrison et al. 2005) Additional methods of elevating IOP and obstructing aqueous outflow include intracameral injections of foreign substances, including hyaluronic acid and latex microspheres.(Urcola et al. 2006; Sappington et al. 2009) In mice, hypertonic saline injection, external laser and injection of microspheres have all been adapted to produce IOP elevation.(Aihara et al. 2003; Sappington et al. 2009; Walsh et al. 2009) Specific features and comparisons of these models have all been discussed elsewhere. (Lindsey & Weinreb 2005; Morrison et al. 2005; Pang & Clark 2007; Morrison et al. 2008; Johnson & Tomarev 2009; McKinnon et al. 2009)
Figure 3.

Histologic section through the limbus of a rat eye, showing sites of experimental aqueous humor outflow obstruction. This section fortuitously demonstrates an aqueous collector channel (CC) traversing the limbal sclera and connecting Schlemm's canal with the limbal blood vessels (*). This clearly shows how a retrograde injection of the limbal vasculature with hypertonic saline can result in scarring of the trabecular meshwork and anterior chamber angle. Similarly, external laser treatment to sclerose the limbal vasculature will also affect the underlying anterior chamber angle. S, sclera; C, cornea.
Another model, using cautery of one or more large external veins located posterior to the rectus muscles, produces immediate pressure elevation, followed by a gradual return to normal after one or more months. (Shareef et al. 1995; Neufeld et al. 1999; Sawada & Neufeld 1999; Mittag et al. 2000; Ahmed et al. 2001; Neufeld et al. 2002; Grozdanic et al. 2003) Although initially proposed to result from increased aqueous outflow resistance due to elevated episcleral venous pressure, an ultrasound study has shown no effects on anterior chamber depth, unlike models with true aqueous outflow obstruction.(Nissirios et al. 2008) It is now felt that the pressure rise with this model results from congestion of the intraocular vasculature.(Grozdanic et al. 2003; Pang & Clark 2007) Because of this, and because the extent and pattern of its optic nerve and retina injury differs fundamentally from that of the hypertonic saline and laser models,(Morrison et al. 2005) this review will not include results from studies using the cautery model.
4. Cellular mechanisms of glaucomatous damage
Rodent models of pressure-induced damage have provided important insights into cellular mechanisms by which IOP elevation contributes to glaucomatous optic nerve damage. They provide broad support for the concept that initial injury in glaucomatous nerve damage occurs within the ONH. Following this, ongoing damage in the ONH and retina most likely occur simultaneously. Experimentally, these phenomena are generally evaluated separately and we will consider the contribution of rodent models to our understanding of the cellular mechanisms of these phenomena in this manner.
4.1 The ONH as the site of initial glaucomatous optic nerve damage
Many lines of evidence point to the ONH as the site of initial damage due to elevated IOP. Among these is the observation that axonal transport obstruction occurs within the ONH, at the level of the lamina cribrosa.(Anderson & Hendrickson 1974; Quigley & Addicks 1980; Quigley 1981) In addition, in most cases of human glaucoma, the greatest degree of cupping appears at the superior and inferior poles of the ONH, with corresponding nerve fiber layer injury and visual field loss.(Quigley & Green 1979; Sommer et al. 1991; Tuulonen 1991) In humans, the density and size of the lamina cribrosa beams is most sparse, and conversely the size of the laminar pores between the beams is greatest, in the superior and inferior ONH.(Quigley & Addicks 1981) These observations have led to the concept that, not only is the ONH the site of initial injury in glaucoma, but the lamina cribrosa plays a pivotal role in this process. Rodent models of chronic pressure elevation support this concept to a degree, but also shed new light on the role of the lamina cribrosa.
Obstructed orthograde and retrograde transport, primarily localized within the ONH, has also been observed in rats with elevated IOP, as well as primates.(Pease et al. 2000; Quigley et al. 2000) In addition, since pressure-dependent models in both rats and mice, discussed above, unequivocally produce axonal degeneration and RGC loss, and neither of these species possesses a collagenous lamina, it appears that the presence of such a structure is not required for chronic IOP elevation to cause either axonal transport obstruction or optic nerve damage.
The ONH still appears to be the site of early pressure-induced optic nerve injury in rodents. In our initial description of a chronic pressure model in rats, we demonstrated axonal degeneration within the ONH.(Morrison et al. 1997) Several groups, working separately with the DBA/2J model, have observed atrophy of discrete nerve fiber bundles, radiating out from the ONH, a pattern that is most consistent with injury within the nerve head.(Jakobs et al. 2005; Schlamp et al. 2006; Howell et al. 2007; Buckingham et al. 2008) Some localized early injury to axons in the astrocyte column region of the ONH (Howell et al. 2007) while others used retrograde tracers to confirm that axonal damage precedes loss of the RGC soma.(Buckingham et al. 2008) This interpretation is further supported by experiments performed in DBA/2J mice crossed with a strain deficient in Bax, which is required for apoptosis of RGC.(Li et al. 2000; Libby et al. 2005a; Howell et al. 2007) Animals with elevated IOP developed focal axonal damage within the glial lamina, but were observed to retain their RGC, along with their proximal axon segments up to, but not posterior to, the ONH. Further experiments, crossing DBA/2J mice with a strain possessing the Wlds allele, which protects axons from insults, resulted in preservation of optic nerve axons along with some protection of RGC soma, further supporting the concept that the initial injury in this model is to axons within the ONH. A subsequent experiment, using laser-induced glaucoma in Wlds transgenic rats, showed that this gene successfully delayed axonal degeneration, although it did not prevent RGC degeneration or loss of dendritic arborization.(Beirowski et al. 2008)
It is also interesting that early injury in rats with experimental aqueous outflow obstruction occurs to a greater degree within the superior region of the ONH. (Figure 4) This results in greater early axonal degeneration in the superior portions of the retrobulbar optic nerve(Morrison et al. 1997) and greater RGC and nerve fiber layer loss in the superior retina.(WoldeMussie et al. 2001; Huang & Knighton 2009) Although the explanation for this injury pattern does not rely on a collagenous lamina cribrosa, we have noted that the retinal artery and vein enter the rat eye inferior to, rather than within, the neural portion of the ONH.(Morrison et al. 1999) (Figure 1) Because of this, only the superior portion of the nerve appears to actually be in contact with the sclera. These observations suggest that peripapillary scleral stresses and strains induced by elevated IOP might be transmitted to the adjacent optic nerve. Although the mechanism by which these forces are transmitted and how they result in axonal injury is currently unknown, this model offers unique opportunities to understand these processes at the cellular level, which are likely to be relevant to regional optic nerve injury in human glaucoma.
Figure 4.

Transmission electron micrograph of the superior portion of a rat ONH with early optic nerve damage from chronic IOP elevation. Note lucent spaces (*) between astrocyte foot processes. The space at the right of the photomicrograph appears to contain debris from a degenerating axon. ‡ indicates abnormal, swollen axon containing numerous vesicles.
While experimental pressure elevation in mice has also demonstrated a predilection for superior RGC injury,(Mabuchi et al. 2004; Salinas-Navarro et al. 2009) studies of early injury in the DBA/2J mouse model do not support any specific regional pattern.(Jakobs et al. 2005; May & Mittag 2006; Schlamp et al. 2006; Howell et al. 2007) Currently, it is not clear whether this is due to the more gradual pressure elevation in these models, strain differences, or to the smaller mouse ONH, which does not appear to present the same regional anatomic relationship between superior optic nerve and the adjacent sclera, and possible differences in vascular supply.(May & Lutjen-Drecoll 2002)
4.2 ONH mechanisms of glaucomatous damage
Rodent models have also demonstrated that ONH damage from elevated IOP is accompanied by numerous cellular responses. The gradual cupping that is characteristic of glaucomatous optic nerve damage is now known to involve remodeling of the ONH, well beyond simple loss of axons and posterior bowing of the lamina cribrosa.(Hernandez 2000; Morrison 2006) This was initially demonstrated in non-human primates with experimental glaucoma, where deposits of collagen and other extracellular matrix (ECM) materials were found within laminar pores.(Morrison et al. 1990) Similar deposits were later described in rat eyes with chronic experimental IOP elevation,(Johnson et al. 1996; Johnson et al. 2000) and also in the DBA/2J mouse model.(May & Mittag 2006) A gene microarray study of the ONH in the hypertonic saline model has found over 2000 genes that were significantly regulated.(Johnson et al. 2007) Of these genes, 225 had a 2-fold or greater change, representing gene classes involving cell proliferation, immune response, ECM, and the cytoskeleton. In addition, specific genes involved in cell proliferation, the ECM, and astrocyte water channels were found to demonstrate their maximal change in eyes with minimal injury, suggesting the possibility that a similar study of eyes with early injury might uncover gene changes responsible for initial axonal injury. Currently, the source of these extensive gene changes is unknown. However, astrocytes of the glial lamina have attracted increasing attention in recent years.(Hernandez 2000; Morgan 2000; Johnson & Morrison 2009) Immunohistochemistry of ONH with antibodies to the cell cycle regulator proliferating cell nuclear antigen (PCNA) shortly after IOP elevation has demonstrated extensive labeling of nuclei in these glial columns.(Johnson et al. 2000) It is likely that it is astrocytes that are proliferating, and they may be the predominant source for many of the gene changes involved.
It has been proposed that astrocytes may become “activated” in glaucomatous nerve damage--a state in which the cells hypertrophy, become hyperplastic and increase their expression of GFAP. One theory suggests that these activated cells may produce substances that are directly injurious to axons. An example of this is the production of inducible nitric oxide synthase (iNOS) that may encourage the production of excess nitric oxide and free radical formation, which leads to axonal injury. This view received initial support from positive immunohistochemical labeling of glial cells in glaucomatous human ONH with antibodies to iNOS,(Neufeld et al. 1997) along with a demonstration that iNOS inhibitors could significantly reduce RGC loss in the rat cautery model.(Neufeld et al. 1999) However, subsequent work failed to document upregulation of iNOS in glaucomatous human ONH and in either the hypertonic saline rat model or the DBA/2J mouse model. In addition, iNOS inhibition was not found to be neuroprotective in either model.(Pang et al. 2005; Libby et al. 2007)
Using the hypertonic saline model, we did not find immunohistochemical evidence for increased expression of GFAP in the ONH shortly after IOP elevation.(Johnson et al. 2000) In addition, early ONH gene regulation for GFAP appears to be, if anything, downregulated, along with aquaporin 4, the principal water channel protein of astrocytes, and the gap junctional protein Cx43.(Johnson et al. 2007). This leaves open the possibility that, instead of actively contributing to axonal injury in glaucoma, astrocytes may simply lose their more specialized axonal support functions, and this increases the vulnerability of axons to IOP-induced stress. Along these lines, elevated IOP and optic nerve damage in the DBA/2J model has been associated with evidence of axonal mitochondrial fission and cristae depletion.(Ju et al. 2008)
4.3 Retinal mechanisms of glaucomatous damage
Breakthroughs in understanding mechanisms of vision loss in glaucoma initially centered on the loss of RGC, when it was determined that this occurred by apoptosis, or programmed cell death.(Quigley et al. 1995) Although originally identified in primate glaucoma, this has now been confirmed in both experimental rat(Johnson et al. 2000; Hanninen et al. 2002; Guo et al. 2005) and mouse glaucoma models.(Gross et al. 2003; Ji et al. 2005; Reichstein et al. 2007) This process may occur by two interrelated pathways and involves a complex series of molecular events that leads to activation of proteases. These pathways, and evidence for their involvement in rodent glaucoma models, are thoroughly described in another article in this issue. (Grosskreutz—EER) Unfortunately, simply preventing RGC death in glaucoma does not always prevent optic nerve degeneration. This is illustrated by the previously mentioned experiment in which Bax-deficient DBA/2J mice demonstrate degeneration of nearly all axons posterior to the globe despite survival of RGCs. (Howell et al. 2007) This suggests that it is also important to understand the mechanisms by which elevated IOP leads to the axonal injury that ultimately initiates RGC apoptotic pathways.
One of the most widely recognized theories for how this occurs is that of neurotrophin (NT) deprivation due to axonal transport obstruction. As with all neuronal cells, RGCs depend on a steady supply of NT for survival and function, and intravitreal injection of brain derived neurotrophic factor (BDNF), one of several members of the nerve growth factor family of NT, has been shown to prolong RGC survival following optic nerve trauma. (Di Polo et al. 1998; Chen & Weber 2001) In order to exert their effects on the RGC, the NT, produced in the superior colliculus, complex with their tyrosine-related kinase (Trk) receptors and are then transported in retrograde fashion to the RGC soma. Recently, injection of BDNF into the cortex as well as the vitreous has been shown to provide additional RGC survival, suggesting that intracellular supply of NT, which must depend on retrograde axonal transport, is critical to achieving their trophic effects. (Weber et al. 2009) Because the Trk receptors themselves originate in the RGC, and must first be transported to the superior colliculus, obstruction of both orthograde and retrograde axonal transport by elevated IOP(Pease et al. 2000; Quigley et al. 2000) could result in critical deficits and apoptotic RGC death.
Several in vivo studies have been performed in rodent glaucoma models to determine the effect of supplementing these deficits. Martin et al, using a viral vector to increase retinal BDNF levels in the rat laser model, demonstrated significantly less axon loss than in optic nerves of model eyes without gene therapy.(Martin et al. 2003) Others have directly evaluated the effect of supplementation with other trophic factors. These include intravitreal delivery of bioerodable microspheres containing glial cell line-derived neurotrophic factor (GDNF), which provides significant RGC layer neuronal survival in the hypertonic saline model.(Jiang et al. 2007) Another study, using intravitreal injection of ciliary neurotrophic factor (CNTF) in the laser model, found significantly less RGC loss as compared to lasered eyes that did not receive CNTF.(Ji et al. 2004) Recently, virally-mediated overexpression of CNTF in the same model has also been shown to reduce optic nerve axon loss by 15%. (Pease et al. 2009) Another group used a similar approach to increase retinal expression of MEK1, an immediate upstream kinase, to increase levels of ERK1/2, which is activated by many trophic factors, to promote RGC survival.(Cheng et al. 2002) They found that animals with increased expression of MEK1 had greater RGC and axon survival than control animals with comparable pressure elevation.(Zhou et al. 2005) This suggests a potential neuroprotective role for activation of the ERK1/2 pathway, independent of neurotrophic input.
In spite of these results, experimental support for the NT theory remains limited. This is due, in part, to the complexity of the NT system itself.(Guo et al. 2009; Johnson et al. 2009) An immunohistochemical study in rat eyes with experimental IOP elevation and different degrees of optic nerve damage has demonstrated that, even though RGC label for BDNF was initially reduced, positive label was seen in eyes with prolonged IOP elevation and extensive optic nerve damage.(Johnson et al. 2000) More recently, in a systematic analysis of the simultaneous retinal responses of the major components of the NT pathways and their interacting proteins to elevated IOP we found no significant changes in mRNA levels for BDNF or other NTs. Although message for TrkB was decreased, there were no changes in protein levels of either TrkB or its phosphorylated form. The lack of change in retinal NT or message for TrkB suggests that retinal sources, demonstrated by other investigators,(Rudzinski et al. 2004; Kim et al. 2006) are capable of producing and maintaining NT levels in the face of elevated IOP and obstructed transport. Interestingly, retinal message for p75NTR receptor was significantly upregulated.(Guo et al. 2009) Although binding of this receptor to NT in association with specific adaptor proteins does promote apoptosis, these proteins were downregulated, suggesting that RGC death may not be the main outcome of p75NTR receptor upregulation. Alternatively, the p75NTR receptor can have pro-survival effects in association with NF-kB, which we did find to be upregulated. We also found an upregulation of retinal Jun and increased immunolabeling of RGC with antibodies to phosphorylated Jun, in agreement with other studies.(Levkovitch-Verbin et al. 2005) Although this could be consistent with p75NTR-mediated cell death via the JNK-p53-Bax pathway, we also uncovered a marked upregulation of the immediate early response gene activating transcription factor 3 (Atf3), which, in addition to promoting apoptosis, may have survival effects when dimerized with Jun.(Takeda et al. 2000) This analysis illustrates the complexity of the NT system,(Johnson et al. 2009) and suggests that the response of many components of the NT system may favor survival over apoptosis, and that simple deficit replacement therapy alone may not provide long term benefits to patients with glaucoma
The discovery of elevated levels of several forms of heat shock proteins in the retinas of experimental glaucoma models brings up the possibility of endogenous protective pathways for glaucoma.(Park et al. 2001; Sakai et al. 2003; Kwong et al. 2006; Huang et al. 2007; Kalesnykas et al. 2007) High molecular weight forms of heat shock proteins, which function as chaperone proteins, were first reported elevated in the laser rat model of glaucoma(Park et al. 2001) and in monkeys with laser-induced IOP elevation,(Sakai et al. 2003) along with a demonstration that their induction could be protective in the former.(Park et al. 2001; Kwong et al. 2003) Subsequent work has shown that retinal levels of low molecular weight heat shock proteins, most notably Hsp27, are also increased in several experimental glaucoma models,(Sakai et al. 2003; Huang et al. 2007) This protein, which is induced in the CNS by a variety of stimuli, was found to be expressed by RGCs and glial cells in eyes with elevated IOP. (Huang et al. 2007; Kalesnykas et al. 2007) In addition, Huang et al, working with both rats and the DBA/2J mouse model, noted increased retinal expression of phosphorylated Hsp27, a post-translational modification that may have an effect on its ability to protect against apoptosis.(Huang et al. 2007)
Additional insights into mechanisms of RGC death from elevated IOP have resulted from applying gene microarray methods to retinas from models of elevated IOP. Studies of monkey retinas, as well as the rat and the DBA/2J model, have shown that elevated IOP alters regulation of many specific genes and gene categories, including upregulation of genes involved in neuroinflammation, apoptosis, glial activation and tissue remodeling, and downregulation of crystallins and cytoskeleton-related genes. (Miyahara et al. 2003; Ahmed et al. 2004; Steele et al. 2006; Yang et al. 2007)
While these observations do support some specific theories of mechanisms involved in RGC death,(Wax & Tezel 2009) they are based on material from whole retina samples and may have overlooked changes specific to the RGC, due to dilution from the highly cellular outer layers of the retina. A recent gene array study comparing results in whole retina to those found in the RGC layer (RGCL) of the retina, isolated by laser capture microdissection, has shown that this may indeed be the case.(Guo et al. 2010) By enriching the sample with RGCs and their associated cells of the RGC layer, we found a 5-fold increase in the number of regulated genes, many with a greater extent of change, as compared to identically analyzed whole retina samples from eyes with comparable IOP exposure and optic nerve injury. This analysis also yielded a much greater number of distinct, upregulated cellular processes, as compared to the whole retina analysis. Interestingly, immune process was not the most likely gene category to be upregulated in response to pressure. It was instead overshadowed by increases in protein biosynthesis and metabolism, as well as stress responsive genes, such as Atf3. Concentrating on the RGCL revealed a much greater number of downregulated genes in response to elevated IOP than seen in the whole retina. These included downregulation of several metabolic processes, protein folding and nucleotide biosynthesis, and reduction in genes involved in axon extension and dendrite morphogenesis. QPCR analysis of specific RGC markers demonstrated a uniform downregulation of transcription factors Brn3a, Brn3b and Brn3c as well as the Thy1 cell surface antigen and the axonal neurofilament proteins Nefl and Nefh. These findings support results in non-human primate and rodent glaucoma models that, although RGCs appear to survive for a period of time in the face of elevated IOP, they undergo specific changes of somal shrinkage, axonal atrophy and dendrite remodeling.(Weber et al. 1998; Schlamp et al. 2001; Weber & Harman 2005; Schlamp et al. 2006; Buckingham et al. 2008) These observations suggest that there may be a window of opportunity in which treatments to reverse these changes might be applied to prolong RGC and axon survival.
5. What is the future of rodent models in understanding glaucomatous optic neuropathy?
Rodent glaucoma models have dramatically improved our appreciation of the cellular mechanisms of glaucomatous optic nerve damage. In spite of the lack of a collagenous lamina cribrosa, elevated IOP in all of the rodent models discussed here still produces optic nerve damage primarily situated at the ONH. To us, this suggests that the lamina is not necessary for elevated IOP to cause axonal injury. Instead, it is likely that a robust collagenous lamina, required in larger eyes to protect optic nerve fibers from ONH stresses and strains caused by fluctuations in IOP, modifies the distribution of these forces through regional variations in its structure. We also believe that these stresses and strains are responsible for the ONH cellular responses we now associate with elevated IOP, many of which are likely to be responsible for axonal injury. The rodent ONH, with its ultrastructural similarities to the primate, can be considered equivalent to axon bundles in a single primate laminar pore. In this way, rodent models offer an excellent opportunity to determine the source of these responses and understand how they injure axons in glaucoma.
The rodent eye offers many advantages for this research. Among these are the ready availability and cost-effectiveness of the animals, which allow enough specimens to be used for statistical analyses adequate to overcome the confounding influence of individual variability. From the above, it is clear that these models allow detailed study with numerous, sophisticated cell biology techniques. An additional advantage, with the increased availability of spontaneous and experimental mouse models of elevated IOP, is the growing use of powerful genetic tools that can be used to visualize specific cell types and responses, as well as test the contribution of specific genes to the events of axonal injury and susceptibility. With all these tools, specific risk factors, such as aging and previous injury, can now be addressed with these models.
However, there are specific limitations in rodent models that we must continue to address. One of the most important of these is that, regardless of the model and mode of pressure elevation, the level of IOP to which the ONH is exposed must be accurately determined in order to have the best understanding of the relationship between pressure and the resulting cellular responses. This is particularly true if these models are to be used to test the effectiveness of neuroprotective interventions in preventing optic nerve damage. Unfortunately, experimental glaucoma models that rely on aqueous humor outflow obstruction and current spontaneous glaucoma models all demonstrate at least some degree of unpredictable pressure fluctuations. For this reason, and because IOP cannot be adequately controlled, frequent, noninvasive pressure measurements are still required. While continuous telemetric monitoring of IOP in unrestrained animals represents the most promising solution to this challenge,(McLaren et al. 1996; Li & Liu 2008) widespread use of this technology in rodents remains a future goal. We continue to advocate frequent tonometry, with animals in the awake state,(Jia et al. 2000b) and a continued awareness that, when animals are housed in a light:dark environment, wide natural fluctuations in IOP do occur during the dark phase(McLaren et al. 1996; Moore et al. 1996) and that these can be markedly accentuated in eyes with obstructed aqueous humor outflow.(Jia et al. 2000a)
Acknowledgments
Supported by NIH grants EY010145, EY016866 and an unrestricted grant from Research to Prevent Blindness, Inc.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- AGIS-Investigators The Advanced Glaucoma Intervention Study (AGIS): 7. The relationship between control of intraocular pressure and visual field deterioration. The AGIS Investigators. Am J Ophthalmol. 2000;130(4):429–40. doi: 10.1016/s0002-9394(00)00538-9. [DOI] [PubMed] [Google Scholar]
- Ahmed F, Brown KM, Stephan DA, Morrison JC, Johnson EC, Tomarev SI. Microarray analysis of changes in mRNA levels in the rat retina after experimental elevation of intraocular pressure. Invest Ophthalmol Vis Sci. 2004;45(4):1247–58. doi: 10.1167/iovs.03-1123. [DOI] [PubMed] [Google Scholar]
- Ahmed FA, Hegazy K, Chaudhary P, Sharma SC. Neuroprotective effect of alpha(2) agonist (brimonidine) on adult rat retinal ganglion cells after increased intraocular pressure. Brain Res. 2001;913(2):133–9. doi: 10.1016/s0006-8993(01)02759-7. [DOI] [PubMed] [Google Scholar]
- Aihara M, Lindsey JD, Weinreb RN. Experimental mouse ocular hypertension: establishment of the model. Invest Ophthalmol Vis Sci. 2003;44(10):4314–20. doi: 10.1167/iovs.03-0137. [DOI] [PubMed] [Google Scholar]
- Anderson DR, Hendrickson A. Effect of intraocular pressure on rapid axoplasmic transport in monkey optic nerve. Invest Ophthalmol. 1974;13(10):771–83. [PubMed] [Google Scholar]
- Beirowski B, Babetto E, Coleman MP, Martin KR. The WldS gene delays axonal but not somatic degeneration in a rat glaucoma model. Eur J Neurosci. 2008;28(6):1166–79. doi: 10.1111/j.1460-9568.2008.06426.x. [DOI] [PubMed] [Google Scholar]
- Buckingham BP, Inman DM, Lambert W, Oglesby E, Calkins DJ, Steele MR, Vetter ML, Marsh-Armstrong N, Horner PJ. Progressive ganglion cell degeneration precedes neuronal loss in a mouse model of glaucoma. J Neurosci. 2008;28(11):2735–44. doi: 10.1523/JNEUROSCI.4443-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Weber AJ. BDNF enhances retinal ganglion cell survival in cats with optic nerve damage. Invest Ophthalmol Vis Sci. 2001;42(5):966–74. [PubMed] [Google Scholar]
- Cheng L, Sapieha P, Kittlerova P, Hauswirth WW, Di Polo A. TrkB gene transfer protects retinal ganglion cells from axotomy-induced death in vivo. J Neurosci. 2002;22(10):3977–86. doi: 10.1523/JNEUROSCI.22-10-03977.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cioffi GA, Van Buskirk EM. Microvasculature of the anterior optic nerve. Surv Ophthalmol. 1994;38(Suppl):S107–16. doi: 10.1016/0039-6257(94)90054-x. discussion S116-7. [DOI] [PubMed] [Google Scholar]
- Di Polo A, Aigner LJ, Dunn RJ, Bray GM, Aguayo AJ. Prolonged delivery of brain-derived neurotrophic factor by adenovirus-infected Muller cells temporarily rescues injured retinal ganglion cells. Proc Natl Acad Sci U S A. 1998;95(7):3978–83. doi: 10.1073/pnas.95.7.3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drance SM. The Collaborative Normal-Tension Glaucoma Study and some of its lessons. Can J Ophthalmol. 1999;34(1):1–6. [PubMed] [Google Scholar]
- Eida H, Bhutto IA, Amemiya T. Corrosion cast demonstration of choroidal vasculature in normal Wistar Kyoto rat. Ital J Anat Embryol. 2001;106(2 Suppl 1):245–50. [PubMed] [Google Scholar]
- Gross RL, Ji J, Chang P, Pennesi ME, Yang Z, Zhang J, Wu SM. A mouse model of elevated intraocular pressure: retina and optic nerve findings. Trans Am Ophthalmol Soc. 2003;101:163–9. discussion 169-71. [PMC free article] [PubMed] [Google Scholar]
- Grozdanic SD, Betts DM, Sakaguchi DS, Kwon YH, Kardon RH, Sonea IM. Temporary elevation of the intraocular pressure by cauterization of vortex and episcleral veins in rats causes functional deficits in the retina and optic nerve. Exp Eye Res. 2003;77(1):27–33. doi: 10.1016/s0014-4835(03)00089-7. [DOI] [PubMed] [Google Scholar]
- Guo L, Moss SE, Alexander RA, Ali RR, Fitzke FW, Cordeiro MF. Retinal ganglion cell apoptosis in glaucoma is related to intraocular pressure and IOP-induced effects on extracellular matrix. Invest Ophthalmol Vis Sci. 2005;46(1):175–82. doi: 10.1167/iovs.04-0832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Johnson E, Cepurna W, Jia L, Dyck J, Morrison JC. Does elevated intraocular pressure reduce retinal TRKB-mediated survival signaling in experimental glaucoma? Exp Eye Res. 2009 doi: 10.1016/j.exer.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Cepurna WO, Dyck J, Doser T, Johnson EC, Morrison JC. Retinal Cell Responses to Elevated Intraocular Pressure: A Gene Array Comparison between the Whole Retina and Retinal Ganglion Cell Layer. Invest Ophthalmol Vis Sci. 2010 doi: 10.1167/iovs.09-4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanninen VA, Pantcheva MB, Freeman EE, Poulin NR, Grosskreutz CL. Activation of caspase 9 in a rat model of experimental glaucoma. Curr Eye Res. 2002;25(6):389–95. doi: 10.1076/ceyr.25.6.389.14233. [DOI] [PubMed] [Google Scholar]
- Heijl A, Leske MC, Bengtsson B, Hyman L, Bengtsson B, Hussein M. Reduction of intraocular pressure and glaucoma progression: results from the Early Manifest Glaucoma Trial. Arch Ophthalmol. 2002;120(10):1268–79. doi: 10.1001/archopht.120.10.1268. [DOI] [PubMed] [Google Scholar]
- Hernandez MR. The optic nerve head in glaucoma: role of astrocytes in tissue remodeling. Prog Retin Eye Res. 2000;19(3):297–321. doi: 10.1016/s1350-9462(99)00017-8. [DOI] [PubMed] [Google Scholar]
- Howell GR, Libby RT, Jakobs TC, Smith RS, Phalan FC, Barter JW, Barbay JM, Marchant JK, Mahesh N, Porciatti V. Axons of retinal ganglion cells are insulted in the optic nerve early in DBA/2J glaucoma. J Cell Biol. 2007;179(7):1523–37. doi: 10.1083/jcb.200706181. and others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Fileta JB, Filippopoulos T, Ray A, Dobberfuhl A, Grosskreutz CL. Hsp27 phosphorylation in experimental glaucoma. Invest Ophthalmol Vis Sci. 2007;48(9):4129–35. doi: 10.1167/iovs.06-0606. [DOI] [PubMed] [Google Scholar]
- Huang XR, Knighton RW. Altered F-actin distribution in retinal nerve fiber layer of a rat model of glaucoma. Exp Eye Res. 2009;88(6):1107–14. doi: 10.1016/j.exer.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobs TC, Libby RT, Ben Y, John SW, Masland RH. Retinal ganglion cell degeneration is topological but not cell type specific in DBA/2J mice. J Cell Biol. 2005;171(2):313–25. doi: 10.1083/jcb.200506099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji J, Chang P, Pennesi ME, Yang Z, Zhang J, Li D, Wu SM, Gross RL. Effects of elevated intraocular pressure on mouse retinal ganglion cells. Vision Res. 2005;45(2):169–79. doi: 10.1016/j.visres.2004.08.008. [DOI] [PubMed] [Google Scholar]
- Ji JZ, Elyaman W, Yip HK, Lee VW, Yick LW, Hugon J, So KF. CNTF promotes survival of retinal ganglion cells after induction of ocular hypertension in rats: the possible involvement of STAT3 pathway. Eur J Neurosci. 2004;19(2):265–72. doi: 10.1111/j.0953-816x.2003.03107.x. [DOI] [PubMed] [Google Scholar]
- Jia L, Cepurna WO, Johnson EC, Morrison JC. Patterns of intraocular pressure elevation after aqueous humor outflow obstruction in rats. Invest Ophthalmol Vis Sci. 2000a;41(6):1380–5. [PubMed] [Google Scholar]
- Jia L, Cepurna WO, Johnson EC, Morrison JC. Effect of general anesthetics on IOP in rats with experimental aqueous outflow obstruction. Invest Ophthalmol Vis Sci. 2000b;41(11):3415–9. [PubMed] [Google Scholar]
- Jiang C, Moore MJ, Zhang X, Klassen H, Langer R, Young M. Intravitreal injections of GDNF-loaded biodegradable microspheres are neuroprotective in a rat model of glaucoma. Mol Vis. 2007;13:1783–92. [PubMed] [Google Scholar]
- John SW. Mechanistic insights into glaucoma provided by experimental genetics the cogan lecture. Invest Ophthalmol Vis Sci. 2005;46(8):2649–61. doi: 10.1167/iovs.05-0205. [DOI] [PubMed] [Google Scholar]
- John SW, Smith RS, Savinova OV, Hawes NL, Chang B, Turnbull D, Davisson M, Roderick TH, Heckenlively JR. Essential iris atrophy, pigment dispersion, and glaucoma in DBA/2J mice. Invest Ophthalmol Vis Sci. 1998;39(6):951–62. [PubMed] [Google Scholar]
- Johnson EC, Morrison JC. Friend or Foe? Resolving the Impact of Glial Responses in Glaucoma. Journal of Glaucoma. 2009;18(5):341–353. doi: 10.1097/IJG.0b013e31818c6ef6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson EC, Guo Y, Cepurna WO, Morrison JC. Neurotrophin roles in retinal ganglion cell survival: lessons from rat glaucoma models. Exp Eye Res. 2009;88(4):808–15. doi: 10.1016/j.exer.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson EC, Deppmeier LM, Wentzien SK, Hsu I, Morrison JC. Chronology of optic nerve head and retinal responses to elevated intraocular pressure. Invest Ophthalmol Vis Sci. 2000;41(2):431–42. [PubMed] [Google Scholar]
- Johnson EC, Jia L, Cepurna WO, Doser TA, Morrison JC. Global changes in optic nerve head gene expression after exposure to elevated intraocular pressure in a rat glaucoma model. Invest Ophthalmol Vis Sci. 2007;48(7):3161–77. doi: 10.1167/iovs.06-1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson EC, Morrison JC, Farrell S, Deppmeier L, Moore CG, McGinty MR. The effect of chronically elevated intraocular pressure on the rat optic nerve head extracellular matrix. Exp Eye Res. 1996;62(6):663–74. doi: 10.1006/exer.1996.0077. [DOI] [PubMed] [Google Scholar]
- Johnson TV, Tomarev SI. Rodent models of glaucoma. Brain Res Bull. 2009;81(2-3):349–58. doi: 10.1016/j.brainresbull.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju WK, Kim KY, Lindsey JD, Angert M, Duong-Polk KX, Scott RT, Kim JJ, Kukhmazov I, Ellisman MH, Perkins GA. Intraocular pressure elevation induces mitochondrial fission and triggers OPA1 release in glaucomatous optic nerve. Invest Ophthalmol Vis Sci. 2008;49(11):4903–11. doi: 10.1167/iovs.07-1661. and others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalesnykas G, Niittykoski M, Rantala J, Miettinen R, Salminen A, Kaarniranta K, Uusitalo H. The expression of heat shock protein 27 in retinal ganglion and glial cells in a rat glaucoma model. Neuroscience. 2007;150(3):692–704. doi: 10.1016/j.neuroscience.2007.09.078. [DOI] [PubMed] [Google Scholar]
- Kass MA, Heuer DK, Higginbotham EJ, Johnson CA, Keltner JL, Miller JP, Parrish RK, 2nd, Wilson MR, Gordon MO. The Ocular Hypertension Treatment Study: a randomized trial determines that topical ocular hypotensive medication delays or prevents the onset of primary open-angle glaucoma. Arch Ophthalmol. 2002;120(6):701–13. doi: 10.1001/archopht.120.6.701. discussion 829-30. [DOI] [PubMed] [Google Scholar]
- Kim CY, Kuehn MH, Clark AF, Kwon YH. Gene expression profile of the adult human retinal ganglion cell layer. Mol Vis. 2006;12:1640–8. [PubMed] [Google Scholar]
- Klein BE, Klein R, Sponsel WE, Franke T, Cantor LB, Martone J, Menage MJ. Prevalence of glaucoma. The Beaver Dam Eye Study. Ophthalmology. 1992;99(10):1499–504. doi: 10.1016/s0161-6420(92)31774-9. [DOI] [PubMed] [Google Scholar]
- Kwong JM, Lam TT, Caprioli J. Hyperthermic pre-conditioning protects retinal neurons from N-methyl-D-aspartate (NMDA)-induced apoptosis in rat. Brain Res. 2003;970(1-2):119–30. doi: 10.1016/s0006-8993(03)02298-4. [DOI] [PubMed] [Google Scholar]
- Kwong JM, Lalezary M, Nguyen JK, Yang C, Khattar A, Piri N, Mareninov S, Gordon LK, Caprioli J. Co-expression of heat shock transcription factors 1 and 2 in rat retinal ganglion cells. Neurosci Lett. 2006;405(3):191–5. doi: 10.1016/j.neulet.2006.06.070. [DOI] [PubMed] [Google Scholar]
- Levkovitch-Verbin H, Quigley HA, Martin KR, Valenta D, Baumrind LA, Pease ME. Translimbal laser photocoagulation to the trabecular meshwork as a model of glaucoma in rats. Invest Ophthalmol Vis Sci. 2002;43(2):402–10. [PubMed] [Google Scholar]
- Levkovitch-Verbin H, Quigley HA, Martin KR, Harizman N, Valenta DF, Pease ME, Melamed S. The transcription factor c-jun is activated in retinal ganglion cells in experimental rat glaucoma. Exp Eye Res. 2005;80(5):663–70. doi: 10.1016/j.exer.2004.11.016. [DOI] [PubMed] [Google Scholar]
- Li R, Liu JH. Telemetric monitoring of 24 h intraocular pressure in conscious and freely moving C57BL/6J and CBA/CaJ mice. Mol Vis. 2008;14:745–9. [PMC free article] [PubMed] [Google Scholar]
- Li Y, Schlamp CL, Poulsen KP, Nickells RW. Bax-dependent and independent pathways of retinal ganglion cell death induced by different damaging stimuli. Exp Eye Res. 2000;71(2):209–13. doi: 10.1006/exer.2000.0873. [DOI] [PubMed] [Google Scholar]
- Libby RT, Li Y, Savinova OV, Barter J, Smith RS, Nickells RW, John SW. Susceptibility to neurodegeneration in a glaucoma is modified by Bax gene dosage. PLoS Genet. 2005a;1(1):17–26. doi: 10.1371/journal.pgen.0010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby RT, Howell GR, Pang IH, Savinova OV, Mehalow AK, Barter JW, Smith RS, Clark AF, John SW. Inducible nitric oxide synthase, Nos2, does not mediate optic neuropathy and retinopathy in the DBA/2J glaucoma model. BMC Neurosci. 2007;8(1):108. doi: 10.1186/1471-2202-8-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby RT, Anderson MG, Pang IH, Robinson ZH, Savinova OV, Cosma IM, Snow A, Wilson LA, Smith RS, Clark AF. Inherited glaucoma in DBA/2J mice: pertinent disease features for studying the neurodegeneration. Vis Neurosci. 2005b;22(5):637–48. doi: 10.1017/S0952523805225130. and others. [DOI] [PubMed] [Google Scholar]
- Lindsey JD, Weinreb RN. Elevated intraocular pressure and transgenic applications in the mouse. J Glaucoma. 2005;14(4):318–20. doi: 10.1097/01.ijg.0000169411.09258.f6. [DOI] [PubMed] [Google Scholar]
- Mabuchi F, Aihara M, Mackey MR, Lindsey JD, Weinreb RN. Regional optic nerve damage in experimental mouse glaucoma. Invest Ophthalmol Vis Sci. 2004;45(12):4352–8. doi: 10.1167/iovs.04-0355. [DOI] [PubMed] [Google Scholar]
- Martin KR, Quigley HA, Zack DJ, Levkovitch-Verbin H, Kielczewski J, Valenta D, Baumrind L, Pease ME, Klein RL, Hauswirth WW. Gene therapy with brain-derived neurotrophic factor as a protection: retinal ganglion cells in a rat glaucoma model. Invest Ophthalmol Vis Sci. 2003;44(10):4357–65. doi: 10.1167/iovs.02-1332. [DOI] [PubMed] [Google Scholar]
- May CA. The optic nerve head region of the aged rat: an immunohistochemical investigation. Curr Eye Res. 2003;26(6):347–54. doi: 10.1076/ceyr.26.5.347.15438. [DOI] [PubMed] [Google Scholar]
- May CA, Lutjen-Drecoll E. Morphology of the murine optic nerve. Invest Ophthalmol Vis Sci. 2002;43(7):2206–12. [PubMed] [Google Scholar]
- May CA, Mittag T. Optic nerve degeneration in the DBA/2NNia mouse: is the lamina cribrosa important in the development of glaucomatous optic neuropathy? Acta Neuropathol. 2006;111(2):158–67. doi: 10.1007/s00401-005-0011-2. [DOI] [PubMed] [Google Scholar]
- McKinnon SJ, Schlamp CL, Nickells RW. Mouse models of retinal ganglion cell death and glaucoma. Exp Eye Res. 2009;88(4):816–24. doi: 10.1016/j.exer.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren JW, Brubaker RF, FitzSimon JS. Continuous measurement of intraocular pressure in rabbits by telemetry. Invest Ophthalmol Vis Sci. 1996;37(6):966–75. [PubMed] [Google Scholar]
- Mittag TW, Danias J, Pohorenec G, Yuan HM, Burakgazi E, Chalmers-Redman R, Podos SM, Tatton WG. Retinal damage after 3 to 4 months of elevated intraocular pressure in a rat glaucoma model. Invest Ophthalmol Vis Sci. 2000;41(11):3451–9. [PubMed] [Google Scholar]
- Miyahara T, Kikuchi T, Akimoto M, Kurokawa T, Shibuki H, Yoshimura N. Gene microarray analysis of experimental glaucomatous retina from cynomologous monkey. Invest Ophthalmol Vis Sci. 2003;44(10):4347–56. doi: 10.1167/iovs.02-1032. [DOI] [PubMed] [Google Scholar]
- Moon JI, Kim IB, Gwon JS, Park MH, Kang TH, Lim EJ, Choi KR, Chun MH. Changes in retinal neuronal populations in the DBA/2J mouse. Cell Tissue Res. 2005;320(1):51–9. doi: 10.1007/s00441-004-1062-8. [DOI] [PubMed] [Google Scholar]
- Moore CG, Johnson EC, Morrison JC. Circadian rhythm of intraocular pressure in the rat. Curr Eye Res. 1996;15(2):185–91. doi: 10.3109/02713689608997412. [DOI] [PubMed] [Google Scholar]
- Morgan JE. Optic nerve head structure in glaucoma: astrocytes as mediators of axonal damage. Eye. 2000;14(Pt 3B):437–44. doi: 10.1038/eye.2000.128. [DOI] [PubMed] [Google Scholar]
- Morrison J, Farrell S, Johnson E, Deppmeier L, Moore CG, Grossmann E. Structure and composition of the rodent lamina cribrosa. Exp Eye Res. 1995;60(2):127–35. doi: 10.1016/s0014-4835(95)80002-6. [DOI] [PubMed] [Google Scholar]
- Morrison JC. Integrins in the optic nerve head: potential roles in glaucomatous optic neuropathy (an American Ophthalmological Society thesis) Trans Am Ophthalmol Soc. 2006;104:453–77. [PMC free article] [PubMed] [Google Scholar]
- Morrison JC, Johnson E, Cepurna WO. Rat models for glaucoma research. Prog Brain Res. 2008;173:285–301. doi: 10.1016/S0079-6123(08)01121-7. [DOI] [PubMed] [Google Scholar]
- Morrison JC, Jerdan JA, Dorman ME, Quigley HA. Structural proteins of the neonatal and adult lamina cribrosa. Arch Ophthalmol. 1989;107(8):1220–4. doi: 10.1001/archopht.1989.01070020286040. [DOI] [PubMed] [Google Scholar]
- Morrison JC, Dorman-Pease ME, Dunkelberger GR, Quigley HA. Optic nerve head extracellular matrix in primary optic atrophy and experimental glaucoma. Arch Ophthalmol. 1990;108(7):1020–4. doi: 10.1001/archopht.1990.01070090122053. [DOI] [PubMed] [Google Scholar]
- Morrison JC, Johnson EC, Cepurna WO, Funk RH. Microvasculature of the rat optic nerve head. Invest Ophthalmol Vis Sci. 1999;40(8):1702–9. [PubMed] [Google Scholar]
- Morrison JC, Johnson EC, Cepurna W, Jia L. Understanding mechanisms of pressure-induced optic nerve damage. Prog Retin Eye Res. 2005;24(2):217–240. doi: 10.1016/j.preteyeres.2004.08.003. [DOI] [PubMed] [Google Scholar]
- Morrison JC, Moore CG, Deppmeier LM, Gold BG, Meshul CK, Johnson EC. A rat model of chronic pressure-induced optic nerve damage. Exp Eye Res. 1997;64(1):85–96. doi: 10.1006/exer.1996.0184. [DOI] [PubMed] [Google Scholar]
- Neufeld AH, Hernandez MR, Gonzalez M. Nitric oxide synthase in the human glaucomatous optic nerve head. Arch Ophthalmol. 1997;115(4):497–503. doi: 10.1001/archopht.1997.01100150499009. [DOI] [PubMed] [Google Scholar]
- Neufeld AH, Sawada A, Becker B. Inhibition of nitric-oxide synthase 2 by aminoguanidine provides neuroprotection of retinal ganglion cells in a rat model of chronic glaucoma. Proc Natl Acad Sci U S A. 1999;96(17):9944–8. doi: 10.1073/pnas.96.17.9944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neufeld AH, Das S, Vora S, Gachie E, Kawai S, Manning PT, Connor JR. A prodrug of a selective inhibitor of inducible nitric oxide synthase is neuroprotective in the rat model of glaucoma. J Glaucoma. 2002;11(3):221–5. doi: 10.1097/00061198-200206000-00010. [DOI] [PubMed] [Google Scholar]
- Nissirios N, Chanis R, Johnson E, Morrison J, Cepurna WO, Jia L, Mittag T, Danias J. Comparison of anterior segment structures in two rat glaucoma models: an ultrasound biomicroscopic study. Invest Ophthalmol Vis Sci. 2008;49(6):2478–82. doi: 10.1167/iovs.07-0965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang IH, Clark AF. Rodent models for glaucoma retinopathy and optic neuropathy. J Glaucoma. 2007;16(5):483–505. doi: 10.1097/IJG.0b013e3181405d4f. [DOI] [PubMed] [Google Scholar]
- Pang IH, Johnson EC, Jia L, Cepurna WO, Shepard AR, Hellberg MR, Clark AF, Morrison JC. Evaluation of inducible nitric oxide synthase in glaucomatous optic neuropathy and pressure-induced optic nerve damage. Invest Ophthalmol Vis Sci. 2005;46(4):1313–21. doi: 10.1167/iovs.04-0829. [DOI] [PubMed] [Google Scholar]
- Park KH, Cozier F, Ong OC, Caprioli J. Induction of heat shock protein 72 protects retinal ganglion cells in a rat glaucoma model. Invest Ophthalmol Vis Sci. 2001;42(7):1522–30. [PubMed] [Google Scholar]
- Pease ME, McKinnon SJ, Quigley HA, Kerrigan-Baumrind LA, Zack DJ. Obstructed axonal transport of BDNF and its receptor TrkB in experimental glaucoma. Invest Ophthalmol Vis Sci. 2000;41(3):764–74. [PubMed] [Google Scholar]
- Pease ME, Zack DJ, Berlinicke C, Bloom K, Cone F, Wang Y, Klein RL, Hauswirth WW, Quigley HA. Effect of CNTF on retinal ganglion cell survival in experimental glaucoma. Invest Ophthalmol Vis Sci. 2009;50(5):2194–200. doi: 10.1167/iovs.08-3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quigley HA, Green WR. The histology of human glaucoma cupping and optic nerve damage: clinicopathologic correlation in 21 eyes. Ophthalmology. 1979;86(10):1803–30. doi: 10.1016/s0161-6420(79)35338-6. [DOI] [PubMed] [Google Scholar]
- Quigley HA, Addicks EM. Chronic experimental glaucoma in primates. II. Effect of extended intraocular pressure elevation on optic nerve head and axonal transport. Invest Ophthalmol Vis Sci. 1980;19(2):137–52. [PubMed] [Google Scholar]
- Quigley HA, Addicks EM. Regional differences in the structure of the lamina cribrosa and their relation to glaucomatous optic nerve damage. Arch Ophthalmol. 1981;99(1):137–43. doi: 10.1001/archopht.1981.03930010139020. [DOI] [PubMed] [Google Scholar]
- Quigley HA, Nickells RW, Kerrigan LA, Pease ME, Thibault DJ, Zack DJ. Retinal ganglion cell death in experimental glaucoma and after axotomy occurs by apoptosis. Invest Ophthalmol Vis Sci. 1995;36(5):774–86. [PubMed] [Google Scholar]
- Quigley HA, McKinnon SJ, Zack DJ, Pease ME, Kerrigan-Baumrind LA, Kerrigan DF, Mitchell RS. Retrograde axonal transport of BDNF in retinal ganglion cells is blocked by acute IOP elevation in rats. Invest Ophthalmol Vis Sci. 2000;41(11):3460–6. [PubMed] [Google Scholar]
- Quigley HAA, E. M, Green WR, Maumenee AE. Optic nerve damage in human glaucoma. II. The site of injury and susceptibility to damage. Arch Ophthalmol. 1981;99(4):635–49. doi: 10.1001/archopht.1981.03930010635009. [DOI] [PubMed] [Google Scholar]
- Reichstein D, Ren L, Filippopoulos T, Mittag T, Danias J. Apoptotic retinal ganglion cell death in the DBA/2 mouse model of glaucoma. Exp Eye Res. 2007;84(1):13–21. doi: 10.1016/j.exer.2006.08.009. [DOI] [PubMed] [Google Scholar]
- Rudzinski M, Wong TP, Saragovi HU. Changes in retinal expression of neurotrophins and neurotrophin receptors induced by ocular hypertension. J Neurobiol. 2004;58(3):341–54. doi: 10.1002/neu.10293. [DOI] [PubMed] [Google Scholar]
- Sakai M, Sakai H, Nakamura Y, Fukuchi T, Sawaguchi S. Immunolocalization of heat shock proteins in the retina of normal monkey eyes and monkey eyes with laser-induced glaucoma. Jpn J Ophthalmol. 2003;47(1):42–52. doi: 10.1016/s0021-5155(02)00627-5. [DOI] [PubMed] [Google Scholar]
- Salinas-Navarro M, Alarcon-Martinez L, Valiente-Soriano FJ, Ortin-Martinez A, Jimenez-Lopez M, Aviles-Trigueros M, Villegas-Perez MP, de la Villa P, Vidal-Sanz M. Functional and morphological effects of laser-induced ocular hypertension in retinas of adult albino Swiss mice. Mol Vis. 2009;15:2578–98. [PMC free article] [PubMed] [Google Scholar]
- Sappington RM, Carlson BJ, Crish SD, Calkins DJ. The microbead occlusion model: a paradigm for induced ocular hypertension in rats and mice. Invest Ophthalmol Vis Sci. 2009;51(1):207–16. doi: 10.1167/iovs.09-3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawada A, Neufeld AH. Confirmation of the rat model of chronic, moderately elevated intraocular pressure. Exp Eye Res. 1999;69(5):525–31. doi: 10.1006/exer.1999.0732. [DOI] [PubMed] [Google Scholar]
- Schlamp CL, Johnson EC, Li Y, Morrison JC, Nickells RW. Changes in Thy1 gene expression associated with damaged retinal ganglion cells. Mol Vis. 2001;7:192–201. [PubMed] [Google Scholar]
- Schlamp CL, Li Y, Dietz JA, Janssen KT, Nickells RW. Progressive ganglion cell loss and optic nerve degeneration in DBA/2J mice is variable and asymmetric. BMC Neurosci. 2006;7(1):66. doi: 10.1186/1471-2202-7-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shareef SR, Garcia-Valenzuela E, Salierno A, Walsh J, Sharma SC. Chronic ocular hypertension following episcleral venous occlusion in rats [letter] Exp Eye Res. 1995;61(3):379–82. doi: 10.1016/s0014-4835(05)80131-9. [DOI] [PubMed] [Google Scholar]
- Sommer A. Intraocular pressure and glaucoma. Am J Ophthalmol. 1989;107(2):186–8. doi: 10.1016/0002-9394(89)90221-3. [DOI] [PubMed] [Google Scholar]
- Sommer A, Katz J, Quigley HA, Miller NR, Robin AL, Richter RC, Witt KA. Clinically detectable nerve fiber atrophy precedes the onset of glaucomatous field loss. Arch Ophthalmol. 1991;109(1):77–83. doi: 10.1001/archopht.1991.01080010079037. [DOI] [PubMed] [Google Scholar]
- Steele MR, Inman DM, Calkins DJ, Horner PJ, Vetter ML. Microarray analysis of retinal gene expression in the DBA/2J model of glaucoma. Invest Ophthalmol Vis Sci. 2006;47(3):977–85. doi: 10.1167/iovs.05-0865. [DOI] [PubMed] [Google Scholar]
- Sugiyama K, Gu ZB, Kawase C, Yamamoto T, Kitazawa Y. Optic nerve and peripapillary choroidal microvasculature of the rat eye. Invest Ophthalmol Vis Sci. 1999;40(13):3084–90. [PubMed] [Google Scholar]
- Sun D, Lye-Barthel M, Masland RH, Jakobs TC. The morphology and spatial arrangement of astrocytes in the optic nerve head of the mouse. J Comp Neurol. 2009;516(1):1–19. doi: 10.1002/cne.22058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda M, Kato H, Takamiya A, Yoshida A, Kiyama H. Injury-specific expression of activating transcription factor-3 in retinal ganglion cells and its colocalized expression with phosphorylated c-Jun. Invest Ophthalmol Vis Sci. 2000;41(9):2412–21. [PubMed] [Google Scholar]
- Tuulonen A, Airaksinen PJ. Initial glaucomatous optic disk and retinal nerve fiber layer abnormalities and their progression. Am J Ophthalmol. 1991;111(4):485–90. doi: 10.1016/s0002-9394(14)72385-2. [DOI] [PubMed] [Google Scholar]
- Ueda J, Sawaguchi S, Hanyu T, Yaoeda K, Fukuchi T, Abe H, Ozawa H. Experimental glaucoma model in the rat induced by laser trabecular photocoagulation after an intracameral injection of India ink. Jpn J Ophthalmol. 1998;42(5):337–44. doi: 10.1016/s0021-5155(98)00026-4. [DOI] [PubMed] [Google Scholar]
- Urcola JH, Hernandez M, Vecino E. Three experimental glaucoma models in rats: comparison of the effects of intraocular pressure elevation on retinal ganglion cell size and death. Exp Eye Res. 2006;83(2):429–37. doi: 10.1016/j.exer.2006.01.025. [DOI] [PubMed] [Google Scholar]
- Walsh MM, Yi H, Friedman J, Cho KI, Tserentsoodol N, McKinnon S, Searle K, Yeh A, Ferreira PA. Gene and protein expression pilot profiling and biomarkers in an experimental mouse model of hypertensive glaucoma. Exp Biol Med (Maywood) 2009;234(8):918–30. doi: 10.3181/0811-RM-344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wax MB, Tezel G. Immunoregulation of retinal ganglion cell fate in glaucoma. Exp Eye Res. 2009;88(4):825–30. doi: 10.1016/j.exer.2009.02.005. [DOI] [PubMed] [Google Scholar]
- Weber AJ, Harman CD. Structure-function relations of parasol cells in the normal and glaucomatous primate retina. Invest Ophthalmol Vis Sci. 2005;46(9):3197–207. doi: 10.1167/iovs.04-0834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber AJ, Kaufman PL, Hubbard WC. Morphology of single ganglion cells in the glaucomatous primate retina. Invest Ophthalmol Vis Sci. 1998;39(12):2304–20. [PubMed] [Google Scholar]
- Weber AJ, Viswanathan S, Ramanathan C, Harman CD. Combined application of BDNF to the eye and brain enhances ganglion cell survival and function in the cat after optic nerve injury. Invest Ophthalmol Vis Sci. 2009;51(1):327–34. doi: 10.1167/iovs.09-3740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WoldeMussie E, Ruiz G, Wijono M, Wheeler LA. Neuroprotection of retinal ganglion cells by brimonidine in rats with laser-induced chronic ocular hypertension. Invest Ophthalmol Vis Sci. 2001;42(12):2849–55. [PubMed] [Google Scholar]
- Yang Z, Quigley HA, Pease ME, Yang Y, Qian J, Valenta D, Zack DJ. Changes in Gene Expression in Experimental Glaucoma and Optic Nerve Transection: The Equilibrium between Protective and Detrimental Mechanisms. Invest Ophthalmol Vis Sci. 2007;48(12):5539–48. doi: 10.1167/iovs.07-0542. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Pernet V, Hauswirth WW, Di Polo A. Activation of the extracellular signal-regulated kinase 1/2 pathway by AAV gene transfer protects retinal ganglion cells in glaucoma. Mol Ther. 2005;12(3):402–12. doi: 10.1016/j.ymthe.2005.04.004. [DOI] [PubMed] [Google Scholar]