

Abstract
ADP-ribosylation is an important post-translational modification involved in processes including cellular replication, DNA repair, and cell death. Despite these roles, the functions of ADP-ribosylation, in particular mono-ADP-ribosylation, remain poorly understood. The development of a technique to generate large amounts of site-specific, ADP-ribosylated peptides would provide a useful tool for deconvoluting the biochemical roles of ADP-ribosylation. Here we demonstrate that synthetic histone H2B tail peptides, incorporating aminooxy or N-methyl aminooxy functionalized amino acids, can be site-specifically conjugated to ADP-ribose. These peptides are recognized as substrates by the ADP-ribosylation biochemical machinery (PARP1), can interact with the ADP-ribose binding proteins macroH2A1.1 and PARP9, and demonstrate superior enzymatic and chemical stability when compared to ester-linked ADP-ribose. In addition, the incorporation of benzophenone photocrosslinkers into these peptides is demonstrated to provide a means to probe for, and enrich ADP-ribose binding proteins.
ADP-ribosylation involves the transfer of ADP-ribose (ADPR) from β-NAD+ onto protein substrates.1 A family of enzymes termed poly(ADP-ribose) polymerases (PARPs), which generate protein-linked ADPR monomers (mono-ADP-ribosylation) or polymers (poly-ADP-ribosylation), catalyze this reaction. ADP-ribosylation occurs on several residues including glutamic acid where the attachment is through an ester linkage.1 The functional roles of ADP-ribosylation, in particular mono-ADP-ribosylation, are poorly understood. The study of ADP-ribosylation has proven difficult for many reasons. For example, the ester-linked ADPR generated by PARPs is unstable at basic pH (t1/2 < 1 h pH 7.51b), and endogenous enzymes rapidly cleave poly-ADP-ribose polymers (e.g. poly(ADP-ribose) glycohydrolase; t1/2 0.6-6 min1b). There is also a lack of commercial antibodies specific for mono-ADP-ribose, mono-ADP-ribose protein-linkages, or poly-ADP-ribose branching.1 The ability to site-specifically attach ADPR to peptides/proteins would provide a useful tool for studying the biochemical effects of this modification. Such a technique, using a protected mono-ADP-ribosylated asparagine building block for peptide synthesis has recently been described.2 In this report, we demonstrate the use of aminooxy-functionalized amino acids to enable site-specific attachment of ADPR onto peptides. Using this technology, we show that an ADP-ribosylated version of the histone H2B tail can interact with the ADPR binding protein, macroH2A1.1 (mH2A1.1), and demonstrate that incorporation of photocrosslinkers into these peptides improves their ability to detect ADPR binding proteins.
We set out to develop a general strategy for the synthesis of mono-ADPR peptide-conjugates. In principle, a chemoselective ligation approach would allow precise control over the ADPR conjugation site using readily accessible building blocks. This would enable large amounts of ADPR conjugated peptides to be synthesized for biochemical studies. Further, the chemical approach would allow the incorporation of linkers featuring improved chemical and enzymatic stability, when compared to enzymatic ADP-ribosylation. Uses for such a method would include the investigation of protein interactions, the potential to generate site-specific poly-ADP-ribosylated peptides through a templated initiator ADPR residue,3 and the generation of ADPR conjugated proteins using expressed protein ligation.4
Under physiological conditions, ADPR (Scheme 1A) reacts with lysine and arginine side-chains to form imine-linked species, which degrade to give a mixture of products.5 While this process does not provide a route to stable, site-specific ADPR-peptide conjugates, it suggested to us that oxime ligation might prove suitable for this purpose. Oxime ligation is performed at ∼ pH 4.5, a pH at which peptide lysine and arginine residues are protonated and unreactive. In addition, oxime ligation is compatible with all natural amino acid functional groups, and the oxime linkage is significantly more stable to hydrolysis compared to imines or the native ADPR ester linkage.6 Ligation of reducing-carbohydrates through an oxime, using an aminooxy group, generates mainly ring-opened carbohydrates, with a small amount of the ring-closed form (Scheme 1B).7 Conversion of the aminooxy group to a secondary alkoxyamine, through N-methylation, results in the attached carbohydrates adopting a ring-closed form exclusively (Scheme 1C).7b Based on these observations, we decided to investigate if aminooxy and N-methyl aminooxy functionalized peptides could undergo site-specific ligation reactions with ADPR.
Scheme 1.

The structure of (A) ADPR and its ligation to (B) aminooxy and (C) N-methyl aminooxy functionalized peptides.
Histone H2B was selected as a model for ADPR conjugation as it has been reported to be mono-ADP-ribosylated on glutamate residue 2 in rat liver.8 In preliminary studies, we found this modification can also occur on glutamate-2 of human H2B in vitro using recombinant PARP10 (Figure S1), which has been shown to mono-ADP-ribosylate histones.9 Moreover, we demonstrated that human H2B is a major acceptor of ADP-ribosylation in HeLa cell nuclei (Figure S2).
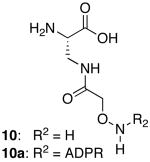
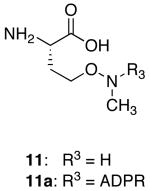
A series of peptides corresponding to the N-terminus of human histone H2B were synthesized on solid-phase (1-8; Table 1). Several of these peptides contained suitably protected versions of aminooxy-containing building block 1010 or N-methyl aminooxy-containing amino acid 1111 substituted for glutamic acid residue 2. Peptides incorporating 10 or 11 were ligated to ADPR in 0.5 M sodium acetate (∼ pH 4.5) to generate mimetics of the mono-ADP-ribosylated H2B amino-terminal tail. Ligation reactions to peptides incorporating 10 were rapid, completing in less than 1 h at 30 °C using 1.2-10 eq. of ADPR (Figure 1A). Ligation to 3, incorporating 11, was slow, required a larger excess of ADPR (≥ 10 eq), and never reached completion (< 40 % by 3 days; Figure 1B). The amount of 3a obtained after purification was not sufficient for in depth biochemical studies. Attempts to improve the reaction rate and yield of 3a through the addition of aniline12 (0.1-100 mM) to ligations were unsuccessful, leading to the formation of multiple ADP-ribose associated products (Figure S3A). Aniline, however, accelerated d-glucose ligation to 3 by 7-fold (Figure S3B), demonstrating that this molecule can accelerate the ligation of carbohydrates to N-methyl aminooxy groups. The site-specific ligation to peptides incorporating 10 was confirmed by analyzing tryptic digests of 2a (Figure S4). Ligation through the ADPR distal ribose was established by treating 9a with snake venom phosphodiesterase and calf intestinal alkaline phosphatase, which left a species identical in mass to the peptide conjugated to the ADPR distal ribose (Figure S5). The E and Z oxime-linked species were observed by NMR in a 2:1 ratio (Figure S6). Peptide 6a was assessed for stability to a variety of conditions (pH, nucleophiles, lysates, enzymes), with the oxime ADPR linkage found to be significantly more stable to conditions known to rapidly cleave the native ADPR-protein ester linkage (Figure S7).8a,9a
Table 1.
Synthetic peptide library.
| Compound | AA1 | AA2 | AA3 | Peptidea | R1 | AA4 |
|---|---|---|---|---|---|---|
| 1 | - | Pro | Glu | H2B(3-19) | Biotin | - |
| 2 | - | Pro | 10 | H2B(3-19) | Biotin | - |
| 2a | - | Pro | 10a | H2B(3-19) | Biotin | - |
| 3 | - | Pro | 11 | H2B(3-19) | Biotin | - |
| 3a | - | Pro | 11a | H2B(3-19) | Biotin | - |
| 4 | - | Pro | Glu | H2B(3-19) | - | - |
| 5 | - | Pro | Ala | H2B(3-19) | - | - |
| 6 | - | Pro | 10 | H2B(3-19) | - | - |
| 6a | - | Pro | 10a | H2B(3-19) | - | - |
| 7 | 12 | Pro | Glu | H2B(3-19) | Biotin | - |
| 8 | 12 | Pro | 10 | H2B(3-19) | Biotin | - |
| 8a | 12 | Pro | 10a | H2B(3-19) | Biotin | - |
| 9 | 10 | Gly | Gly | - | Biotin | Gly |
| 9a | 10a | Gly | Gly | - | Biotin | Gly |
 |
 |
 |
||||
H2B(3-19) Sequence: PAK SAPAP KKGSK KAVT
Figure 1.

Ligation of ADPR to (a) 2 or (b) 3, with representative RP-HPLC and ESI-MS data for starting peptides and their respective products (2a and 3a) at specified times. The products absorb at 259 nm due to the presence of the ADPR associated adenine.
We were keen to assess whether an ADPR-peptide conjugate could be used to nucleate the site-specific generation of poly-ADP-ribosylated peptides using recombinant PARPs and β-NAD+. Indeed, we found that PARP1 can transfer ADP-ribose on to ADPR-containing peptide 6a, but not on to control peptides 4 or 5 which lack the ADPR template (Figure S8). Thus, site-specific poly-ADP-ribose peptide conjugates can be accessed using this chemoenzymatic strategy.
Next we sought to demonstrate that the peptide ADPR conjugates could interact with a known ADPR binding protein. Several so-called macro domains have been shown to bind ADPR,13 and it is postulated that ADP-ribosylated proteins may also interact with these domains.13b,14 Indeed, a recent report suggests that the macro domain containing histone, mH2A1.1, can bind poly-ADP-ribosylated PARP1.14c Since histones H2B and mH2A1.1 exist in the same nuclear environment (i.e. within nucleosome core particles of chromatin), we asked if mH2A1.1 could bind the synthetic ADPR conjugated peptides using a peptide pull-down assay. A splice-variant, mH2A1.2, which does not bind ADPR,13c was used as a negative control in this experiment. Peptides 1, 2a and 9a were assessed for their ability to interact with mH2A1.1 and mH2A1.2 (Figure 2). ADPR-peptide conjugates 2a and 9a were able to pull-down mH2A1.1, but not mH2A1.2. Control peptide 1 did not interact with mH2A1.1, suggesting that the ADPR moiety is important for the interaction. Consistent with this, peptide 2a did not pull-down with a point-mutant of mH2A1.1 (G224E) (Figure S9) which abolishes ADPR binding.13c Interestingly, peptide 2a pulls down more mH2A1.1 than peptide 9a, suggesting that the H2B peptide sequence in 2a also contributes to the interaction. Additional studies will be needed to confirm this. The key result here is that the ADPR-peptide conjugates afforded by our strategy are able to interact with the macro domain. To our knowledge, this is the first demonstration that an ADPR binding domain can engage the modification when it is covalently linked to a peptide carrier.
Figure 2.
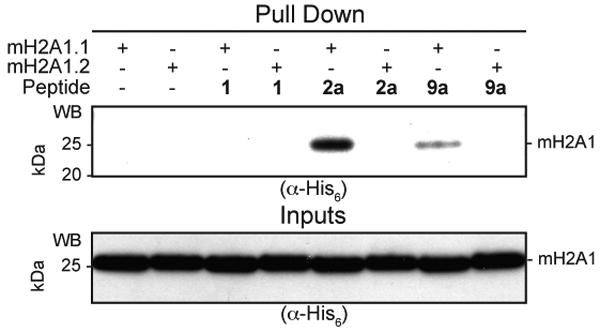
Binding of synthetic biotinylated peptides to mH2A1.1. Indicated peptides were incubated with either mH2A1.1 or, as a control, mH2A1.2. Peptides were then immobilized using streptavidin-beads and captured complexes analyzed by SDS-PAGE followed by western blotting against the His6-tag on the protein. Equal loading of peptides was shown by RP-HPLC.
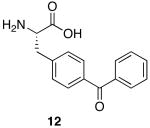
We imagined that our ADPR-peptide conjugates could be useful in the identification of novel ADPR-binding proteins from crude cellular lysates. The affinity of free ADPR for mH2A1.1 is modest, in the μM range.13c Thus, we were concerned that affinity-capture methods might not be fruitful. Indeed, peptide 2a was unable to pull-down low concentrations of mH2A1.1 doped into HeLa S3 nuclear lysates (Figure 3, lane 6). One approach to overcome this problem would be to incorporate a photocrosslinker into the conjugate, thereby covalently linking the probe to the target.15 With this in mind, we synthesized ADPR-peptide conjugate 8a featuring benzophenone crosslinker 12 appended to the N-terminus of the H2B sequence (Table 1). We also synthesized peptide 7, which contains the crosslinker but lacks the ADPR moiety. Incubation of peptide 8a, but not 7, with HeLa S3 nuclear lysates doped with mH2A1.1 followed by UV irradiation, resulted in the generation of a robust crosslink to the macro domain (Figure 3). This effect was observed at two macro domain concentrations, differing by 5-fold. At the higher concentration, peptide 8a was able to pull down both crosslinked mH2A1.1 and non-crosslinked mH2A1.1 (upper and lower bands, respectively, in the α-His blot of the precipitated material, Figure 3). Since the mH2A1.1 macro domain can form dimers,16 this data suggests that the crosslinked species may co-precipitate interacting proteins in their native state. In addition, PARP9, a known ADP-ribose binding protein,13b could be enriched from Farage nuclear lysates after crosslinking with peptide 8a, but not with 7 (Figure S10). Based on these observations, incorporation of photocrosslinkers and ADPR into peptides could be useful for enriching, or probing for ADPR binding proteins.
Figure 3.
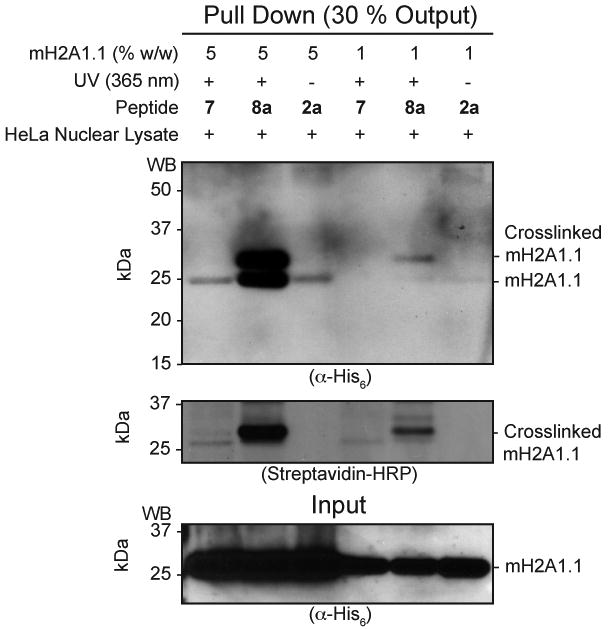
Photocrosslinking studies. Indicated peptides were incubated with HeLa cell nuclear lysates doped with mH2A1.1 at two concentrations. Mixtures were then irradiated with UV light (or not in the case of peptide 2a) and then immobilized using streptavidin-beads. Captured complexes were analyzed by SDS-PAGE followed by western blotting against mH2A1.1 (α-His6) or the peptide (streptavidin-HRP).
Research into ADP-ribosylation has proven difficult due to the paucity of tools available for studying this modification. Several β-NAD+-based chemical probes have been described.17 These analogues, if recognized by ADP-ribosylating enzymes, offer the potential to detect and enrich ADP-ribosylated proteins. The sites of modification, however, depend on the enzyme used, and unstable ADPR-protein linkages may be generated. In contrast, the methodology described in this study enables multi-milligram amounts of highly pure, stable and site-specific ADPR peptide-conjugates to be generated. The ADPR peptide-conjugates can bind to the mH2A1.1 and PARP9 macro domains, the first demonstration that a macro domain can engage ADPR when linked to a peptide. Moreover, the ADPR peptide-conjugates could be poly-ADP-ribosylated by PARP1, and incorporation of photocrosslinkers allowed the capture of ADPR-binding proteins from lysates. Consequently, this technique may be useful for identifying and enriching ADPR binding proteins, and may enable the generation of site-specifically poly-ADP-ribosylated peptides and proteins for biochemical assays.
Supplementary Material
Acknowledgments
This work was funded by the US National Institutes of Health (GM086868 & RC2CA148354). P.M.M. was supported by the American Australian Association and the Australian National Health and Medical Research Council. We thank C.D. Allis, H. Kleine, X. Li, S. Lockless, B. Lüscher, R. McGinty, M. Pratt, A. Ruthenburg, R. Subramanian, J. Weinger and members of the Muir laboratory.
Footnotes
Supporting Information Available: Experimental procedures, characterization data, complete ref 9b, and supporting figures. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Hottiger MO, Hassa PO, Luscher B, Schuler H, Koch-Nolte F. Trends Biochem Sci. 2010;35:208–219. doi: 10.1016/j.tibs.2009.12.003. [DOI] [PubMed] [Google Scholar]; (b) Hassa PO, Haenni SS, Elser M, Hottiger MO. Microbiol Mol Biol Rev. 2006;70:789–829. doi: 10.1128/MMBR.00040-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van der Heden van Noort GJ, van der Horst MG, Overkleeft HS, van der Marel GA, Filippov DV. J Am Chem Soc. 2010;132:5236–5240. doi: 10.1021/ja910940q. [DOI] [PubMed] [Google Scholar]
- 3.Ueda K, Kawaichi M, Okayama H, Hayaishi O. J Biol Chem. 1979;254:679–687. [PubMed] [Google Scholar]
- 4.Muir TW, Sondhi D, Cole PA. Proc Natl Acad Sci U S A. 1998;95:6705–6710. doi: 10.1073/pnas.95.12.6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cervantes-Laurean D, Jacobson EL, Jacobson MK. J Biol Chem. 1996;271:10461–10469. doi: 10.1074/jbc.271.18.10461. [DOI] [PubMed] [Google Scholar]
- 6.Kalia J, Raines RT. Angew Chem Int Ed Engl. 2008;47:7523–7526. doi: 10.1002/anie.200802651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Cervigni SE, Dumy P, Mutter M. Angew Chem Int Ed Engl. 1996;35:1230–1232. [Google Scholar]; (b) Peri F, Dumy P, Mutter M. Tetrahedron. 1998;54:12269–12278. [Google Scholar]
- 8.(a) Burzio LO, Riquelme PT, Koide SS. J Biol Chem. 1979;254:3029–3037. [PubMed] [Google Scholar]; (b) Ogata N, Ueda K, Hayaishi O. J Biol Chem. 1980;255:7610–7615. [PubMed] [Google Scholar]
- 9.(a) Kleine H, Poreba E, Lesniewicz K, Hassa PO, Hottiger MO, Litchfield DW, Shilton BH, Luscher B. Mol Cell. 2008;32:57–69. doi: 10.1016/j.molcel.2008.08.009. [DOI] [PubMed] [Google Scholar]; (b) Yu M, et al. Oncogene. 2005;24:1982–1993. doi: 10.1038/sj.onc.1208410. [DOI] [PubMed] [Google Scholar]
- 10.Wahl F, Mutter M. Tetrahedron Lett. 1996;37:6861–6864. [Google Scholar]
- 11.Carrasco MR, Brown RT. J Org Chem. 2003;68:8853–8858. doi: 10.1021/jo034984x. [DOI] [PubMed] [Google Scholar]
- 12.Dirksen A, Hackeng TM, Dawson PE. Angew Chem Int Ed Engl. 2006;45:7581–7584. doi: 10.1002/anie.200602877. [DOI] [PubMed] [Google Scholar]
- 13.(a) Egloff MP, Malet H, Putics A, Heinonen M, Dutartre H, Frangeul A, Gruez A, Campanacci V, Cambillau C, Ziebuhr J, Ahola T, Canard B. J Virol. 2006;80:8493–8502. doi: 10.1128/JVI.00713-06. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Karras GI, Kustatscher G, Buhecha HR, Allen MD, Pugieux C, Sait F, Bycroft M, Ladurner AG. EMBO J. 2005;24:1911–1920. doi: 10.1038/sj.emboj.7600664. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kustatscher G, Hothorn M, Pugieux C, Scheffzek K, Ladurner AG. Nat Struct Mol Biol. 2005;12:624–625. doi: 10.1038/nsmb956. [DOI] [PubMed] [Google Scholar]
- 14.(a) Dani N, Stilla A, Marchegiani A, Tamburro A, Till S, Ladurner AG, Corda D, Di Girolamo M. Proc Natl Acad Sci U S A. 2009;106:4243–4248. doi: 10.1073/pnas.0900066106. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Till S, Ladurner AG. Front Biosci. 2009;14:3246–3258. doi: 10.2741/3448. [DOI] [PubMed] [Google Scholar]; (c) Timinszky G, Till S, Hassa PO, Hothorn M, Kustatscher G, Nijmeijer B, Colombelli J, Altmeyer M, Stelzer EHK, Scheffzek K, Hottiger MO, Ladurner AG. Nat Struct Mol Biol. 2009;16:923–U941. doi: 10.1038/nsmb.1664. [DOI] [PubMed] [Google Scholar]
- 15.Kauer JC, Erickson-Viitanen S, Wolfe HR, Degrado WF. J Biol Chem. 1986;261:10695–10700. [PubMed] [Google Scholar]
- 16.Vijay-Kumar S, Chandra N, Dharia C, Pehrson JR. Proteins. 1995;22:290–292. doi: 10.1002/prot.340220311. [DOI] [PubMed] [Google Scholar]
- 17.(a) Du JT, Jiang H, Lin HN. Biochemistry. 2009;48:2878–2890. doi: 10.1021/bi802093g. [DOI] [PubMed] [Google Scholar]; (b) Kim HT, Haley BE. J Biol Chem. 1990;265:3636–3641. [PubMed] [Google Scholar]; (c) Zhang J, Snyder SH. Biochemistry. 1993;32:2228–2233. doi: 10.1021/bi00060a014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.