

Abstract
Although the ability to monitor specific molecules in vivo in real-time could revolutionize many aspects of healthcare, the technological challenges that stand in the way of reaching this goal are considerable and are poorly met by most existing analytical approaches. Nature, however, has already solved the problem of real-time molecular detection in complex media by employing “biomolecular switches.” That is, protein and nucleic acids that sense chemical cues and, by undergoing specific, binding-induced conformational changes, transduce this recognition into high-gain signal outputs. Here, we argue that devices employing such switches represent a promising route towards versatile, real-time molecular monitoring in vivo.
Introduction
Imagine a world in which your doctor could slip a fine-needle sensor into your arm and use it to continuously monitor the concentration of almost any molecule in real-time as it circulates through your peripheral blood vessels. Such technology would revolutionize many areas of diagnostics and could enable the development of a broad suite of new therapies. In the diagnostic arena, for example, the continuous, quantitative measurement of serum creatinine and other molecular markers could be used to monitor kidney function [1], and the ability to monitor both glucose and insulin would, by preventing hyperinsulinemia, improve diabetes management and advance the development of an artificial pancreas [2]. More broadly, continuous in vivo monitoring could fuel future advances in personalized medicine; although this is often described as the technology to deliver the right drug to the right patient, in order to achieve the most effective therapeutic outcome, it is just as important to deliver the right dose. Even within a single patient, however, the optimal dose of a given drug varies over the course of hours or days in concert with changes in metabolism or health status. By closing the loop between a drug’s administration and its in vivo concentration, the ability to continuously monitor serum drug levels would overcome such time-dependence, enabling precise, feedback-controlled drug delivery. This, in turn, would open the door to an entirely new wave of “quantitative therapeutics” in which drugs with complex dosing regimens or hitherto uncomfortably narrow therapeutic indices could be easily and effectively administered. Unfortunately, though, while the potential applications of continuous, real-time molecular detection have been recognized for decades [3, 4], the capability to perform such detection remains limited. Here, we examine the state-of-the-art and describe what we view as a promising potential route toward this important goal.
The majority of existing technologies for molecular analysis, including chromatography, mass spectrometry and immunochemical techniques, are multi-step “batch” processes that cannot support continuous, real-time detection. Conversely, with few exceptions [5–7], the various real-time methods by which specific molecules can be monitored, such as spectroscopy, fail when implemented in vivo owing to the high molecular complexity and challenging physical properties (e.g. the scattering and absorbance of light) of cells, blood and tissues. Indeed, in order to achieve continuous, real-time detection in vivo, a number of key challenges must be surmounted: (i) the technology must be selective enough to reject false signals arising from interferants present in the complex environments found in vivo; (ii) it should operate without requiring any exogenous reagents beyond those provided in situ by the organism; (iii) it must operate continuously and cannot rely on batch process steps, such as separations, washing or incubation; and (iv) it must be reversible, such that the sensor’s response rises and falls in concert with changing target concentrations. Unfortunately, although conventional technologies (e.g. chromatography, spectroscopy, immunochemical approaches) achieve one or more of these goals, currently there is no general approach to molecular detection that is reagent-free, continuous and selective enough to work directly within living organisms.
Biosensors
Although there is no universal platform that supports real-time, continuous monitoring, a limited number of successes for a few specific molecular targets illustrate potential routes to a more versatile solution. For example, the most commonly employed real-time molecular detection deployed in hospitals today is the pulse oximeter, which monitors blood oxygenation optically through the skin by employing the patient’s own hemoglobin as an indicator of oxygen concentration. Perhaps a more compelling example is the glucose-oxidase-based sensor, which now achieves the continuous, real-time measurement of interstitial glucose concentrations with sub-minute time resolution (reviewed in Ref. [8]). The common denominator in these examples is the use of biomolecular recognition elements as the basis for the sensing technology – an approach that harnesses the exquisite specificity of such “receptor-ligand” interactions in order to detect their target molecules.
Despite the broad range of potential specificities afforded by biomolecular recognition, the above-described examples of in vivo “biosensors” do not easily translate to the detection of other classes of molecular targets. The oxygen sensor, for example, monitors oxygenation of the patient’s own hemoglobin by measuring the absorbance of visible light by red blood cells. Thus, this technology is generally not transferrable to the detection of other molecular analytes. Likewise, the glucose sensor relies on the specific catalytic activity of glucose oxidase, an approach that has been extended to the detection of a limited number of other analytes because the relevant oxidase enzymes are only available for a few specific molecular targets; to date, only lactate [9], glutamate (reviewed in Ref. [10]) and cholesterol [11] have seen similar in vivo sensors described in the literature. Indeed, while biomolecular recognition itself is enormously versatile, there is not yet any general method of adapting such recognition into sensors that support real-time, in vivo detection.
The fundamental difficulty in developing biosensors is linking biomolecular binding to a specific, measurable output. In other words, an antibody does not emit light or electrons upon binding its target antigen. A common solution to this problem has been to attach the receptor to a surface and then measure a physical property, such as refractive index [e.g. surface plasmon resonance (SPR)], mass [e.g. quartz crystal microbalance (QCM)], steric bulk (e.g. static microcantilever, impedance spectroscopy) or charge (e.g. field effect transistors), which changes when the receptor is occupied. A notable disadvantage of these approaches is that, because they monitor the molecular recognition event by measuring differences in basic physical properties, they cannot distinguish between specific binding of target analyte and the non-specific adsorption of contaminants. For example, SPR has proven useful in measuring molecular interactions of purified samples (often achieving detection limits up to an order of magnitude or more below the dissociation constant of the relevant ligand-receptor interaction [12]); however, the approach largely fails when challenged with complex samples that contain excess background molecules, such as blood serum, which contains more than 3000 different proteins at a total concentration of ~70 mg/ml [13]. Thus, the core challenge in applying biosensors to in vivo monitoring remains: how can we link specific molecular recognition events to measurable output signals in the presence of complex backgrounds, without invoking reagents or batch processing?
Sensors based on biomolecular switches
Nature has solved the problem of signal transduction against complex backgrounds by coupling molecular recognition to a second, equally important feature: binding-induced “switching”. That is, the biomolecules that perform chemoperception in vivo invariably couple molecular recognition to a binding-induced change in conformation or oligomerization state. These switching events, in turn, trigger specific output signals, such as the opening of an ion channel or the activation of an enzyme. Inspired by the speed, specificity and versatility of these naturally occurring sensors, recent years have witnessed significant efforts to create artificial biosensors based on these principles (Figure 1). We believe that these efforts represent a promising route towards versatile, real-time molecular monitoring in vivo.
Figure 1.



Recent years have seen the development of a large number of optical and electrochemical biosensors based on binding-induced conformational switching. Examples include: (a) A large family of optical sensors is based on the binding-induced conformational changes in proteins in the periplasmic binding protein family; this motion, in turn, modulates the emission of an attached fluorophore or fluorophores. Reproduced with permission from Ref. [15]. (b) Molecular beacons, which are stem-loop DNA molecules, open, and thus fluoresce, upon hybridization to a complementary target oligonucleotide. Reproduced with permission from Ref. [40]. (c) Electrochemical aptamer-beacons, undergo binding-induced “folding” of specific RNA or DNA aptamers, which can be monitored electrochemically via changes in electron transfer from an attached redox tag to a supporting electrode.
Although the field is little more than a decade old, examples of switch-based biosensors are compelling and growing. These biosensors broadly fall into two classes [14]. The first employs naturally occurring biomolecular switches to detect either their natural ligands or new molecular targets created via the rational or selection-driven re-design of their binding sites (reviewed in Refs. [14, 15]). Specific examples include a large number of sensors that employ binding-induced conformational change in the two-domain periplasmic-binding-protein superfamily (reviewed in Ref. [15]; see also Ref. [16] for a discussion of the precise sensing mechanism), including sensors for the detection of a wide range of specific sugars, amino acids, and inorganic ions. The second class focuses on recognition elements that do not undergo binding-induced conformational changes, which are much more common. These “non-responsive” receptors are then re-engineered to undergo a large-scale conformational change upon binding their target molecules; examples include devices based on the binding-induced folding of polypeptides (reviewed in Ref. [17]), proteins [18, 19] and aptamers (reviewed in Ref. [20]). Aptamers, in particular, can be readily re-engineered to undergo a large-scale conformation change upon target binding, either by destabilization, which couples binding to a change in the folding equilibrium constant, or by introduction of a short auxiliary sequence complementary to the aptamer, forcing it to undergo a double-stranded-to-aptamer fold transition upon target binding. Irrespective of which approach is taken in their design, all binding-induced switches couple target recognition with a large-scale conformational change that is well-suited for sensing applications [21].
An equally important aspect of creating sensors out of such structure-switching molecules is that their conformational state must be linked to a readily detectable output. To date, structure-switching sensors have been described that respond via fluorescence emission (for optical detection), electron transfer (for electrochemical detection) or biochemical activity (typically linked to catalytically amplified detection) (reviewed in Ref. [14]). Although still challenging because photons and electrons can be conducted outside of the body more readily than the products of binding-activated catalytic domains, we focus here on optical and electrochemical output mechanisms. Perhaps the most widely used approach for linking structure switching to a specific output has been to employ the conformation-linked modulation of fluorescence (Figures 1a, b). This is achieved via distance-dependent changes in Förster resonance energy transfer (FRET) (Figure 1a). Another method is the more strongly distance-dependent photoelectron transfer (PET) (Figure 1b) either between two optical reporter groups [17] or via conformation-linked changes in the chemical environment around a single, structure-sensitive fluorophore [22].
Switch-based electrochemical sensors are similarly engineered by attaching an electroactive reporter (e.g. ferrocene or methylene blue) to one position of the biomolecule that is in turn attached to the surface of an interrogating electrode via a second, distal position (Figure 1c) (reviewed in Ref. [23]). Binding-induced conformational changes thus alter the ability of the redox reporter to strike the surface, changing the current it produces when interrogated via, for example, AC voltammetry. To date, several research groups have described such structure-switching electrochemical sensors that employ a range of nucleic acid- [24], polypeptide- [25] and protein- [26] based biomolecular switches. Nucleic acid switches (reviewed in Ref. [23]) have proven particularly amenable to this approach: electrochemical aptamer-based (E-AB) sensors, for example, have been described to date directed against a number of specific proteins, inorganic ions and small molecules (reviewed in Refs. [20, 23]).
The use of switch-based sensors in vivo
Structure-switching sensors appear to be particularly suitable for application in chemically complex environments. For example, binding-induced switching has been used to design genetically encoded maltose [27] and calcium ion [28] sensors composed of three components: a target-responsive switching domain, which undergoes a large-scale conformational change upon target binding, and two fluorescent protein FRET reporters (Figure 2a). Other pioneering examples include the use of FRET-reporting DNA switches, termed molecular beacons [40], to monitor intracellular mRNA levels (Figure 2b) [29], and the demonstration a FRET-reporting, proton-responsive DNA switch that supports intracellular measurements of pH [30].
Figure 2.
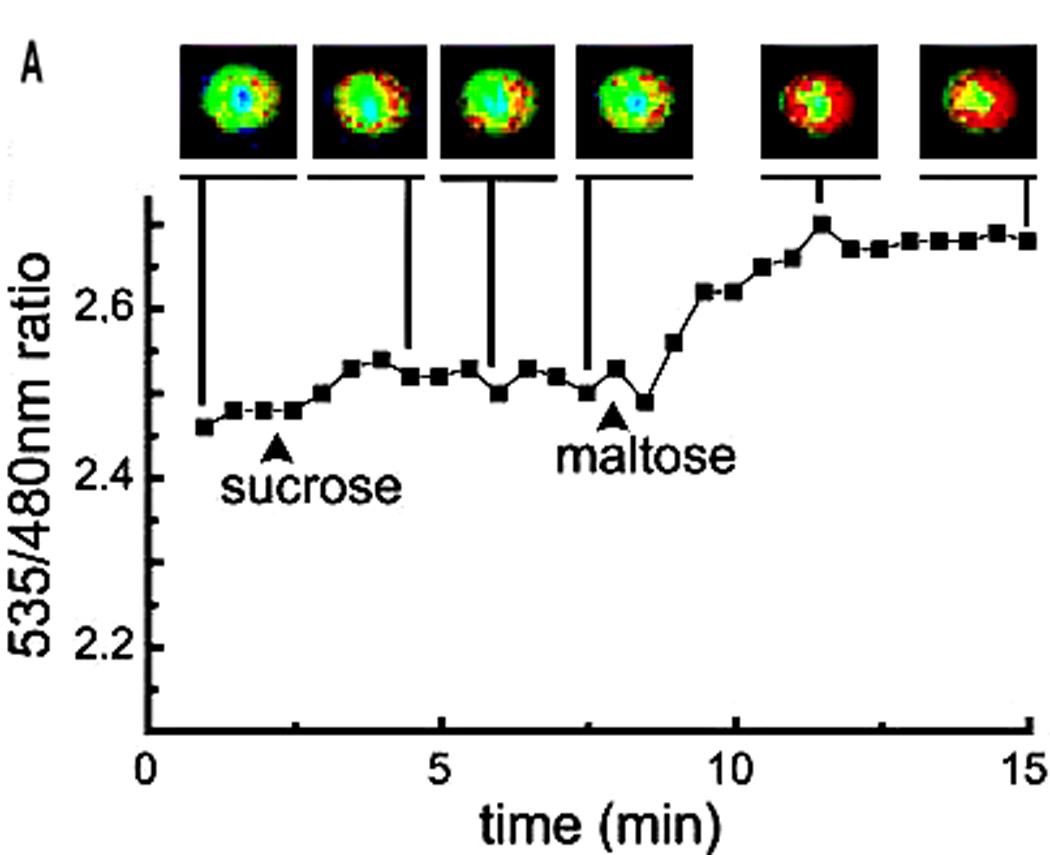
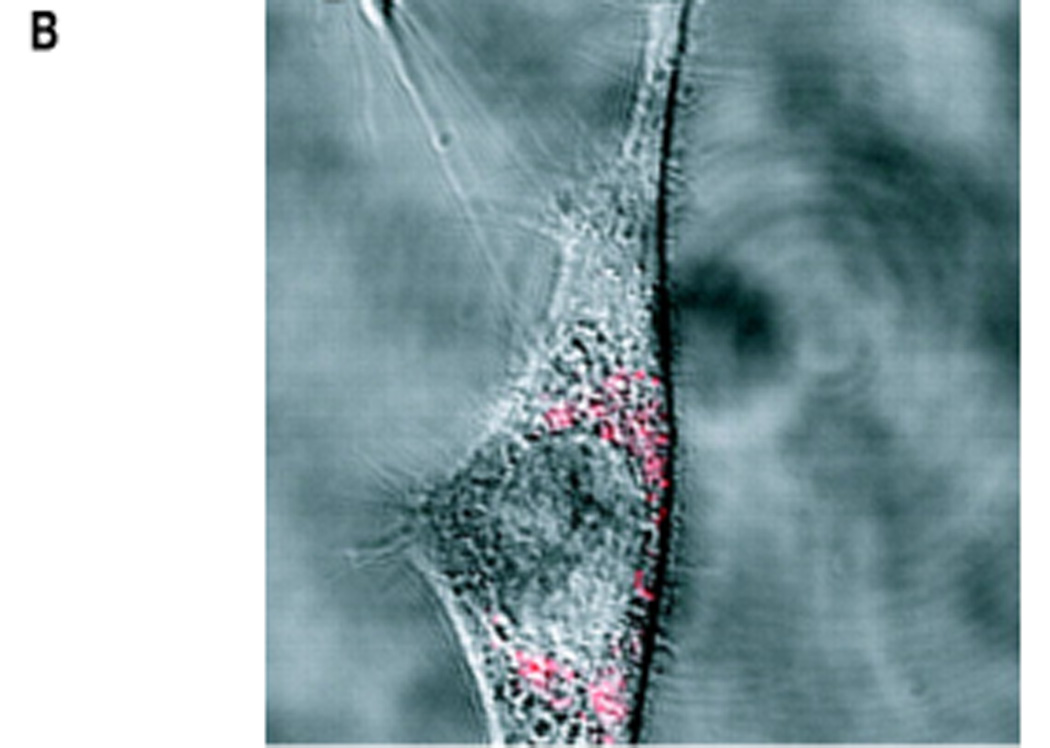
Switch-based sensors are finding increasing application in intracellular studies. (a) A maltose sensor based on the binding-induced switching of maltose-binding protein (a schematic of which is shown in Figure 1a) supports rapid, intracellular measurements of the sugar. The optical reporter groups are two florescent protein variants that are fused to the switching domain and that serve as a RET pair. As all of the components of this system are contained within a single polypeptide chain, this sensor can be expressed in situ. Reproduced with permission from Ref. [27]. (b) Molecular beacons (schematic shown in Figure 1b), which support rapid fluorescence detection of specific oligonucleotide sequences, have been employed to localize and quantify endogenous mRNA levels within living cells, including the detection of mRNA encoding the protein survivin in live human dermal fibroblast cells. In this example, the molecular beacon is delivered into the cell by coupling it with the TAT polypeptide, a signaling sequence that leads to rapid and efficient internalization.
A key advantage that improves the utility of these approaches in vivo is that FRET is a ratiometric measurement (Figure 2a). That is, the concentration of the target molecule is determined by the ratio of the fluorescence emitted by the FRET donor fluorophore to that emitted by the FRET acceptor fluorophore, thus largely obviating the need for precise knowledge of the concentration of the reporting biomolecule (see, however, Ref. [31] for various technical considerations). This approach is therefore quantitative, despite cell-to-cell variability in switch delivery, degradation or expression. Nevertheless, optical read-out mechanisms suffer from significant drawbacks for applications in multi-cellular organisms: the strong optical background arising from the scattering and absorbance of blood and tissues significantly limits the utility of optical approaches for real-time monitoring within, for example, a human patient.
Electro-active contaminants are rare in vivo; therefore, electrochemical methods for monitoring biomolecular switching often perform better than their optical counterparts when challenged in complex environments [32]. Indeed, other than the biosensors described above, the only real-time, in vivo molecular sensing technologies in use in living animals are electrochemical in nature, involving either ion-specific electrodes or relatively abundant, redox-active targets (e.g. the detection of dopamine [33] and nitric oxide [34] in the brain). It has been shown that E-AB sensors perform well, even when challenged directly in whole blood [35]. Moreover, despite employing a potentially labile DNA probe as the recognition element, these sensors are quite stable under such conditions; when stored directly in room-temperature blood serum for periods of more than a week, only minor signal degradation has been noted [36]. Finally, electrochemical sensors can be made quite small (i.e. this class of sensors requires only micron-scale electrodes), rendering them suitable for both intravenous and interstitial applications.
Microfluidic EA-B sensor
Consistent with the attributes described above, EA-B sensors have been used for the continuous, real-time monitoring of molecular analytes in unmodified, undiluted biological samples, at least in vitro (Figure 3). Specifically, a microfluidic EA-B sensor (MECAS) has been described in which a target-specific DNA aptamer folds in response to the target analyte (cocaine) (Figure 1c), thereby generating an electrochemical signal [37]. The MECAS chip (Figure 3a), which incorporates gold working electrodes, a reference electrode and a platinum counter-electrode within a sub-microliter detection chamber, responds sensitively and selectively to its target analyte at physiological concentrations (Figure 3b): when challenged with cocaine at micromolar concentrations in serum, the current increases significantly. The chip operates continuously and is also rapid, exhibiting an equilibration time constant of just 90 s. The MECAS chip exhibits baseline stability in continuously flowing blood serum, and the signaling current of the device rapidly returns to baseline values when the flow of cocaine-doped serum is replaced with cocaine-free serum (Figure 3b), and these baselines remain constant even after 60 min of exposure to nucleases in the flowing serum that might be expected to degrade the sensing aptamer. The unexpectedly high stability of the sensing aptamer might be a result of its surface immobilization and its terminal modification with a redox reporter [38].
Figure 3.

An E-AB sensor for the detection of cocaine (schematic shown in Figure 1c) supports real-time monitoring of cocaine directly in undiluted blood serum as it flows through a sub-microliter microfluidic chamber. (a) The MECAS chip incorporates three electrodes: “R” is the reference electrode, which provides a standard by which voltages can be accurately measured; “W” is the “working electrode”, which interrogates the attached aptamer and is the heart of the biosensor; “C” is the counter-electrode, which completes the circuit. (b) The system continuously monitors the presence of cocaine. When challenged with cocaine in serum (10, 50 and 100 µM), the current (Ip) increases before dropping back to baseline after washing with cocaine-free serum. Figure adapted with permission from Ref. [37].
Perspectives
Of course, detection in flowing blood serum in vitro is not the same as detection in whole blood in vivo. Indeed, to date, there have been no reports of continuous, in vivo detection with any electrochemical, switch-based sensor. Some of the hurdles that stand in the way of achieving this goal are well-known [4, 32] and briefly include biocompatibility, stability and calibration issues. Biofouling can inhibit transport of the target analyte, even if it does not alter the responsiveness of the sensor [32, 39]. Likewise, the calibration of in vivo devices against baseline drift represents a serious challenge [32]; although, in theory, ratiometric measurements analogous to those made with in vivo FRET-based sensors [31] could be performed using control sensors that do not respond to the target analyte, this approach has not been explored for electrochemical sensors to the best of our knowledge.
Thus, quite simply, we are not there yet: there is no universal approach by which an arbitrary molecular analyte can be monitored continuously and in real-time in the body. Nevertheless, we believe that the reagentless, regenerable nature of switch-based biosensors and the selectivity of electrochemical detection offer a promising route towards this goal. Time – and further experimentation – will tell.
Acknowledgments
The authors wish to thank the members of their research groups for their contributions to these ideas and for their editorial suggestions. The Soh and Plaxco group efforts towards achieving the goals described here are funded by the NIH, especially through grants R01EB007689 and R01EB002046.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lisowski-Myjak B. Serum and urinary biomarkers of acute kidney injury. Blood Purification. 2010;29:357–365. doi: 10.1159/000309421. [DOI] [PubMed] [Google Scholar]
- 2.Brown L, Edelman ER. Optimal control of blood glucose: the diabetic patient or the machine? Sci. Trans. Med. 2010;2 doi: 10.1126/scitranslmed.3001083. 27ps18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Turner APF. Chemical sensors for in vivo monitoring; Advances in biomedical engineering: results of the 4th EC Medical and Health Conference; Amsterdam V: IOS Press; 1993. pp. 99–120. [Google Scholar]
- 4.Frost MC, Meyerhoff ME. Implantable chemical sensors for real-time clinical monitoring: progress and challenges. Curr. Opin. Chem. Biol. 2002;6:633–641. doi: 10.1016/s1367-5931(02)00371-x. [DOI] [PubMed] [Google Scholar]
- 5.Hollywood K, Brison DR, Goodacre R. Metabolomics: Current technologies and future trends. Proteomics. 2006;6:4716–4723. doi: 10.1002/pmic.200600106. [DOI] [PubMed] [Google Scholar]
- 6.De Leon-Rodriguez LM, Lubag AJM, Malloy CR, Martinez GV, Gillies RJ, Sherry AD. Responsive MRI agents for sensing metabolism in vivo. Acc. Chem. Research. 2009;42:948–957. doi: 10.1021/ar800237f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bai C, Graham TL, Arnold MA. Assessing and advancing technology for the noninvasive measurement of clinical glucose. Analytical Letters. 2008;41:2773–2793. [Google Scholar]
- 8.Wolpert HA. Continuous glucose monitoring - Coming of age. New England Journal of Medicine. 2010;363:383–384. doi: 10.1056/NEJMe1006098. [DOI] [PubMed] [Google Scholar]
- 9.Baker DA, Gough A. Continuous, implantable lactate sensor. Anal. Chem. 1995;67:1536–1540. [Google Scholar]
- 10.Wilson GS, Hu YB. Enzyme-based biosensors for in vivo measurements. Chem. Rev. 2009;100:2693–2704. doi: 10.1021/cr990003y. [DOI] [PubMed] [Google Scholar]
- 11.Yoneyama Y, Yonemore Y, Murata M, Ohnuki H, Hibi K, Hayashi T, Ren HF, Endo H. Wireless biosensor system for real-time cholesterol monitoring in fish ‘Nile tilapia’. Talanta. 2009;80:909–915. doi: 10.1016/j.talanta.2009.08.014. [DOI] [PubMed] [Google Scholar]
- 12.Boozer C, Kim G, Cong SX, Guan HW, Londergan T. Looking towards label-free biomolecular interaction analysis in a high-throughput format: a review of new surface plasmon resonance technologies. Curr. Op. Biotech. 2006;17:400–405. doi: 10.1016/j.copbio.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 13.Tang HY, Ali-Khan N, Echan LA, Levenkova N, Rux JJ, Speicher DW. A novel four-dimensional strategy combining protein and peptide separation methods enables detection of low-abundance proteins in human plasma and serum proteomes. Proteomics. 2005;8:3329–3342. doi: 10.1002/pmic.200401275. [DOI] [PubMed] [Google Scholar]
- 14.Vallée-Bélisle A, Plaxco KW. Structure-switching biosensors: inspired by nature. Curr. Op. Struc. Biol. 2010;20:518–526. doi: 10.1016/j.sbi.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Medintz IL, Deschamps JR. Maltose-binding protein: a versatile platform for prototyping biosensing. Curr Opin Biotechnol. 2006;17:17–27. doi: 10.1016/j.copbio.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 16.Okumoto S, Takanaga H, Frommer WB. Quantitative imaging for discovery and assembly of the metabo-regulome. New Phytologist. 2008;180:271–295. doi: 10.1111/j.1469-8137.2008.02611.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oh KJ, Cash KJ, Plaxco KW. Beyond molecular beacons: optical sensors based on the binding-induced folding of proteins and polypeptides. Chem. Europ. J. 2009;15:2244–2251. doi: 10.1002/chem.200701748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kohn JE, Plaxco KW. Engineering of a signal-transduction mechanism for protein-based biosensors. Proc. Natl. Acad. Sci. USA. 2005;102:10841–10845. doi: 10.1073/pnas.0503055102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stratton MM, Metrea DM, Loh SN. A Ca2+-sensing molecular switch based on alternate frame protein folding. ACS Chem. Biol. 2008;3:723–732. doi: 10.1021/cb800177f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xiao Y, Plaxco KW. Electrochemical aptamer sensors. In: Lu Y, Li Y, editors. Functional nucleic acids for sensing and other analytical applications. New York: Springer; 2009. pp. 179–198. [Google Scholar]
- 21.Li D, Song S, Fan CH. Target-responsive structural switching for nucleic acid-based sensors. Acc Chem. Res. 2010;43:631–641. doi: 10.1021/ar900245u. [DOI] [PubMed] [Google Scholar]
- 22.De Lorimier RM, Tian YJ, Hellinga HW. Binding and signaling of surface-immobilized reagentless fluorescent biosensors derived from periplasmic binding proteins. Prot. Sci. 2006;15:1936–1944. doi: 10.1110/ps.062261606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lubin AA, Plaxco KW. Folding-based electrochemical biosensors: the case for responsive nucleic acid architectures. Acc. Chem. Res. 2010;43:496–505. doi: 10.1021/ar900165x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fan C, Plaxco KW, Heeger AJ. Electrochemical interrogation of conformational changes as a reagentless method for the sequence-specific detection of DNA. Proc. Natl. Acad. Sci. USA. 2003;100:9134–9137. doi: 10.1073/pnas.1633515100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gerasimov JY, Lai RY. An electrochemical peptide-based biosensing platform for HIV detection. Chem. Comm. 2010;46:395–397. doi: 10.1039/b919070h. [DOI] [PubMed] [Google Scholar]
- 26.Benson DE, Conrad DW, de Lorimer RM, Trammell SA, Hellinga HW. Design of bioelectronic interfaces by exploiting hinge-bending motions in proteins. Science. 2001;293:1641–1644. doi: 10.1126/science.1062461. [DOI] [PubMed] [Google Scholar]
- 27.Fehr M, Frommer WB, Lalonde S. Visualization of maltose uptake in living yeast cells by fluorescent nanosensors. Proc Natl Acad Sci USA. 2002;99:9846–9851. doi: 10.1073/pnas.142089199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Palmer AE, Tsien RY. Measuring calcium signaling using genetically targetable fluorescent indicators. Nat Protoc. 2006;1:1057–1065. doi: 10.1038/nprot.2006.172. [DOI] [PubMed] [Google Scholar]
- 29.Nitin N, Santangelo PJ, Kim G, Nie S, Bao G. Peptide-linked molecular beacons for efficient delivery and rapid mRNA detection in living cells. Nuc. Acid Res. 2004;32:e58. doi: 10.1093/nar/gnh063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Modi S, Swetha MG, Goswami D, Gupta GD, Mayor S, Krishnan Y. A DNA nanomachine that maps spatial and temporal pH changes inside living cells. Nature Nanotechnology. 2009;4:325–330. doi: 10.1038/nnano.2009.83. [DOI] [PubMed] [Google Scholar]
- 31.Lalonde S, Ehrhardt DW, Loque D, Chen J, Rhee SY, Frommer WB. Molecular and cellular approaches for the detection of protein-protein interactions: latest techniques and current limitations. Plant J. 2008;53:610–635. doi: 10.1111/j.1365-313X.2007.03332.x. [DOI] [PubMed] [Google Scholar]
- 32.Wilson GS, Gifford R. Biosensors for real-time in vivo measurements. Biosens. Bioelc. 2005;20:2388–2403. doi: 10.1016/j.bios.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 33.Robinson DL, Herman A, Seipel AT, Wightman RM. Monitoring rapid chemical communication in the brain. Chem. Rev. 2008;108:2554–2584. doi: 10.1021/cr068081q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barbosa RM, Lourenco CF, Santos RM, Pomerleau F, Huettl P, Gerhardt GA, Laranjinha J. Nitric Oxide, Pt. G: Oxidative and Nitrosative Stress in Redox Regulation of Cell Signaling, Book Series: Methods in Enzymology. Vol. 441. 2008. pp. 351–367. [DOI] [PubMed] [Google Scholar]
- 35.White RJ, Plaxco KW. Engineering new aptamer geometries for electrochemical aptamer-based sensors. In: Fell NF, Swaminathan VS, editors. Proc. SPIE Bio-Inspired/Biomimetic Sensor Technologies and Applications. Vol. 7321. 2009. pp. 5.1–5.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lai RY, Seferos DS, Heeger AJ, Bazan GC, Plaxco KW. Comparison of the signaling and stability of electrochemical DNA sensors fabricated from 6- or 11-carbon self-assembled monolayers. Langmuir. 2006;22:10796–10800. doi: 10.1021/la0611817. [DOI] [PubMed] [Google Scholar]
- 37.Swenson JS, Xiao Y, Ferguson BS, Lai RY, Heeger AJ, Plaxco KW, Soh T. Continuous, real-time monitoring of cocaine in undiluted, unmodified blood serum via a microfluidic, aptamer-based sensor. J. Am. Chem. Soc. 2009;131:4262–4266. doi: 10.1021/ja806531z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Truong VL, Walsh SM, Schweibert E, Mao HQ, Guggino WB, August JT, Leong KW. Gene transfer by DNA-gelatin nanospheres. Arch. Biochem. Biophys. 1999;361:47–56. doi: 10.1006/abbi.1998.0975. [DOI] [PubMed] [Google Scholar]
- 39.Wisniewski N, Moussy F, Reichert WM. Characterization of implantable biosensor membrane biofouling. Fresenius J. Anal. Chem. 2000;366:611–621. doi: 10.1007/s002160051556. [DOI] [PubMed] [Google Scholar]
- 40.Tyagi S, Kramer FR. Molecular beacons: Probes that fluoresce upon hybridization. Nat. Biotechnol. 1996;14:303–308. doi: 10.1038/nbt0396-303. [DOI] [PubMed] [Google Scholar]