

Abstract
The biochemical sequelae that follow traumatic brain injury (TBI) are numerous and affect many different brain functions at different points of time as the secondary cascades progress. The complexity of the resulting pathophysiology is such that a singular therapeutic intervention may not provide adequate benefit and a combination of drugs targeting different pathways may be needed. Two of the most widely studied injury mechanisms are oxidative stress and inflammation. Numerous studies have suggested that pharmacological agents targeting either of these pathways may produce an improvement in histological and functional outcome measures. We hypothesized that combining melatonin, a potent antioxidant, with minocycline, a bacteriostatic agent that also inhibit microglia, would provide better neuroprotection than either agent used alone. To test this hypothesis, we subjected anesthetized adult male rats to a 1.5 mm controlled cortical impact and administered melatonin or vehicle in the acute post-injury period followed by daily minocycline or vehicle injections beginning the following day in a 2×2 study design. The animals were allowed to recover for 5 days before undergoing Morris water maze (MWM) testing to assess cognitive functioning following injury. There was no significant difference in MWM performance between the vehicle, melatonin, minocycline, or combination treatments. Following sacrifice and histological examination for neuroprotection, we did not observe a significant difference between the groups in the amount of cortical tissue that was spared nor was there a significant difference in [3H]-PK11195 binding, a marker for activated microglia. These results suggest that neither drug has therapeutic efficacy, however dosing and/or administration issues may have played a role.
Keywords: Autoradiography, Morris water maze, controlled cortical impact, rat
Introduction
There are numerous biochemical processes set into place after a traumatic brain injury (TBI) that present multiple opportunities for neuroprotection or enhancement of functional recovery. However, the “golden bullet” strategy has not proved successful in clinical trials [1]. The failure of monotherapy suggests that this approach is too restricted in mechanism, timing, and magnitude of neuroprotection. A more effective strategy may be to target multiple downstream pathways such as required for diabetes mellitus, hypertension, hyperlipidemia, HIV/AIDS and numerous cancers.
Two well studied mechanisms of neuronal damage following TBI are oxidative/nitrosative stress and inflammation. Within minutes following a traumatic event, a dramatic increase in free radicals saturates endogenous scavenging mechanisms leading to the breakdown of membrane lipids, essential proteins, DNA ultimately leading to cell death [9]. Almost simultaneously, microglia assume an activated state and phagocytize cellular debris [14, 19]. Activated microglia undergo multiple changes, including the up-regulation of the mitochondrial cholesterol transporter which is associated with the 18 kDa translocator protein (TPSO). This protein binds the isoquinoline carboxamide PK11195 with nanomolar affinity and previous studies have used autoradiographic localization of [3H]-PK11195 binding to detect the presence of presumed activated microglia in TBI [12, 13, 22].
Melatonin (MEL), known for its role in regulation of circadian rhythms, is a pleiotropic compound that exerts multiple physiological actions such as antioxidant properties [24]. As an antioxidant, MEL can directly scavenge free radicals while also acting indirectly to increase the expression of endogenous antioxidant enzymes [reviewed in 24] and may be superior to glutathione, mannitol, and vitamin E [23]. Additionally, due to its amphiphilic structure, MEL has no barriers to its distribution and may have the advantages of having a lower side effect profile and producing fewer pharmacokinetic or pharmacodynamic interactions compared to xenobiotic antioxidants. Therefore, MEL could be highly effective in protecting the traumatized brain from oxidative damage.
Minocycline (MIN) is a tetracycline-derived bacteriostatic agent shown to be neuroprotective in rodent models of ischemic brain injury [28, 29]. The neuroprotective potential of MIN in experimental stroke studies have led to its recent use in an open-label clinical trial [16]. Several mechanisms by which MIN may exert neuroprotective properties have been proposed including inhibition of metalloproteases, anti-apoptotic action, and its ability to potently inhibit microglia proliferation and activation [reviewed in 7].
The complexity of TBI pathology led us to hypothesize that modulation of a single pathway would be inferior to pharmacological strategies targeting multiple pathways. Ideally, drug candidates would fit the selected criteria: 1) the broad utility of these drugs in animal models of neurodegenerative disease, 2) both drugs are currently approved for use in humans, 3) each drug is safe and associated with minimal toxicity and 4) both compounds are highly lipophilic, and readily cross the blood-brain barrier (BBB). Our hypothesis is that combination therapy targeting free radical biology using MEL and CNS inflammation using MIN will improve cognitive and histological outcomes following TBI greater than either agent administered alone.
Materials and Methods
Animal Subjects
These experiments utilized 50 adult, male Sprague-Dawley rats (225 - 275 g; Harlan Laboratories, Indianapolis, IN; n = 6 – 9 per treatment group) and were housed 1 – 2 animals per cage in a climate controlled room, and maintained on a 12-hour light/dark schedule. Animals were allowed free access to food and water throughout the duration of the experiments. All experimental procedures were reviewed and approved by the Institutional Animal Care and Use Committee at the University of Kentucky and conducted in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals.
Surgical Methods
All rats were exposed to a controlled cortical impact (CCI) as previously described [8]. Anesthesia was induced with 5% inhaled isoflurane and the animals were weighed for drug dosing. The animals were secured in a Kopf stereotaxic frame (David Kopf Instruments, Tujunga, CA) and anesthesia was maintained with 2% inhaled isoflurane. Following a midline incision, a 6 mm craniotomy was performed using a Michele trephine to expose the underlying somatosensory cortex, taking care to not disturb the dura. The Precision Systems and Instrumentation TBI-0310 (Fairfax Station, VA) administered either a 1.5 mm or 2 mm cortical compression (5 mm impactor diameter, 3.5 m/s velocity, 400 msec dwell time) to the surface of the exposed cortex. Following injury, Surgicel (Johnson & Johnson, Dallas, TX) was placed over the injury site; the skull cap was replaced and sealed with dental acrylic. The skin incision was closed with wound clips and the animal was returned to its home cage to recover 5 days before further experimentation.
Drug Treatment Regimens
Two experiments were conducted to test the effects of MEL and/or MIN following TBI. In Experiment 1, animals injured with a 1.5 mm CCI were randomized to receive equal volume of vehicle (VEH, 2% ethanol/phosphate buffered saline), MEL (5 mg/kg; Sigma, St. Louis, MO), MIN (40 mg/kg; MP Biomedicals, Solon, OH), or both drugs (MEL/MIN) according to the dosing schedule indicated in Figure 1. The immediate dosing schedule for melatonin was selected based on the short time frame of free radical production following TBI [9] while the delayed schedule for minocycline was selected so as not to interfere with the initial microglial response, thought to have a protective role by isolating the region of damage [5, 19]. The acute study (Experiment 2) was performed to determine if minocycline administration was necessary in the acute post-injury period. Animals in the acute administration study were injured with a 2 mm CCI and randomized to receive an equal volume of vehicle (VEH, 2% ethanol/phosphate buffered saline), MEL (5 mg/kg; Sigma, St. Louis, MO) or MIN (45 mg/kg; MP Biomedicals, Solon, OH) at 5 minutes and again at 90 minutes after injury. A more severe injury was used to illicit a greater microglial response and the dose was adjusted to more closely match other literature reports [3]. The MEL+MIN group was omitted in this study to reduce the number of animals used.
FIGURE 1.
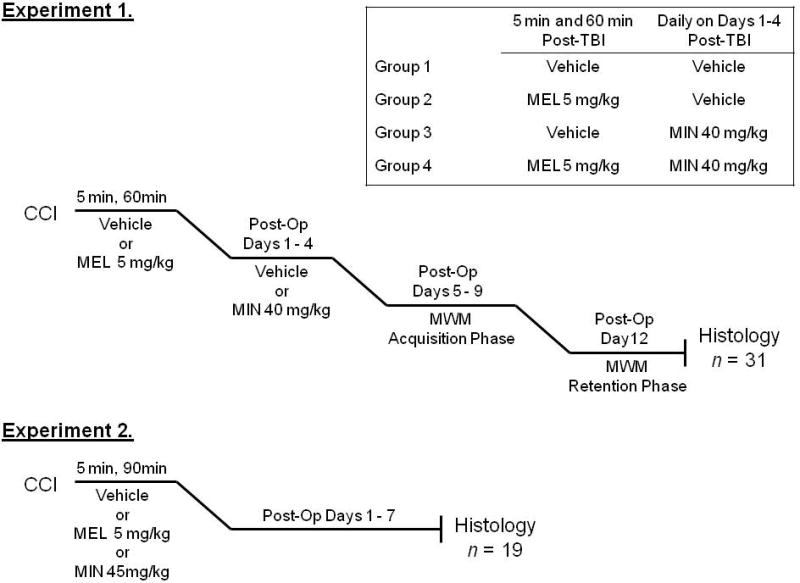
Experimental design.
Post-injury Cognitive Assessment
The animals were tested in the Morris water maze (MWM) to determine changes in spatial learning ability as previously described [8]. Briefly, animals were tested in a 127 cm diameter × 56 cm tall circular pool with a 13.5 cm diameter circular platform submerged 1 cm below the waterline and located in the middle of the SE quadrant. Black, non-toxic powdered paint was added to obscure the platform. The lighting of the room and various spatial cues were constant throughout the acquisition period. The animals were given 4 – 60 second acquisition trials/day over 5 days with their entry points (N, S, E, W) being randomized. If the animal did not find the platform in 60 seconds, it was placed there by the handler and allowed to rest for 15 sec. The animals were allowed a 5-minute rest between each trial. Three days following the last acquisition trial, a retention trial was performed. In this phase, the platform was removed from the pool and the animals entered the pool at a novel location (NW). The animals were allowed to swim for 60 seconds and returned to their home cages. During all phases of the MWM, the animals' performance was recorded and analyzed with Accuscan Instruments EzVideoDV Automated Tracking System (Columbus, OH) that allowed quantification of escape latency and swim speed for the acquisition tests and swimming speed, time spent in target quadrant, distance swam in target quadrant, and target quadrant crossings during the retention test.
Cortical Tissue Sparing Analysis
Following cognitive testing, the animals were euthanatized by rapid decapitation, their brains rapidly removed, flash frozen in chilled isopentane, and stored at -80°C until processing. The frozen tissue was sectioned at 16 μm on a Leica 1850M cryostat (Nussloch, Germany) and was thaw mounted onto Fisher SuperFrost Plus® slides. The slides were stored under desiccation overnight at 4°C then transferred to a -80°C freezer until time of experimentation.
To assess the potential neuroprotective effect of drug administration, a tissue sparing analysis was performed. One set of slides were removed from -80°C and allowed to thaw overnight. The slides were then stained with cresyl violet. Images of the slices were obtained between -1.80 mm and -5.30 mm from bregma according the coordinates of Paxinos and Watson [21]. Quantitative assessment of spared tissue employed the Cavalieri method as described previously [26]. The amount of tissue sparing was expressed as a percentage of the total cortical volume of the injured hemisphere compared to the uninjured hemisphere.
[3H]-PK11195 Autoradiography
Microglial activation was assessed by TSPO autoradiography as used previously with mouse and rat TBI sections in our lab [8, 12, 13]. The slides were removed from -80°C and allowed to thaw overnight and loaded into binding racks. The slides were incubated in the following buffers at 4°C: 50 mM Tris HCl (pH 7.4) for 15 minutes, 50 mM Tris HCl and 2 nM [3H]-PK11195 (PerkinElmer, Boston, MA, specific activity = 73.6 Ci/mmol) for 2 hours, 3 washes in 50 mM Tris HCl (pH 7.4) for 3 minutes each, and a brief wash in ddH2O. The slides were left overnight to dry at room temperature, placed into autoradiography cassettes and exposed to Kodak BioMax film for approximately 5 weeks. Brain regions analyzed were selected based on previous studies that have shown increased microglial activation following brain injury [12, 13, 22].
Statistical Methods
All data were tested for outliers and are reported as the mean ± standard deviation (SD). For the acquisition phase of the MWM, the data was analyzed using a two-way (treatment × day), repeated measures (day) ANOVA. Data collected from the target quadrant during the retention phase of the MWM was analyzed using a one-way (treatment) ANOVA. Data from the tissue sparing analysis was analyzed by one-way ANOVA. [3H]-PK11195 binding data was analyzed using a two-way (treatment × hemisphere), repeated measures (hemisphere) ANOVA. Statistical significance, defined as α ≤ 0.05, as detected by ANOVA was further analyzed using Student Neuman-Keulls (SNK) posthoc test.
Results
Post-injury Cognitive Assessment
We did not observe any mortality as a result of the injury or drug treatment. No significant group effects were observed in the acquisition phase of the MWM between animals receiving MEL, animals receiving MIN, or the MEL+MIN combination (Figure 2). Two-way, repeated measures ANOVA results of the total distance swam revealed a significant effect of day [F(4,100) = 42.27, P < 0.0001] suggesting that each group was equally capable of learning the task over the 5-day testing period. Treatment groups ended the 5-day testing period with slightly lower latencies than VEH treated animals, but there was no significant effect of treatment and no significant interaction between the two terms. Likewise, no significant differences were seen in any measure of the retention phase of the MWM (data not shown). These results suggest that neither drug, either alone or in combination, had any effect on the ability for the animal to remember the target quadrant and navigate to that quadrant.
FIGURE 2.

Average escape latency travelled during the acquisition phase of the MWM following a 1.5 mm CCI for animals treated with MEL, MIN, or MEL+MIN.
Acquisition data for VEH, MEL 5mg/kg, MIN 40 mg/kg, and MEL+MIN treated animals beginning on the sixth experimental day (5 days following a 1.5 mm injury). No significant treatment effects were seen in the acquisition phase of this task. Data represented as mean ± SD.
Cortical Tissue Sparing Analysis
We did not detect a significant difference between the VEH, MEL, MIN, or MEL+MIN groups with the amount of cortical tissue spared following injury suggesting that neither drug alone or in combination had an effect on lesion size following CCI (Fig. 3). Similarly, we did not find a difference compared to vehicle in the amount of cortical tissue spared following acute administration of MEL or MIN following a more severe 2 mm CCI (data not shown).
FIGURE 3.

Cortical tissue spared nine days following a 1.5 mm CCI to rats administered MEL, MIN, or MEL+MIN.
Cortical tissue sparing effect of VEH, MEL 5 mg/kg, MIN 40 mg/kg, and MEL+MIN at 14 days following a 1.5 mm CCI. ANOVA results did not detect a significant treatment effect. Data represented as mean ± SD.
[3H]-PK11195 Autoradiography
ANOVA results of [3H]-PK11195 binding did not detect a significant treatment × hemisphere interaction or a significant effect of treatment, but did show a significant hemisphere effect (Supplement 1). Post hoc analysis showed a significant increase in [3H]-PK11195 binding in the injured hemisphere over the uninjured hemisphere suggesting the presence of more activated microglia. This finding was consistent in each brain region analyzed with the exception of the dentate gyrus of the VEH treated animals where no significant difference was observed between the uninjured and injured hemispheres. These data suggest that neither MEL, MIN, nor MEL+MIN had any effect on presumable glia cell activation in these brain regions. Similar findings were observed in the acute administration study where neither MEL nor MIN attenuated [3H]-PK11195 binding in any brain region compared to vehicle following the 2 mm CCI (data not shown).
Discussion
MEL and MIN are approved by the U.S. Food and Drug Administration and widely used in clinical practice. Additionally, both compounds have shown neuroprotective potential in multiple models of experimental brain injury. The primary aim of the current study was to test the hypothesis that MEL and MIN administration would improve cognitive and histopathological outcomes following TBI to a greater extent than either drug administered alone.
MEL appears to be an ideal candidate molecule for the treatment of TBI with its antioxidant potency, ability to cross the BBB, and low toxicity. However, our research failed to demonstrate a neuroprotective effect. Our studies were limited in terms of dose-effect relationships with MEL and it is possible that administration of higher doses would have been more efficacious. The doses that were used in this study were selected based on research reports using this agent in models of both traumatic and ischemic brain injury. Although the doses of MEL reported for TBI treatment vary significantly (0.625 mg/kg to 200 mg/kg), two TBI studies used a 5 mg/kg MEL dose following a weight-drop TBI with positive results [2, 20]. However, it is possible that the pineal gland sustained damage due to the relatively uncontrolled nature of the weight-drop injury compared to the CCI. Additionally, some authors report MEL administered during night hours, while endogenous MEL levels are high, was effective whereas administration during daylight hours was not [25, 27]. More research into the pharmacology and pharmacokinetics of these compounds following the CCI and other TBI models would be useful.
Similar issues exist with regards to MIN treatment of neurodegenerative disease. Although MIN has been shown in multiple studies to promote neuroprotection, there are also studies that show either no effect or detrimental outcomes following its administration. In a recent example, MIN treatment administered to mice failed to improve Neurological Severity Score, pericontusional lesion volume, or TUNEL staining at four days post-injury [3]. Results such as this using other models of neurodegeneration have led to a discussion about the utility of MIN for this purpose and the need for publication of negative results [6].
Perhaps one issue is the varying doses and appropriate therapeutic windows reported in the literature. MIN doses of 1 mg/kg were reported to attenuate BBB breakdown and the formation of cerebral edema following injury [11]. While many studies administered drug shortly after injury, some have been published reporting MIN administration beginning at least one day following experimental ischemic injury and continuing daily for an extended period of time [10, 17] including a human stroke study that reported efficacy when MIN was administered up to 24 hours after the ischemic event, though few patients received drug at this time [16]. We chose a delayed MIN administration design based on preliminary studies on microglia activation conducted in our laboratory that showed peak [3H]-PK11195 binding at 24 hours and to avoiding any potential pharmacokinetic interactions with MEL.
The debate about the well known neuroinflammatory effects versus the emerging neuroprotective role of microglia in response to brain injury may also help explain the apparent disparity about the benefits of MIN therapy. These cells respond very rapidly to brain injury, extending cellular processes towards the injured site serving to isolate the area and protect the surrounding tissue [5, 19]. Recent studies have shown that microglia are capable of producing neurotrophic factors and that the amounts produced of each may depend on the type and severity of the brain injury [15]. Therefore, while microglia remain a potential therapeutic target for the amelioration of brain injury, it is not known where in the activation cycle or whether inhibition or enhancement of their functions is the proper strategy.
Interestingly, MIN had no effect on [3H]-PK11195 binding in our study though others have shown a decrease in immunohistochemical markers of microglia activation [3, 17]. Investigations of immunohistochemical markers of microglia activation and [3H]-PK11195 autoradiograms have shown a close correlation between the two, leading to the conclusion that [3H]-PK11195 binding is primarily to activated microglia [18]. It is possible that since the MIN treated animals were euthanized 7 – 9 days after the last dose of MIN, that activated microglia increased following the discontinuation of therapy. All animals were sacrificed and tissue harvested within 14 days following injury, a time frame where activated microglia are found in large quantities in the injured hemisphere [4] and [3H]-PK11195 binding is increased [22]. The pharmacodynamic effect of MIN on [3H]-PK11195 binding should be evaluated when the final drug administration is followed shortly thereafter with sacrifice and tissue collection to fully determine if there is a relationship.
Based on the findings of our studies, it does not appear that either MEL, MIN, or MEL+MIN are effective for the improvement of functional and histopathological deficits following TBI in rats, using the model, dose, timing, and outcome measures that were selected.
Supplementary Material
Acknowledgments
We acknowledge the technical expertise of Deann Hopkins and Alex McFarland-Watts. These experiments were made possible by NS42196 to J.R.P. and AG21981 to S.W.S.
Abbreviations
- ANOVA
analysis of variance
- BBB
blood-brain barrier
- CCI
controlled cortical impact
- MEL
melatonin
- MIN
minocycline
- MWM
Morris water maze
- TBI
traumatic brain injury
- TSPO
translocator protein
- VEH
vehicle
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Beauchamp K, Mutlak H, Smith WR, Shohami E, Stahel PF. Pharmacology of traumatic brain injury: where is the “golden bullet”? Mol Med. 2008;14:731–740. doi: 10.2119/2008-00050.Beauchamp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beni SM, Kohen R, Reiter RJ, Tan DX, Shohami E. Melatonin-induced neuroprotection after closed head injury is associated with increased brain antioxidants and attenuated late-phase activation of NF-kappaB and AP-1. Faseb J. 2004;18:149–151. doi: 10.1096/fj.03-0323fje. [DOI] [PubMed] [Google Scholar]
- 3.Bye N, Habgood MD, Callaway JK, Malakooti N, Potter A, Kossmann T, Morganti-Kossmann MC. Transient neuroprotection by minocycline following traumatic brain injury is associated with attenuated microglial activation but no changes in cell apoptosis or neutrophil infiltration. Exp Neurol. 2007;204:220–233. doi: 10.1016/j.expneurol.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 4.Chen S, Pickard JD, Harris NG. Time course of cellular pathology after controlled cortical impact injury. Experimental neurology. 2003;182:87–102. doi: 10.1016/s0014-4886(03)00002-5. [DOI] [PubMed] [Google Scholar]
- 5.Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan WB. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8:752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- 6.Diguet E, Gross CE, Tison F, Bezard E. Rise and fall of minocycline in neuroprotection: need to promote publication of negative results. Experimental neurology. 2004;189:1–4. doi: 10.1016/j.expneurol.2004.05.016. [DOI] [PubMed] [Google Scholar]
- 7.Domercq M, Matute C. Neuroprotection by tetracyclines. Trends Pharmacol Sci. 2004;25:609–612. doi: 10.1016/j.tips.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 8.Guseva MV, Hopkins DM, Scheff SW, Pauly JR. Dietary Choline Supplementation Improves Behavioral, Histological, and Neurochemical Outcomes in a Rat Model of Traumatic Brain Injury. J Neurotrauma. 2008 doi: 10.1089/neu.2008.0516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hall ED, Vaishnav RA, Mustafa AG. Antioxidant therapies for traumatic brain injury. Neurotherapeutics. 2010;7:51–61. doi: 10.1016/j.nurt.2009.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hayakawa K, Mishima K, Nozako M, Hazekawa M, Mishima S, Fujioka M, Orito K, Egashira N, Iwasaki K, Fujiwara M. Delayed treatment with minocycline ameliorates neurologic impairment through activated microglia expressing a high-mobility group box1-inhibiting mechanism. Stroke; a journal of cerebral circulation. 2008;39:951–958. doi: 10.1161/STROKEAHA.107.495820. [DOI] [PubMed] [Google Scholar]
- 11.Higashida T, Kreipke CW, Rafols JA, Peng C, Schafer S, Schafer P, Ding JY, Dornbos D, Li X, Guthikonda M, Rossi NF, Ding Y. The role of hypoxia-inducible factor-1alpha, aquaporin-4, and matrix metalloproteinase-9 in blood-brain barrier disruption and brain edema after traumatic brain injury. Journal of neurosurgery. 2010 doi: 10.3171/2010.6.JNS10207. [DOI] [PubMed] [Google Scholar]
- 12.Kelso ML, Scheff SW, Pauly JR, Loftin CD. Effects of genetic deficiency of cyclooxygenase-1 or cyclooxygenase-2 on functional and histological outcomes following traumatic brain injury in mice. BMC Neurosci. 2009;10:108. doi: 10.1186/1471-2202-10-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kelso ML, Wehner JM, Collins AC, Scheff SW, Pauly JR. The pathophysiology of traumatic brain injury in alpha7 nicotinic cholinergic receptor knockout mice. Brain Res. 2006;1083:204–210. doi: 10.1016/j.brainres.2006.01.127. [DOI] [PubMed] [Google Scholar]
- 14.Koizumi S, Shigemoto-Mogami Y, Nasu-Tada K, Shinozaki Y, Ohsawa K, Tsuda M, Joshi BV, Jacobson KA, Kohsaka S, Inoue K. UDP acting at P2Y6 receptors is a mediator of microglial phagocytosis. Nature. 2007;446:1091–1095. doi: 10.1038/nature05704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lai AY, Todd KG. Differential regulation of trophic and proinflammatory microglial effectors is dependent on severity of neuronal injury. Glia. 2008;56:259–270. doi: 10.1002/glia.20610. [DOI] [PubMed] [Google Scholar]
- 16.Lampl Y, Boaz M, Gilad R, Lorberboym M, Dabby R, Rapoport A, Anca-Hershkowitz M, Sadeh M. Minocycline treatment in acute stroke: an open-label, evaluator-blinded study. Neurology. 2007;69:1404–1410. doi: 10.1212/01.wnl.0000277487.04281.db. [DOI] [PubMed] [Google Scholar]
- 17.Liu Z, Fan Y, Won SJ, Neumann M, Hu D, Zhou L, Weinstein PR, Liu J. Chronic treatment with minocycline preserves adult new neurons and reduces functional impairment after focal cerebral ischemia. Stroke; a journal of cerebral circulation. 2007;38:146–152. doi: 10.1161/01.STR.0000251791.64910.cd. [DOI] [PubMed] [Google Scholar]
- 18.Myers R, Manjil LG, Cullen BM, Price GW, Frackowiak RS, Cremer JE. Macrophage and astrocyte populations in relation to [3H]PK 11195 binding in rat cerebral cortex following a local ischaemic lesion. J Cereb Blood Flow Metab. 1991;11:314–322. doi: 10.1038/jcbfm.1991.64. [DOI] [PubMed] [Google Scholar]
- 19.Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- 20.Ozdemir D, Tugyan K, Uysal N, Sonmez U, Sonmez A, Acikgoz O, Ozdemir N, Duman M, Ozkan H. Protective effect of melatonin against head trauma-induced hippocampal damage and spatial memory deficits in immature rats. Neurosci Lett. 2005;385:234–239. doi: 10.1016/j.neulet.2005.05.055. [DOI] [PubMed] [Google Scholar]
- 21.Paxinos G, Watson C. The Rat Brain in Sterotaxic Coordinates. Academic Press; Sydney: 1986. [Google Scholar]
- 22.Raghavendra Rao VL, Dogan A, Bowen KK, Dempsey RJ. Traumatic brain injury leads to increased expression of peripheral-type benzodiazepine receptors, neuronal death, and activation of astrocytes and microglia in rat thalamus. Experimental neurology. 2000;161:102–114. doi: 10.1006/exnr.1999.7269. [DOI] [PubMed] [Google Scholar]
- 23.Reiter RJ, Carneiro RC, Oh CS. Melatonin in relation to cellular antioxidative defense mechanisms. Horm Metab Res. 1997;29:363–372. doi: 10.1055/s-2007-979057. [DOI] [PubMed] [Google Scholar]
- 24.Reiter RJ, Tan DX, Osuna C, Gitto E. Actions of melatonin in the reduction of oxidative stress. A review. J Biomed Sci. 2000;7:444–458. doi: 10.1007/BF02253360. [DOI] [PubMed] [Google Scholar]
- 25.Sarrafzadeh AS, Thomale UW, Kroppenstedt SN, Unterberg AW. Neuroprotective effect of melatonin on cortical impact injury in the rat. Acta neurochirurgica. 2000;142:1293–1299. doi: 10.1007/s007010070028. [DOI] [PubMed] [Google Scholar]
- 26.Sullivan PG, Bruce-Keller AJ, Rabchevsky AG, Christakos S, Clair DK, Mattson MP, Scheff SW. Exacerbation of damage and altered NF-kappaB activation in mice lacking tumor necrosis factor receptors after traumatic brain injury. J Neurosci. 1999;19:6248–6256. doi: 10.1523/JNEUROSCI.19-15-06248.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ucar T, Ozkaya G, Demir N, Gurer I, Akyuz M, Onal MZ. The effects of environmental light--dark changes on experimental mild traumatic brain injury. Acta Neurol Scand. 2005;112:163–172. doi: 10.1111/j.1600-0404.2005.00463.x. [DOI] [PubMed] [Google Scholar]
- 28.Yrjanheikki J, Keinanen R, Pellikka M, Hokfelt T, Koistinaho J. Tetracyclines inhibit microglial activation and are neuroprotective in global brain ischemia. Proc Natl Acad Sci U S A. 1998;95:15769–15774. doi: 10.1073/pnas.95.26.15769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yrjanheikki J, Tikka T, Keinanen R, Goldsteins G, Chan PH, Koistinaho J. A tetracycline derivative, minocycline, reduces inflammation and protects against focal cerebral ischemia with a wide therapeutic window. Proc Natl Acad Sci U S A. 1999;96:13496–13500. doi: 10.1073/pnas.96.23.13496. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.